
Metformin Actions on the Liver: Protection Mechanisms Emerging in Hepatocytes and Immune Cells against NASH-Related HCC


{kind=link}
{kind=link}
Abstract
:1. Introduction
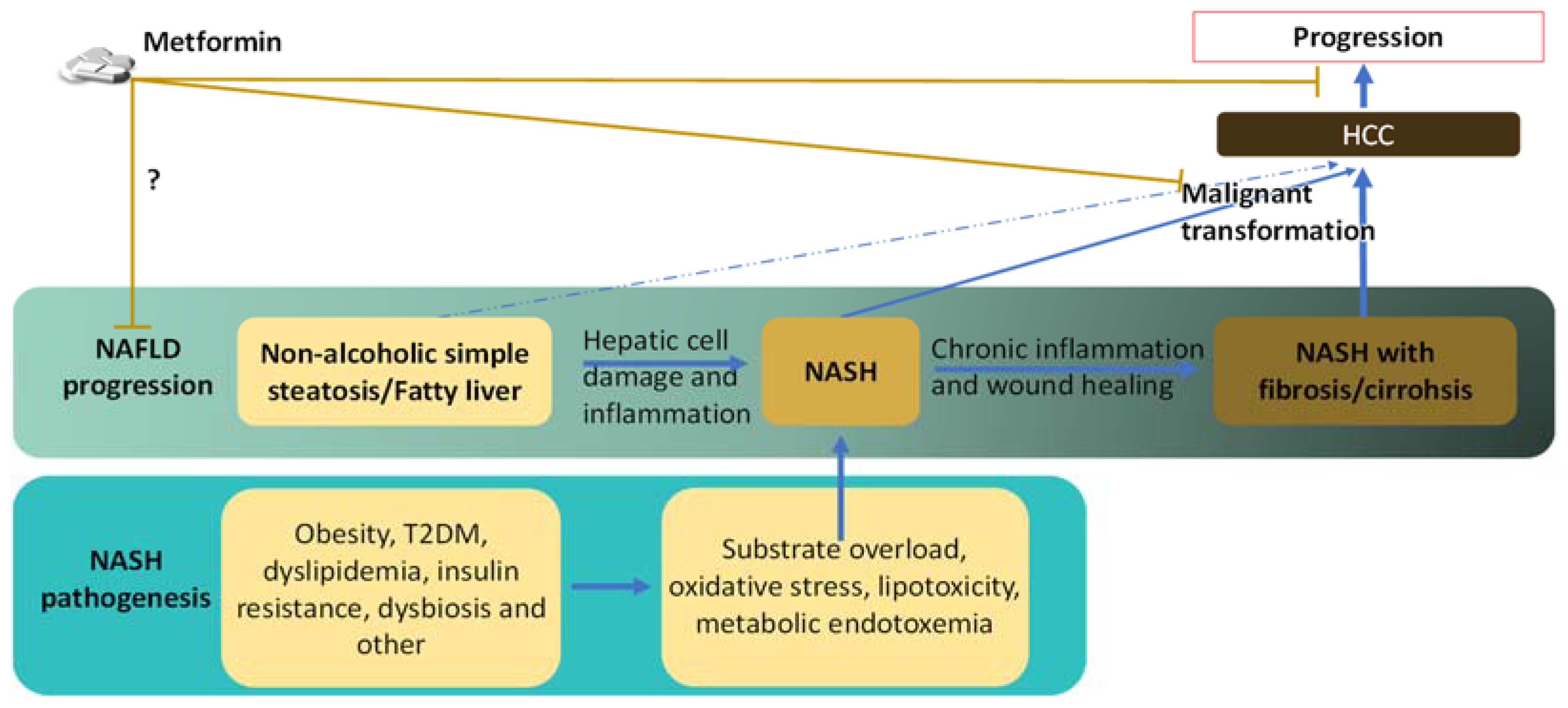
2. Reduced Risk and Progression of HCC in NAFLD/NASH Patients by Metformin
3. Links between NASH and HCC
3.1. Risk Factors and Preconditions That Link NASH to HCC
3.2. Links between the Pathological Abnormalities of Established NASH and the Development of HCC
3.2.1. DNA Damage
3.2.2. Hepatic Inflammation and Dysregulated Immunity
3.2.3. Cirrhosis
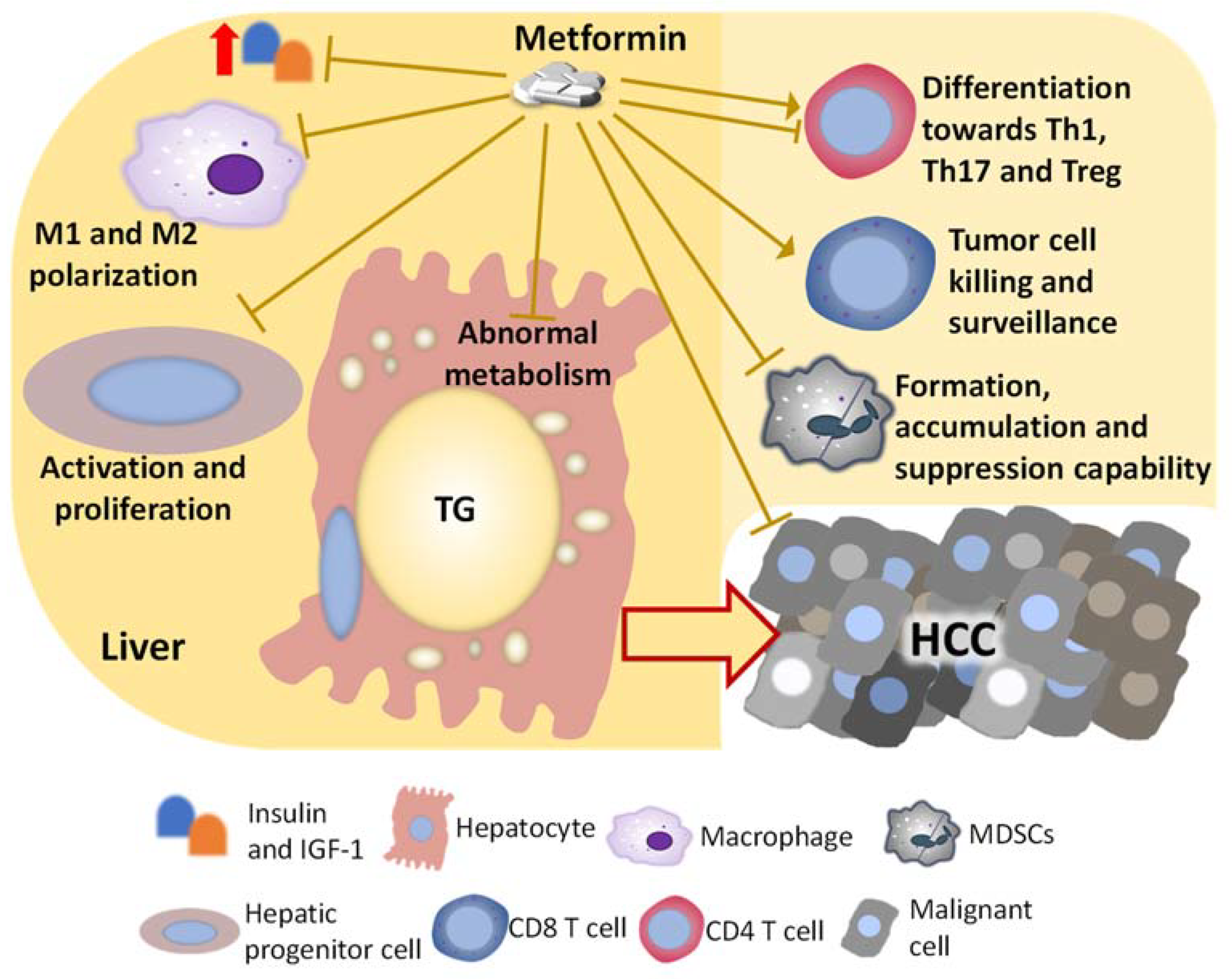
4. Direct Effects and Underlying Mechanisms of Metformin on Hepatocytes or Malignant Cells That May Inhibit NASH-Related HCC
4.1. Metformin and Substrate Overload in Hepatocyte
4.2. Metformin and AMPK Signaling
4.3. Metformin and Other Factors Regulated by AMP
4.4. Metformin and Oxidative Stress
4.5. Metformin and Hepatic Progenitor Cells
5. Metformin on the Immune Population That May Indirectly Inhibit NASH-Related HCC Development
5.1. Metformin on Macrophages
5.2. Metformin on MDSCs
5.3. Metformin on T Cells
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Singh, S.; Allen, A.M.; Wang, Z.; Prokop, L.J.; Murad, M.H.; Loomba, R. Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: A systematic review and meta-analysis of paired-biopsy studies. Clin. Gastroenterol. Hepatol. 2015, 13, 643–654.e9. [Google Scholar] [CrossRef] [Green Version]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Campigotto, M.; Giuffre, M.; Colombo, A.; Visintin, A.; Aversano, A.; Budel, M.; Masutti, F.; Abazia, C.; Croce, L.S. Comparison between hepatocellular carcinoma prognostic scores: A 10-year single-center experience and brief review of the current literature. World J. Hepatol. 2020, 12, 1239–1257. [Google Scholar] [CrossRef]
- Mittal, S.; El-Serag, H.B.; Sada, Y.H.; Kanwal, F.; Duan, Z.; Temple, S.; May, S.B.; Kramer, J.R.; Richardson, P.A.; Davila, J.A. Hepatocellular Carcinoma in the Absence of Cirrhosis in United States Veterans is Associated With Nonalcoholic Fatty Liver Disease. Clin. Gastroenterol. Hepatol. 2016, 14, 124–131.e1. [Google Scholar] [CrossRef] [Green Version]
- Henley, S.J.; Ward, E.M.; Scott, S.; Ma, J.; Anderson, R.N.; Firth, A.U.; Thomas, C.C.; Islami, F.; Weir, H.K.; Lewis, D.R.; et al. Annual report to the nation on the status of cancer, part I: National cancer statistics. Cancer 2020, 126, 2225–2249. [Google Scholar] [CrossRef] [PubMed]
- Brar, G.; Greten, T.F.; Graubard, B.I.; McNeel, T.S.; Petrick, J.L.; McGlynn, K.A.; Altekruse, S.F. Hepatocellular Carcinoma Survival by Etiology: A SEER-Medicare Database Analysis. Hepatol. Commun. 2020, 4, 1541–1551. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.A.; Majumdar, S.R.; Simpson, S.H.; Toth, E.L. Decreased mortality associated with the use of metformin compared with sulfonylurea monotherapy in type 2 diabetes. Diabetes Care 2002, 25, 2244–2248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, J.M.; Donnelly, L.A.; Emslie-Smith, A.M.; Alessi, D.R.; Morris, A.D. Metformin and reduced risk of cancer in diabetic patients. BMJ 2005, 330, 1304–1305. [Google Scholar] [CrossRef] [Green Version]
- Della Corte, C.M.; Ciaramella, V.; Di Mauro, C.; Castellone, M.D.; Papaccio, F.; Fasano, M.; Sasso, F.C.; Martinelli, E.; Troiani, T.; De Vita, F.; et al. Metformin increases antitumor activity of MEK inhibitors through GLI1 downregulation in LKB1 positive human NSCLC cancer cells. Oncotarget 2016, 7, 4265–4278. [Google Scholar] [CrossRef] [Green Version]
- Morgillo, F.; Fasano, M.; Della Corte, C.M.; Sasso, F.C.; Papaccio, F.; Viscardi, G.; Esposito, G.; Di Liello, R.; Normanno, N.; Capuano, A.; et al. Results of the safety run-in part of the METAL (METformin in Advanced Lung cancer) study: A multicentre, open-label phase I-II study of metformin with erlotinib in second-line therapy of patients with stage IV non-small-cell lung cancer. ESMO Open 2017, 2, e000132. [Google Scholar] [CrossRef] [Green Version]
- Zheng, L.; Yang, W.; Wu, F.; Wang, C.; Yu, L.; Tang, L.; Qiu, B.; Li, Y.; Guo, L.; Wu, M.; et al. Prognostic significance of AMPK activation and therapeutic effects of metformin in hepatocellular carcinoma. Clin. Cancer Res. 2013, 19, 5372–5380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, M.; Zhou, J.; Gorak, E.J.; Quddus, F. Metformin is associated with survival benefit in cancer patients with concurrent type 2 diabetes: A systematic review and meta-analysis. Oncologist 2013, 18, 1248–1255. [Google Scholar] [CrossRef] [Green Version]
- Cunha, V.; Cotrim, H.P.; Rocha, R.; Carvalho, K.; Lins-Kusterer, L. Metformin in the prevention of hepatocellular carcinoma in diabetic patients: A systematic review. Ann. Hepatol. 2020, 19, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Zheng, Y.; Xiao, Y.; Zhou, P.; Tan, H. Meta-analysis of studies using metformin as a reducer for liver cancer risk in diabetic patients. Medicine 2017, 96, e6888. [Google Scholar] [CrossRef]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef]
- Akshintala, D.; Chugh, R.; Amer, F.; Cusi, K. Nonalcoholic Fatty Liver Disease: The Overlooked Complication of Type 2 Diabetes. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dhatariya, K., Dungan, K., Grossman, A., Hershman, J.M., Hofland, J., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2019. [Google Scholar]
- Nkontchou, G.; Cosson, E.; Aout, M.; Mahmoudi, A.; Bourcier, V.; Charif, I.; Ganne-Carrie, N.; Grando-Lemaire, V.; Vicaut, E.; Trinchet, J.C.; et al. Impact of metformin on the prognosis of cirrhosis induced by viral hepatitis C in diabetic patients. J. Clin. Endocrinol. Metab. 2011, 96, 2601–2608. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, E.; Tokushige, K.; Ludwig, J. Diagnosis and classification of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis: Current concepts and remaining challenges. Hepatol. Res. 2015, 45, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Kasmari, A.J.; Welch, A.; Liu, G.; Leslie, D.; McGarrity, T.; Riley, T. Independent of Cirrhosis, Hepatocellular Carcinoma Risk Is Increased with Diabetes and Metabolic Syndrome. Am. J. Med. 2017, 130, 746.e1–746.e7. [Google Scholar] [CrossRef] [Green Version]
- Tseng, C.H. Metformin and risk of hepatocellular carcinoma in patients with type 2 diabetes. Liver Int. 2018, 38, 2018–2027. [Google Scholar] [CrossRef]
- Vilar-Gomez, E.; Vuppalanchi, R.; Desai, A.P.; Gawrieh, S.; Ghabril, M.; Saxena, R.; Cummings, O.W.; Chalasani, N. Long-term metformin use may improve clinical outcomes in diabetic patients with non-alcoholic steatohepatitis and bridging fibrosis or compensated cirrhosis. Aliment. Pharmacol. Ther. 2019, 50, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Vilar-Gomez, E.; Calzadilla-Bertot, L.; Wong, V.W.; Castellanos, M.; Aller-de la Fuente, R.; Eslam, M.; Wong, G.L.; George, J.; Romero-Gomez, M.; Adams, L.A. Type 2 Diabetes and Metformin Use Associate With Outcomes of Patients With Nonalcoholic Steatohepatitis-Related, Child-Pugh A Cirrhosis. Clin. Gastroenterol. Hepatol. 2021, 19, 136–145.e6. [Google Scholar] [CrossRef] [PubMed]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Masarone, M.; Rosato, V.; Aglitti, A.; Bucci, T.; Caruso, R.; Salvatore, T.; Sasso, F.C.; Tripodi, M.F.; Persico, M. Liver biopsy in type 2 diabetes mellitus: Steatohepatitis represents the sole feature of liver damage. PLoS ONE 2017, 12, e0178473. [Google Scholar] [CrossRef] [PubMed]
- Feingold, K.R. Obesity and Dyslipidemia. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dhatariya, K., Dungan, K., Grossman, A., Hershman, J.M., Hofland, J., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2018. [Google Scholar]
- Samuel, V.T.; Shulman, G.I. Nonalcoholic Fatty Liver Disease as a Nexus of Metabolic and Hepatic Diseases. Cell Metab. 2018, 27, 22–41. [Google Scholar] [CrossRef] [Green Version]
- Chaurasia, B.; Summers, S.A. Ceramides—Lipotoxic Inducers of Metabolic Disorders. Trends Endocrinol. Metab. 2015, 26, 538–550. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Ferre, P.; Foufelle, F. Hepatic steatosis: A role for de novo lipogenesis and the transcription factor SREBP-1c. Diabetes Obes. Metab. 2010, 12 (Suppl. 2), 83–92. [Google Scholar] [CrossRef] [PubMed]
- Mendez-Sanchez, N.; Cruz-Ramon, V.C.; Ramirez-Perez, O.L.; Hwang, J.P.; Barranco-Fragoso, B.; Cordova-Gallardo, J. New Aspects of Lipotoxicity in Nonalcoholic Steatohepatitis. Int. J. Mol. Sci. 2018, 19, 2034. [Google Scholar] [CrossRef] [Green Version]
- Mouzaki, M.; Wang, A.Y.; Bandsma, R.; Comelli, E.M.; Arendt, B.M.; Zhang, L.; Fung, S.; Fischer, S.E.; McGilvray, I.G.; Allard, J.P. Bile Acids and Dysbiosis in Non-Alcoholic Fatty Liver Disease. PLoS ONE 2016, 11, e0151829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yerushalmi, B.; Dahl, R.; Devereaux, M.W.; Gumpricht, E.; Sokol, R.J. Bile acid-induced rat hepatocyte apoptosis is inhibited by antioxidants and blockers of the mitochondrial permeability transition. Hepatology 2001, 33, 616–626. [Google Scholar] [CrossRef]
- Liu, B.; Zhang, Z.; Hu, Y.; Lu, Y.; Li, D.; Liu, J.; Liao, S.; Hu, M.; Wang, Y.; Zhang, D.; et al. Sustained ER stress promotes hyperglycemia by increasing glucagon action through the deubiquitinating enzyme USP14. Proc. Natl. Acad. Sci. USA 2019, 116, 21732–21738. [Google Scholar] [CrossRef] [Green Version]
- Morgantini, C.; Jager, J.; Li, X.; Levi, L.; Azzimato, V.; Sulen, A.; Barreby, E.; Xu, C.; Tencerova, M.; Naslund, E.; et al. Liver macrophages regulate systemic metabolism through non-inflammatory factors. Nat. Metab. 2019, 1, 445–459. [Google Scholar] [CrossRef] [PubMed]
- Azzimato, V.; Jager, J.; Chen, P.; Morgantini, C.; Levi, L.; Barreby, E.; Sulen, A.; Oses, C.; Willerbrords, J.; Xu, C.; et al. Liver macrophages inhibit the endogenous antioxidant response in obesity-associated insulin resistance. Sci. Transl. Med. 2020, 12, eaaw9709. [Google Scholar] [CrossRef] [PubMed]
- Frasca, F.; Pandini, G.; Scalia, P.; Sciacca, L.; Mineo, R.; Costantino, A.; Goldfine, I.D.; Belfiore, A.; Vigneri, R. Insulin receptor isoform A, a newly recognized, high-affinity insulin-like growth factor II receptor in fetal and cancer cells. Mol. Cell Biol. 1999, 19, 3278–3288. [Google Scholar] [CrossRef] [Green Version]
- Belfiore, A.; Malaguarnera, R.; Vella, V.; Lawrence, M.C.; Sciacca, L.; Frasca, F.; Morrione, A.; Vigneri, R. Insulin Receptor Isoforms in Physiology and Disease: An Updated View. Endocr. Rev. 2017, 38, 379–431. [Google Scholar] [CrossRef]
- Leung, K.C.; Doyle, N.; Ballesteros, M.; Waters, M.J.; Ho, K.K. Insulin regulation of human hepatic growth hormone receptors: Divergent effects on biosynthesis and surface translocation. J. Clin. Endocrinol. Metab. 2000, 85, 4712–4720. [Google Scholar] [CrossRef] [PubMed]
- Calle, E.E.; Kaaks, R. Overweight, obesity and cancer: Epidemiological evidence and proposed mechanisms. Nat. Rev. Cancer 2004, 4, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, X.; Gao, P. Diabetes Mellitus and Risk of Hepatocellular Carcinoma. Biomed. Res. Int. 2017, 2017, 5202684. [Google Scholar] [CrossRef] [Green Version]
- White, D.L.; Kanwal, F.; El-Serag, H.B. Association between nonalcoholic fatty liver disease and risk for hepatocellular cancer, based on systematic review. Clin. Gastroenterol. Hepatol. 2012, 10, 1342–1359.e2. [Google Scholar] [CrossRef] [Green Version]
- Saitta, C.; Pollicino, T.; Raimondo, G. Obesity and liver cancer. Ann. Hepatol. 2019, 18, 810–815. [Google Scholar] [CrossRef]
- El-Serag, H.B.; Hampel, H.; Javadi, F. The association between diabetes and hepatocellular carcinoma: A systematic review of epidemiologic evidence. Clin. Gastroenterol. Hepatol. 2006, 4, 369–380. [Google Scholar] [CrossRef]
- Tanaka, S.; Miyanishi, K.; Kobune, M.; Kawano, Y.; Hoki, T.; Kubo, T.; Hayashi, T.; Sato, T.; Sato, Y.; Takimoto, R.; et al. Increased hepatic oxidative DNA damage in patients with nonalcoholic steatohepatitis who develop hepatocellular carcinoma. J. Gastroenterol. 2013, 48, 1249–1258. [Google Scholar] [CrossRef] [PubMed]
- Raghunath, A.; Sundarraj, K.; Arfuso, F.; Sethi, G.; Perumal, E. Dysregulation of Nrf2 in Hepatocellular Carcinoma: Role in Cancer Progression and Chemoresistance. Cancers 2018, 10, 481. [Google Scholar] [CrossRef] [Green Version]
- Kouroumalis, E.; Voumvouraki, A.; Augoustaki, A.; Samonakis, D.N. Autophagy in liver diseases. World J. Hepatol. 2021, 13, 6–65. [Google Scholar] [CrossRef] [PubMed]
- Schults, M.A.; Nagle, P.W.; Rensen, S.S.; Godschalk, R.W.; Munnia, A.; Peluso, M.; Claessen, S.M.; Greve, J.W.; Driessen, A.; Verdam, F.J.; et al. Decreased nucleotide excision repair in steatotic livers associates with myeloperoxidase-immunoreactivity. Mutat. Res. 2012, 736, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Cornell, L.; Munck, J.M.; Alsinet, C.; Villanueva, A.; Ogle, L.; Willoughby, C.E.; Televantou, D.; Thomas, H.D.; Jackson, J.; Burt, A.D.; et al. DNA-PK-A candidate driver of hepatocarcinogenesis and tissue biomarker that predicts response to treatment and survival. Clin. Cancer Res. 2015, 21, 925–933. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Dong, Z.; Wang, C.J.; Barlow, L.J.; Fako, V.; Serrano, M.A.; Zou, Y.; Liu, J.Y.; Zhang, J.T. FASN regulates cellular response to genotoxic treatments by increasing PARP-1 expression and DNA repair activity via NF-kappaB and SP1. Proc. Natl. Acad. Sci. USA 2016, 113, E6965–E6973. [Google Scholar] [CrossRef] [Green Version]
- Remmerie, A.; Martens, L.; Scott, C.L. Macrophage Subsets in Obesity, Aligning the Liver and Adipose Tissue. Front. Endocrinol. 2020, 11, 259. [Google Scholar] [CrossRef]
- McGettigan, B.; McMahan, R.; Orlicky, D.; Burchill, M.; Danhorn, T.; Francis, P.; Cheng, L.L.; Golden-Mason, L.; Jakubzick, C.V.; Rosen, H.R. Dietary Lipids Differentially Shape Nonalcoholic Steatohepatitis Progression and the Transcriptome of Kupffer Cells and Infiltrating Macrophages. Hepatology 2019, 70, 67–83. [Google Scholar] [CrossRef]
- Cai, D.; Yuan, M.; Frantz, D.F.; Melendez, P.A.; Hansen, L.; Lee, J.; Shoelson, S.E. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat. Med. 2005, 11, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Ambade, A.; Satishchandran, A.; Saha, B.; Gyongyosi, B.; Lowe, P.; Kodys, K.; Catalano, D.; Szabo, G. Hepatocellular carcinoma is accelerated by NASH involving M2 macrophage polarization mediated by hif-1alphainduced IL-10. Oncoimmunology 2016, 5, e1221557. [Google Scholar] [CrossRef] [Green Version]
- Tsunashima, H.; Tsuneyama, K.; Moritoki, Y.; Hara, M.; Kikuchi, K. Accumulated myeloid-derived suppressor cells demonstrate distinct phenotypes and functions in two non-alcoholic steatohepatitis mouse models. Hepatobiliary Surg. Nutr. 2015, 4, 313–319. [Google Scholar]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.C.; Chang, C.J.; Hsu, C.H. Targeting myeloid-derived suppressor cells in the treatment of hepatocellular carcinoma: Current state and future perspectives. J. Hepatocell. Carcinoma 2019, 6, 71–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacotte, S.; Slits, F.; Orci, L.A.; Meyer, J.; Oldani, G.; Delaune, V.; Gonelle-Gispert, C.; Morel, P.; Toso, C. Impact of myeloid-derived suppressor cell on Kupffer cells from mouse livers with hepatocellular carcinoma. Oncoimmunology 2016, 5, e1234565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Liu, M.; Sun, H.; Feng, Y.; Xu, L.; Chan, A.W.H.; Tong, J.H.; Wong, J.; Chong, C.C.N.; Lai, P.B.S.; et al. Hepatoma-intrinsic CCRK inhibition diminishes myeloid-derived suppressor cell immunosuppression and enhances immune-checkpoint blockade efficacy. Gut 2018, 67, 931–944. [Google Scholar] [CrossRef]
- Wolf, M.J.; Adili, A.; Piotrowitz, K.; Abdullah, Z.; Boege, Y.; Stemmer, K.; Ringelhan, M.; Simonavicius, N.; Egger, M.; Wohlleber, D.; et al. Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross-talk with hepatocytes. Cancer Cell 2014, 26, 549–564. [Google Scholar] [CrossRef] [Green Version]
- Van Herck, M.A.; Weyler, J.; Kwanten, W.J.; Dirinck, E.L.; De Winter, B.Y.; Francque, S.M.; Vonghia, L. The Differential Roles of T Cells in Non-alcoholic Fatty Liver Disease and Obesity. Front. Immunol. 2019, 10, 82. [Google Scholar] [CrossRef] [Green Version]
- Ye, Y.; Xie, X.; Yu, J.; Zhou, L.; Xie, H.; Jiang, G.; Yu, X.; Zhang, W.; Wu, J.; Zheng, S. Involvement of Th17 and Th1 effector responses in patients with Hepatitis B. J. Clin. Immunol. 2010, 30, 546–555. [Google Scholar] [CrossRef]
- Tang, Y.; Bian, Z.; Zhao, L.; Liu, Y.; Liang, S.; Wang, Q.; Han, X.; Peng, Y.; Chen, X.; Shen, L.; et al. Interleukin-17 exacerbates hepatic steatosis and inflammation in non-alcoholic fatty liver disease. Clin. Exp. Immunol. 2011, 166, 281–290. [Google Scholar] [CrossRef]
- Ma, C.; Kesarwala, A.H.; Eggert, T.; Medina-Echeverz, J.; Kleiner, D.E.; Jin, P.; Stroncek, D.F.; Terabe, M.; Kapoor, V.; ElGindi, M.; et al. NAFLD causes selective CD4+ T lymphocyte loss and promotes hepatocarcinogenesis. Nature 2016, 531, 253–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; Hua, J.; Mohamood, A.R.; Hamad, A.R.; Ravi, R.; Li, Z. A high-fat diet and regulatory T cells influence susceptibility to endotoxin-induced liver injury. Hepatology 2007, 46, 1519–1529. [Google Scholar] [CrossRef]
- Lan, Y.T.; Fan, X.P.; Fan, Y.C.; Zhao, J.; Wang, K. Change in the Treg/Th17 cell imbalance in hepatocellular carcinoma patients and its clinical value. Medicine 2017, 96, e7704. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.H.; Yamagiwa, S.; Ichida, T.; Matsuda, Y.; Sugahara, S.; Watanabe, H.; Sato, Y.; Abo, T.; Horwitz, D.A.; Aoyagi, Y. Increase of CD4+ CD25+ regulatory T-cells in the liver of patients with hepatocellular carcinoma. J. Hepatol. 2006, 45, 254–262. [Google Scholar] [CrossRef]
- Mikulak, J.; Bruni, E.; Oriolo, F.; Di Vito, C.; Mavilio, D. Hepatic Natural Killer Cells: Organ-Specific Sentinels of Liver Immune Homeostasis and Physiopathology. Front. Immunol. 2019, 10, 946. [Google Scholar] [CrossRef] [Green Version]
- Tosello-Trampont, A.; Surette, F.A.; Ewald, S.E.; Hahn, Y.S. Immunoregulatory Role of NK Cells in Tissue Inflammation and Regeneration. Front. Immunol. 2017, 8, 301. [Google Scholar] [CrossRef] [Green Version]
- Cepero-Donates, Y.; Lacraz, G.; Ghobadi, F.; Rakotoarivelo, V.; Orkhis, S.; Mayhue, M.; Chen, Y.G.; Rola-Pleszczynski, M.; Menendez, A.; Ilangumaran, S.; et al. Interleukin-15-mediated inflammation promotes non-alcoholic fatty liver disease. Cytokine 2016, 82, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Santos, L.; Luka, Z.; Wagner, C.; Fernandez-Alvarez, S.; Lu, S.C.; Mato, J.M.; Martinez-Chantar, M.L.; Beraza, N. Inhibition of natural killer cells protects the liver against acute injury in the absence of glycine N-methyltransferase. Hepatology 2012, 56, 747–759. [Google Scholar] [CrossRef] [Green Version]
- Tosello-Trampont, A.C.; Krueger, P.; Narayanan, S.; Landes, S.G.; Leitinger, N.; Hahn, Y.S. NKp46(+) natural killer cells attenuate metabolism-induced hepatic fibrosis by regulating macrophage activation in mice. Hepatology 2016, 63, 799–812. [Google Scholar] [CrossRef] [Green Version]
- Stiglund, N.; Strand, K.; Cornillet, M.; Stal, P.; Thorell, A.; Zimmer, C.L.; Naslund, E.; Karlgren, S.; Nilsson, H.; Mellgren, G.; et al. Retained NK Cell Phenotype and Functionality in Non-alcoholic Fatty Liver Disease. Front. Immunol. 2019, 10, 1255. [Google Scholar] [CrossRef]
- Sun, C.; Sun, H.; Zhang, C.; Tian, Z. NK cell receptor imbalance and NK cell dysfunction in HBV infection and hepatocellular carcinoma. Cell Mol. Immunol. 2015, 12, 292–302. [Google Scholar] [CrossRef] [Green Version]
- Hoechst, B.; Voigtlaender, T.; Ormandy, L.; Gamrekelashvili, J.; Zhao, F.; Wedemeyer, H.; Lehner, F.; Manns, M.P.; Greten, T.F.; Korangy, F. Myeloid derived suppressor cells inhibit natural killer cells in patients with hepatocellular carcinoma via the NKp30 receptor. Hepatology 2009, 50, 799–807. [Google Scholar] [CrossRef]
- Tan, S.; Xu, Y.; Wang, Z.; Wang, T.; Du, X.; Song, X.; Guo, X.; Peng, J.; Zhang, J.; Liang, Y.; et al. Tim-3 Hampers Tumor Surveillance of Liver-Resident and Conventional NK Cells by Disrupting PI3K Signaling. Cancer Res. 2020, 80, 1130–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bi, J.; Tian, Z. NK Cell Exhaustion. Front. Immunol. 2017, 8, 760. [Google Scholar] [CrossRef]
- Nishina, S.; Yamauchi, A.; Kawaguchi, T.; Kaku, K.; Goto, M.; Sasaki, K.; Hara, Y.; Tomiyama, Y.; Kuribayashi, F.; Torimura, T.; et al. Dipeptidyl Peptidase 4 Inhibitors Reduce Hepatocellular Carcinoma by Activating Lymphocyte Chemotaxis in Mice. Cell Mol. Gastroenterol. Hepatol. 2019, 7, 115–134. [Google Scholar] [CrossRef] [Green Version]
- Kurien, B.T.; Scofield, R.H. Autoimmunity and oxidatively modified autoantigens. Autoimmun. Rev. 2008, 7, 567–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeFuria, J.; Belkina, A.C.; Jagannathan-Bogdan, M.; Snyder-Cappione, J.; Carr, J.D.; Nersesova, Y.R.; Markham, D.; Strissel, K.J.; Watkins, A.A.; Zhu, M.; et al. B cells promote inflammation in obesity and type 2 diabetes through regulation of T-cell function and an inflammatory cytokine profile. Proc. Natl. Acad. Sci. USA 2013, 110, 5133–5138. [Google Scholar] [CrossRef] [Green Version]
- Davis, B.K.; Wen, H.; Ting, J.P. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu. Rev. Immunol. 2011, 29, 707–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McPherson, S.; Henderson, E.; Burt, A.D.; Day, C.P.; Anstee, Q.M. Serum immunoglobulin levels predict fibrosis in patients with non-alcoholic fatty liver disease. J. Hepatol. 2014, 60, 1055–1062. [Google Scholar] [CrossRef] [Green Version]
- Brunner, S.M.; Itzel, T.; Rubner, C.; Kesselring, R.; Griesshammer, E.; Evert, M.; Teufel, A.; Schlitt, H.J.; Fichtner-Feigl, S. Tumor-infiltrating B cells producing antitumor active immunoglobulins in resected HCC prolong patient survival. Oncotarget 2017, 8, 71002–71011. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Ma, L.; Goswami, S.; Ma, J.; Zheng, B.; Duan, M.; Liu, L.; Zhang, L.; Shi, J.; Dong, L.; et al. Landscape of infiltrating B cells and their clinical significance in human hepatocellular carcinoma. Oncoimmunology 2019, 8, e1571388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, X.; Lao, X.M.; Chen, M.M.; Liu, R.X.; Wei, Y.; Ouyang, F.Z.; Chen, D.P.; Zhao, X.Y.; Zhao, Q.; Li, X.F.; et al. PD-1hi Identifies a Novel Regulatory B-cell Population in Human Hepatoma That Promotes Disease Progression. Cancer Discov. 2016, 6, 546–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouyang, F.Z.; Wu, R.Q.; Wei, Y.; Liu, R.X.; Yang, D.; Xiao, X.; Zheng, L.; Li, B.; Lao, X.M.; Kuang, D.M. Dendritic cell-elicited B-cell activation fosters immune privilege via IL-10 signals in hepatocellular carcinoma. Nat. Commun. 2016, 7, 13453. [Google Scholar] [CrossRef]
- Shalapour, S.; Lin, X.J.; Bastian, I.N.; Brain, J.; Burt, A.D.; Aksenov, A.A.; Vrbanac, A.F.; Li, W.; Perkins, A.; Matsutani, T.; et al. Inflammation-induced IgA+ cells dismantle anti-liver cancer immunity. Nature 2017, 551, 340–345. [Google Scholar] [CrossRef]
- Koo, S.Y.; Park, E.J.; Lee, C.W. Immunological distinctions between nonalcoholic steatohepatitis and hepatocellular carcinoma. Exp. Mol. Med. 2020, 52, 1209–1219. [Google Scholar] [CrossRef]
- Kanwal, F.; Kramer, J.R.; Mapakshi, S.; Natarajan, Y.; Chayanupatkul, M.; Richardson, P.A.; Li, L.; Desiderio, R.; Thrift, A.P.; Asch, S.M.; et al. Risk of Hepatocellular Cancer in Patients With Non-Alcoholic Fatty Liver Disease. Gastroenterology 2018, 155, 1828–1837.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giuffre, M.; Giuricin, M.; Bonazza, D.; Rosso, N.; Giraudi, P.J.; Masutti, F.; Palmucci, S.; Basile, A.; Zanconati, F.; de Manzini, N.; et al. Optimization of Point-Shear Wave Elastography by Skin-to-Liver Distance to Assess Liver Fibrosis in Patients Undergoing Bariatric Surgery. Diagnostics 2020, 10, 795. [Google Scholar] [CrossRef]
- Foschi, F.G.; Domenicali, M.; Giacomoni, P.; Dall’Aglio, A.C.; Conti, F.; Borghi, A.; Bevilacqua, V.; Napoli, L.; Mirici, F.; Cucchetti, A.; et al. Is there an association between commonly employed biomarkers of liver fibrosis and liver stiffness in the general population? Ann. Hepatol. 2020, 19, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Garrido, A.; Djouder, N. Cirrhosis: A Questioned Risk Factor for Hepatocellular Carcinoma. Trends Cancer 2021, 7, 29–36. [Google Scholar] [CrossRef]
- Ozdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef] [Green Version]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foretz, M.; Guigas, B.; Viollet, B. Understanding the glucoregulatory mechanisms of metformin in type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2019, 15, 569–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madiraju, A.K.; Qiu, Y.; Perry, R.J.; Rahimi, Y.; Zhang, X.M.; Zhang, D.; Camporez, J.G.; Cline, G.W.; Butrico, G.M.; Kemp, B.E.; et al. Metformin inhibits gluconeogenesis via a redox-dependent mechanism in vivo. Nat. Med. 2018, 24, 1384–1394. [Google Scholar] [CrossRef] [PubMed]
- Griss, T.; Vincent, E.E.; Egnatchik, R.; Chen, J.; Ma, E.H.; Faubert, B.; Viollet, B.; DeBerardinis, R.J.; Jones, R.G. Metformin Antagonizes Cancer Cell Proliferation by Suppressing Mitochondrial-Dependent Biosynthesis. PLoS Biol. 2015, 13, e1002309. [Google Scholar] [CrossRef] [Green Version]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. eLife 2014, 3, e02242. [Google Scholar] [CrossRef]
- Hsu, C.C.; Wu, L.C.; Hsia, C.Y.; Yin, P.H.; Chi, C.W.; Yeh, T.S.; Lee, H.C. Energy metabolism determines the sensitivity of human hepatocellular carcinoma cells to mitochondrial inhibitors and biguanide drugs. Oncol. Rep. 2015, 34, 1620–1628. [Google Scholar] [CrossRef]
- Zhang, C.S.; Li, M.; Ma, T.; Zong, Y.; Cui, J.; Feng, J.W.; Wu, Y.Q.; Lin, S.Y.; Lin, S.C. Metformin Activates AMPK through the Lysosomal Pathway. Cell Metab. 2016, 24, 521–522. [Google Scholar] [CrossRef] [Green Version]
- Hardie, D.G.; Pan, D.A. Regulation of fatty acid synthesis and oxidation by the AMP-activated protein kinase. Biochem. Soc. Trans. 2002, 30 Pt 6, 1064–1070. [Google Scholar] [CrossRef]
- Fullerton, M.D.; Galic, S.; Marcinko, K.; Sikkema, S.; Pulinilkunnil, T.; Chen, Z.P.; O’Neill, H.M.; Ford, R.J.; Palanivel, R.; O’Brien, M.; et al. Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nat. Med. 2013, 19, 1649–1654. [Google Scholar] [CrossRef] [Green Version]
- Boudaba, N.; Marion, A.; Huet, C.; Pierre, R.; Viollet, B.; Foretz, M. AMPK Re-Activation Suppresses Hepatic Steatosis but its Downregulation Does Not Promote Fatty Liver Development. EBioMedicine 2018, 28, 194–209. [Google Scholar] [CrossRef] [Green Version]
- Luizon, M.R.; Eckalbar, W.L.; Wang, Y.; Jones, S.L.; Smith, R.P.; Laurance, M.; Lin, L.; Gallins, P.J.; Etheridge, A.S.; Wright, F.; et al. Genomic Characterization of Metformin Hepatic Response. PLoS Genet. 2016, 12, e1006449. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Liu, Y.; Li, M.; Wu, H.; Wang, Y.; You, Y.; Li, P.; Ding, X.; Liu, C.; Gong, J. Predictive and preventive significance of AMPK activation on hepatocarcinogenesis in patients with liver cirrhosis. Cell Death Dis. 2018, 9, 264. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Hu, J.; Sheng, L.; Yuan, M.; Wu, Y.; Chen, L.; Zheng, G.; Qiu, Z. Metformin delays AKT/c-Met-driven hepatocarcinogenesis by regulating signaling pathways for de novo lipogenesis and ATP generation. Toxicol. Appl. Pharmacol. 2019, 365, 51–60. [Google Scholar] [CrossRef]
- Jiang, X.; Tan, H.Y.; Teng, S.; Chan, Y.T.; Wang, D.; Wang, N. The Role of AMP-Activated Protein Kinase as a Potential Target of Treatment of Hepatocellular Carcinoma. Cancers 2019, 11, 647. [Google Scholar] [CrossRef] [Green Version]
- Zhao, P.; Sun, X.; Chaggan, C.; Liao, Z.; In Wong, K.; He, F.; Singh, S.; Loomba, R.; Karin, M.; Witztum, J.L.; et al. An AMPK-caspase-6 axis controls liver damage in nonalcoholic steatohepatitis. Science 2020, 367, 652–660. [Google Scholar] [CrossRef]
- Valentine, R.J.; Coughlan, K.A.; Ruderman, N.B.; Saha, A.K. Insulin inhibits AMPK activity and phosphorylates AMPK Ser(4)(8)(5)/(4)(9)(1) through Akt in hepatocytes, myotubes and incubated rat skeletal muscle. Arch. Biochem. Biophys. 2014, 562, 62–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, R.W.; Hughey, C.C.; Lantier, L.; Sundelin, E.I.; Peggie, M.; Zeqiraj, E.; Sicheri, F.; Jessen, N.; Wasserman, D.H.; Sakamoto, K. Metformin reduces liver glucose production by inhibition of fructose-1-6-bisphosphatase. Nat. Med. 2018, 24, 1395–1406. [Google Scholar] [CrossRef] [Green Version]
- Miller, R.A.; Chu, Q.; Xie, J.; Foretz, M.; Viollet, B.; Birnbaum, M.J. Biguanides suppress hepatic glucagon signalling by decreasing production of cyclic AMP. Nature 2013, 494, 256–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Wang, C.; Zhao, F.; Luo, X.; Qin, M.; Arunachalam, E.; Ge, Z.; Wang, N.; Deng, X.; Jin, G.; et al. Loss of FBP1 facilitates aggressive features of hepatocellular carcinoma cells through the Warburg effect. Carcinogenesis 2017, 38, 134–143. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Huangyang, P.; Burrows, M.; Guo, K.; Riscal, R.; Godfrey, J.; Lee, K.E.; Lin, N.; Lee, P.; Blair, I.A.; et al. FBP1 loss disrupts liver metabolism and promotes tumorigenesis through a hepatic stellate cell senescence secretome. Nat. Cell Biol. 2020, 22, 728–739. [Google Scholar] [CrossRef]
- Massimi, M.; Ragusa, F.; Cardarelli, S.; Giorgi, M. Targeting Cyclic AMP Signalling in Hepatocellular Carcinoma. Cells 2019, 8, 1511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conde de la Rosa, L.; Vrenken, T.E.; Buist-Homan, M.; Faber, K.N.; Moshage, H. Metformin protects primary rat hepatocytes against oxidative stress-induced apoptosis. Pharmacol. Res. Perspect. 2015, 3, e00125. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Jun, C.; Lu, Y.; Jiangmei, S. Combination of metformin and luteolin synergistically protects carbon tetrachloride-induced hepatotoxicity: Mechanism involves antioxidant, anti-inflammatory, antiapoptotic, and Nrf2/HO-1 signaling pathway. Biofactors 2019, 45, 598–606. [Google Scholar] [CrossRef]
- Rahimi, G.; Heydari, S.; Rahimi, B.; Abedpoor, N.; Niktab, I.; Safaeinejad, Z.; Peymani, M.; Seyed Forootan, F.; Derakhshan, Z.; Esfahani, M.H.N.; et al. A combination of herbal compound (SPTC) along with exercise or metformin more efficiently alleviated diabetic complications through down-regulation of stress oxidative pathway upon activating Nrf2-Keap1 axis in AGE rich diet-induced type 2 diabetic mice. Nutr. Metab. 2021, 18, 14. [Google Scholar] [CrossRef] [PubMed]
- Joo, M.S.; Kim, W.D.; Lee, K.Y.; Kim, J.H.; Koo, J.H.; Kim, S.G. AMPK Facilitates Nuclear Accumulation of Nrf2 by Phosphorylating at Serine 550. Mol. Cell Biol. 2016, 36, 1931–1942. [Google Scholar] [CrossRef] [Green Version]
- Prasad, S.; Sajja, R.K.; Kaisar, M.A.; Park, J.H.; Villalba, H.; Liles, T.; Abbruscato, T.; Cucullo, L. Role of Nrf2 and protective effects of Metformin against tobacco smoke-induced cerebrovascular toxicity. Redox. Biol. 2017, 12, 58–69. [Google Scholar] [CrossRef]
- Kitamura, H.; Motohashi, H. NRF2 addiction in cancer cells. Cancer Sci. 2018, 109, 900–911. [Google Scholar] [CrossRef] [Green Version]
- Do, M.T.; Kim, H.G.; Khanal, T.; Choi, J.H.; Kim, D.H.; Jeong, T.C.; Jeong, H.G. Metformin inhibits heme oxygenase-1 expression in cancer cells through inactivation of Raf-ERK-Nrf2 signaling and AMPK-independent pathways. Toxicol. Appl. Pharmacol. 2013, 271, 229–238. [Google Scholar] [CrossRef]
- Yu, C.; Jiao, Y.; Xue, J.; Zhang, Q.; Yang, H.; Xing, L.; Chen, G.; Wu, J.; Zhang, S.; Zhu, W.; et al. Metformin Sensitizes Non-small Cell Lung Cancer Cells to an Epigallocatechin-3-Gallate (EGCG) Treatment by Suppressing the Nrf2/HO-1 Signaling Pathway. Int. J. Biol. Sci. 2017, 13, 1560–1569. [Google Scholar] [CrossRef]
- Huang, S.; He, T.; Yang, S.; Sheng, H.; Tang, X.; Bao, F.; Wang, Y.; Lin, X.; Yu, W.; Cheng, F.; et al. Metformin reverses chemoresistance in non-small cell lung cancer via accelerating ubiquitination-mediated degradation of Nrf2. Transl. Lung Cancer Res. 2020, 9, 2337–2355. [Google Scholar] [CrossRef]
- Zhang, J.; Jiao, K.; Liu, J.; Xia, Y. Metformin reverses the resistance mechanism of lung adenocarcinoma cells that knocks down the Nrf2 gene. Oncol. Lett. 2018, 16, 6071–6080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, M.; Yang, L.; Liao, H.; Liang, X.; Xie, B.; Xiong, J.; Tao, X.; Chen, X.; Cheng, Y.; Chen, X.; et al. Metformin sensitizes endometrial cancer cells to chemotherapy through IDH1-induced Nrf2 expression via an epigenetic mechanism. Oncogene 2018, 37, 5666–5681. [Google Scholar] [CrossRef]
- Overi, D.; Carpino, G.; Franchitto, A.; Onori, P.; Gaudio, E. Hepatocyte Injury and Hepatic Stem Cell Niche in the Progression of Non-Alcoholic Steatohepatitis. Cells 2020, 9, 590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sia, D.; Villanueva, A.; Friedman, S.L.; Llovet, J.M. Liver Cancer Cell of Origin, Molecular Class, and Effects on Patient Prognosis. Gastroenterology 2017, 152, 745–761. [Google Scholar] [CrossRef] [Green Version]
- DePeralta, D.K.; Wei, L.; Ghoshal, S.; Schmidt, B.; Lauwers, G.Y.; Lanuti, M.; Chung, R.T.; Tanabe, K.K.; Fuchs, B.C. Metformin prevents hepatocellular carcinoma development by suppressing hepatic progenitor cell activation in a rat model of cirrhosis. Cancer 2016, 122, 1216–1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, S.; Li, A.; Liu, F.; Chen, F.; Williams, M.; Zhang, C.; Kelley, Z.; Wu, C.L.; Luo, R.; Xiao, H. NCOA5 haploinsufficiency results in glucose intolerance and subsequent hepatocellular carcinoma. Cancer Cell 2013, 24, 725–737. [Google Scholar] [CrossRef] [Green Version]
- Williams, M.; Liu, X.; Zhang, Y.; Reske, J.; Bahal, D.; Gohl, T.G.; Hollern, D.; Ensink, E.; Kiupel, M.; Luo, R.; et al. NCOA5 deficiency promotes a unique liver protumorigenic microenvironment through p21(WAF1/CIP1) overexpression, which is reversed by metformin. Oncogene 2020, 39, 3821–3836. [Google Scholar] [CrossRef] [PubMed]
- Molnar, Z.; Millward, A.B.; Tse, W.; Demaine, A.G. p21(WAF1/CIP1) Expression is Differentially Regulated by Metformin and Rapamycin. Int. J. Chronic Dis. 2014, 2014, 327640. [Google Scholar]
- Wu, H.; Wade, M.; Krall, L.; Grisham, J.; Xiong, Y.; Van Dyke, T. Targeted in vivo expression of the cyclin-dependent kinase inhibitor p21 halts hepatocyte cell-cycle progression, postnatal liver development and regeneration. Genes Dev. 1996, 10, 245–260. [Google Scholar] [CrossRef] [Green Version]
- McNelis, J.C.; Olefsky, J.M. Macrophages, immunity, and metabolic disease. Immunity 2014, 41, 36–48. [Google Scholar] [CrossRef] [Green Version]
- de Oliveira, S.; Houseright, R.A.; Graves, A.L.; Golenberg, N.; Korte, B.G.; Miskolci, V.; Huttenlocher, A. Metformin modulates innate immune-mediated inflammation and early progression of NAFLD-associated hepatocellular carcinoma in zebrafish. J. Hepatol. 2019, 70, 710–721. [Google Scholar] [CrossRef] [Green Version]
- Montfort, A.; Colacios, C.; Levade, T.; Andrieu-Abadie, N.; Meyer, N.; Segui, B. The TNF Paradox in Cancer Progression and Immunotherapy. Front. Immunol. 2019, 10, 1818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arai, M.; Uchiba, M.; Komura, H.; Mizuochi, Y.; Harada, N.; Okajima, K. Metformin, an antidiabetic agent, suppresses the production of tumor necrosis factor and tissue factor by inhibiting early growth response factor-1 expression in human monocytes in vitro. J. Pharmacol. Exp. Ther. 2010, 334, 206–213. [Google Scholar] [CrossRef]
- Kim, J.; Kwak, H.J.; Cha, J.Y.; Jeong, Y.S.; Rhee, S.D.; Kim, K.R.; Cheon, H.G. Metformin suppresses lipopolysaccharide (LPS)-induced inflammatory response in murine macrophages via activating transcription factor-3 (ATF-3) induction. J. Biol. Chem. 2014, 289, 23246–23255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasamsetti, S.B.; Karnewar, S.; Kanugula, A.K.; Thatipalli, A.R.; Kumar, J.M.; Kotamraju, S. Metformin inhibits monocyte-to-macrophage differentiation via AMPK-mediated inhibition of STAT3 activation: Potential role in atherosclerosis. Diabetes 2015, 64, 2028–2041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, L.; Liang, G.; Yao, Z.; Zhang, J.; Liu, R.; Chen, H.; Zhou, Y.; Wu, H.; Yang, B.; He, Q. Metformin prevents cancer metastasis by inhibiting M2-like polarization of tumor associated macrophages. Oncotarget 2015, 6, 36441–36455. [Google Scholar] [CrossRef] [Green Version]
- Wei, Z.; Zhang, X.; Yong, T.; Bie, N.; Zhan, G.; Li, X.; Liang, Q.; Li, J.; Yu, J.; Huang, G.; et al. Boosting anti-PD-1 therapy with metformin-loaded macrophage-derived microparticles. Nat. Commun. 2021, 12, 440. [Google Scholar] [CrossRef]
- Chiang, C.F.; Chao, T.T.; Su, Y.F.; Hsu, C.C.; Chien, C.Y.; Chiu, K.C.; Shiah, S.G.; Lee, C.H.; Liu, S.Y.; Shieh, Y.S. Metformin-treated cancer cells modulate macrophage polarization through AMPK-NF-kappaB signaling. Oncotarget 2017, 8, 20706–20718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, G.; Lian, J.; Huang, L.; Zhao, Q.; Liu, S.; Zhang, Z.; Chen, X.; Yue, D.; Li, L.; Li, F.; et al. Metformin blocks myeloid-derived suppressor cell accumulation through AMPK-DACH1-CXCL1 axis. Oncoimmunology 2018, 7, e1442167. [Google Scholar] [CrossRef] [Green Version]
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. AMPK activation inhibits the functions of myeloid-derived suppressor cells (MDSC): Impact on cancer and aging. J. Mol. Med. 2019, 97, 1049–1064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Wang, L.; Li, J.; Fan, Z.; Yang, L.; Zhang, Z.; Zhang, C.; Yue, D.; Qin, G.; Zhang, T.; et al. Metformin-Induced Reduction of CD39 and CD73 Blocks Myeloid-Derived Suppressor Cell Activity in Patients with Ovarian Cancer. Cancer Res. 2018, 78, 1779–1791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, P.; Yin, K.; Tang, X.; Tian, J.; Zhang, Y.; Ma, J.; Xu, H.; Xu, Q.; Wang, S. Metformin inhibits the function of granulocytic myeloid-derived suppressor cells in tumor-bearing mice. Biomed. Pharmacother. 2019, 120, 109458. [Google Scholar] [CrossRef] [PubMed]
- Eikawa, S.; Nishida, M.; Mizukami, S.; Yamazaki, C.; Nakayama, E.; Udono, H. Immune-mediated antitumor effect by type 2 diabetes drug, metformin. Proc. Natl. Acad. Sci. USA 2015, 112, 1809–1814. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Li, F.; Tian, Y.; Cao, L.; Gao, Q.; Zhang, C.; Zhang, K.; Shen, C.; Ping, Y.; Maimela, N.R.; et al. Metformin Enhances the Antitumor Activity of CD8(+) T Lymphocytes via the AMPK-miR-107-Eomes-PD-1 Pathway. J. Immunol. 2020, 204, 2575–2588. [Google Scholar] [CrossRef]
- Cha, J.H.; Yang, W.H.; Xia, W.; Wei, Y.; Chan, L.C.; Lim, S.O.; Li, C.W.; Kim, T.; Chang, S.S.; Lee, H.H.; et al. Metformin Promotes Antitumor Immunity via Endoplasmic-Reticulum-Associated Degradation of PD-L1. Mol. Cell 2018, 71, 606–620.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, R.; Tan, Z.; Deng, L.; Chen, Y.; Xia, Y.; Gao, Y.; Wang, X.; Sun, B. Interleukin-22 promotes human hepatocellular carcinoma by activation of STAT3. Hepatology 2011, 54, 900–909. [Google Scholar] [CrossRef]
- Zhao, D.; Long, X.D.; Lu, T.F.; Wang, T.; Zhang, W.W.; Liu, Y.X.; Cui, X.L.; Dai, H.J.; Xue, F.; Xia, Q. Metformin decreases IL-22 secretion to suppress tumor growth in an orthotopic mouse model of hepatocellular carcinoma. Int. J. Cancer 2015, 136, 2556–2565. [Google Scholar] [CrossRef] [PubMed]
- Kunisada, Y.; Eikawa, S.; Tomonobu, N.; Domae, S.; Uehara, T.; Hori, S.; Furusawa, Y.; Hase, K.; Sasaki, A.; Udono, H. Attenuation of CD4+CD25+ Regulatory T Cells in the Tumor Microenvironment by Metformin, a Type 2 Diabetes Drug. EBioMedicine 2017, 25, 154–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, H.J.; Lee, J.; Lee, S.Y.; Kim, E.K.; Park, M.J.; Kim, K.W.; Park, S.H.; Cho, M.L. Metformin attenuates experimental autoimmune arthritis through reciprocal regulation of Th17/Treg balance and osteoclastogenesis. Mediat. Inflamm. 2014, 2014, 973986. [Google Scholar] [CrossRef] [Green Version]
- Duan, W.; Ding, Y.; Yu, X.; Ma, D.; Yang, B.; Li, Y.; Huang, L.; Chen, Z.; Zheng, J.; Yang, C. Metformin mitigates autoimmune insulitis by inhibiting Th1 and Th17 responses while promoting Treg production. Am. J. Transl. Res. 2019, 11, 2393–2402. [Google Scholar] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Wang, H.; Xiao, H. Metformin Actions on the Liver: Protection Mechanisms Emerging in Hepatocytes and Immune Cells against NASH-Related HCC. Int. J. Mol. Sci. 2021, 22, 5016. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22095016
Zhang Y, Wang H, Xiao H. Metformin Actions on the Liver: Protection Mechanisms Emerging in Hepatocytes and Immune Cells against NASH-Related HCC. International Journal of Molecular Sciences. 2021; 22(9):5016. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22095016
Chicago/Turabian StyleZhang, Yueqi, Hongbing Wang, and Hua Xiao. 2021. "Metformin Actions on the Liver: Protection Mechanisms Emerging in Hepatocytes and Immune Cells against NASH-Related HCC" International Journal of Molecular Sciences 22, no. 9: 5016. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22095016