
Heterogeneity of Colorectal Cancer Progression: Molecular Gas and Brakes

 , , ,
, , ,
Abstract
:1. Heterogeneous Gene Damage and Different Progression of Colorectal Cancer
The Discovery of the Main Patterns of Gene Damage, CIN and MSI
2. DNA Hypermethylation and CIMP
3. The Advent of Next-Generation Sequencing and the Evidence of Widespread Genetic Heterogeneity
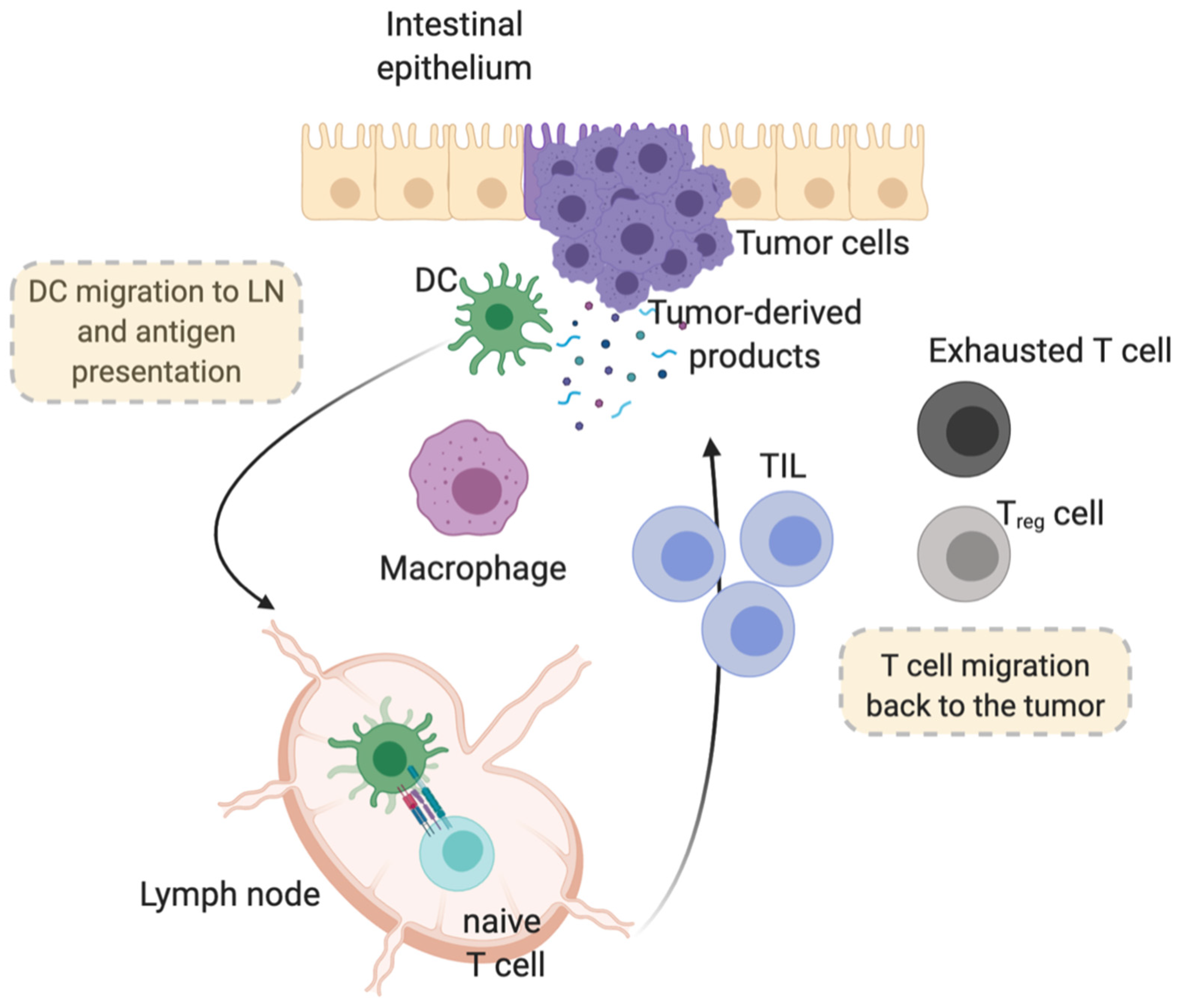
4. Tumor-Host Immune Response as Switcher on the Routes of Cancer Progression
5. Cellular and Molecular Players in the Tumor Microenvironment: Meaningful Links
5.1. Soluble Mediators
5.2. Immune Cell Players
6. Links between Genetic Changes and the Immune-Contexture
7. Inflammation as an Accelerator of Carcinogenesis in Inflammatory Bowel Diseases
8. Micronutrients and Molecular Tuning of Colorectal Carcinogenesis
8.1. Vitamin D
8.2. Calcium
9. Attempting Pharmacological Interference with CRC Development: Chemoprevention
10. Microbial Hosts: Fusobacterium Nucleatum
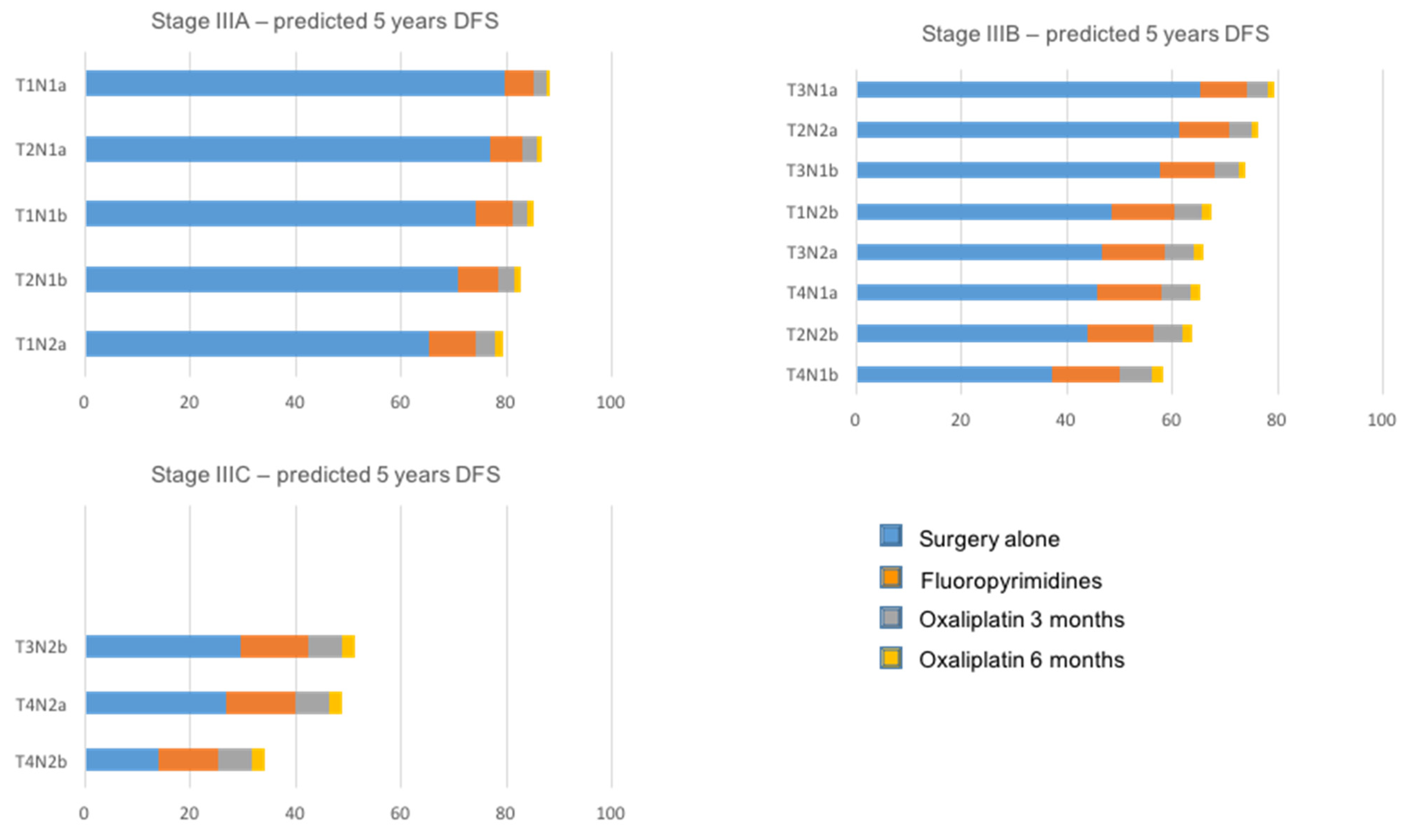
11. The Frame for Biomarker Actionability: TNM Staging System Turning in the 21st Century
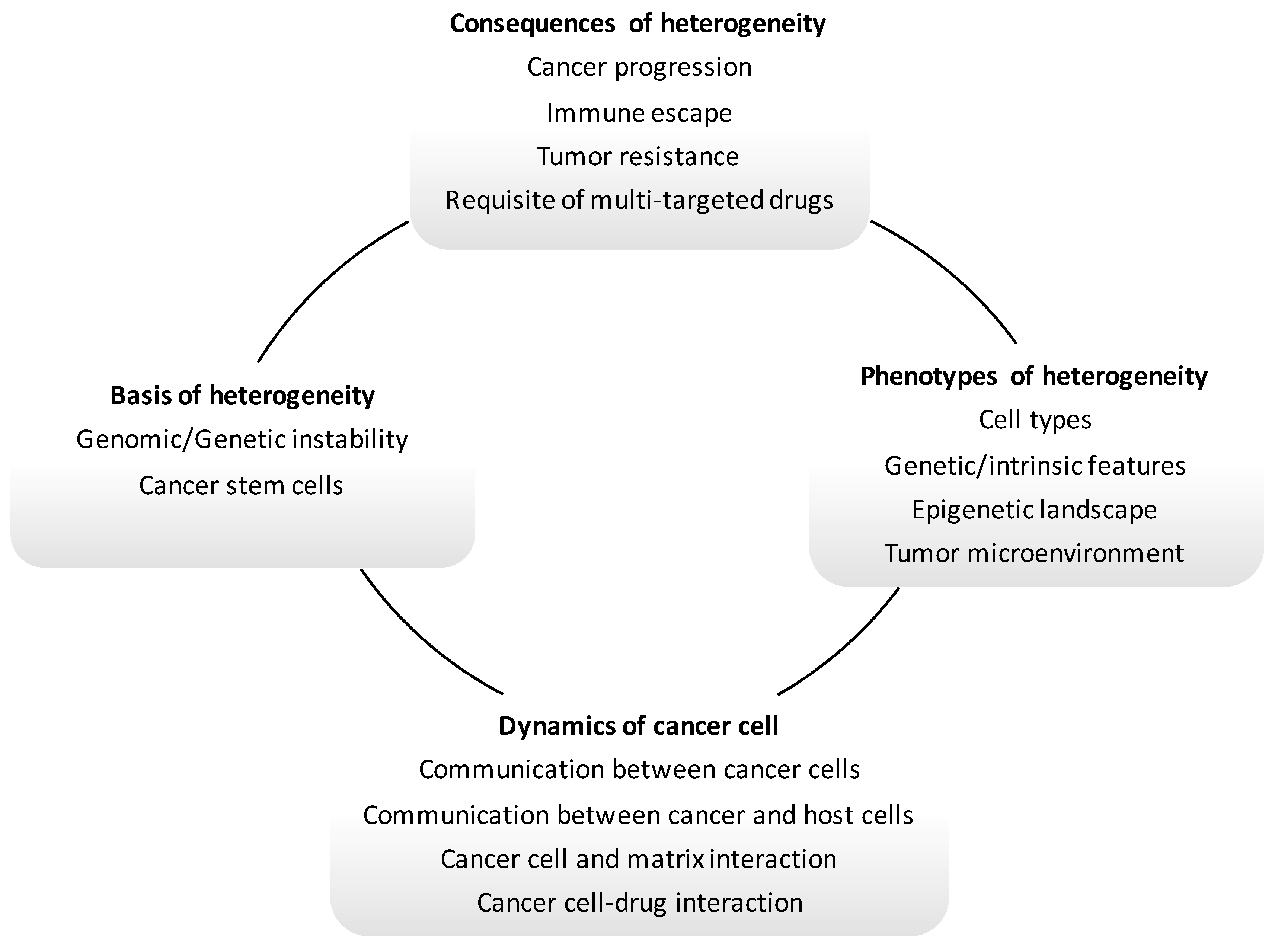
12. Molecular Heterogeneity and Metastatic Seeding
13. Molecular Heterogeneity and the Emergence of Resistance to Target Treatment in Metastatic CRC
14. Restraining the Progression of Metastatic CRC: The Frontier
Author Contributions
Funding
Conflicts of Interest
References
- Leppert, M.; Dobbs, M.; Scambler, P.; O’Connell, P.; Nakamura, Y.; Stauffer, D.; Woodward, S.; Burt, R.; Hughes, J.; Gardner, E.; et al. The gene for familial polyposis coli maps to the long arm of chromosome 5. Science 1987, 238, 1411–1413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solomon, E.; Voss, R.; Hall, V.; Bodmer, W.F.; Jass, J.R.; Jeffreys, A.J.; Lucibello, F.C.; Patel, I.; Rider, S.H. Chromosome 5 allele loss in human colorectal carcinomas. Nature 1987, 328, 616–619. [Google Scholar] [CrossRef] [PubMed]
- Bodmer, W.F.; Bailey, C.J.; Bodmer, J.; Bussey, H.J.; Ellis, A.; Gorman, P.; Lucibello, F.C.; Murday, V.A.; Rider, S.H.; Scambler, P.; et al. Localization of the gene for familial adenomatous polyposis on chromosome 5. Nature 1987, 328, 614–616. [Google Scholar] [CrossRef] [Green Version]
- Knudson, A.G., Jr. Mutation and cancer: Statistical study of retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef] [Green Version]
- Fearnhead, N.S.; Britton, M.P.; Bodmer, W.F. The ABC of APC. Hum. Mol. Genet. 2001, 10, 721–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bos, J.L.; Fearon, E.R.; Hamilton, S.R.; Verlaan-de Vries, M.; van Boom, J.H.; van der Eb, A.J.; Vogelstein, B. Prevalence of ras gene mutations in human colorectal cancers. Nature 1987, 327, 293–297. [Google Scholar] [CrossRef]
- Fearon, E.R.; Hamilton, S.R.; Vogelstein, B. Clonal analysis of human colorectal tumors. Science 1987, 238, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.J.; Fearon, E.R.; Nigro, J.M.; Hamilton, S.R.; Preisinger, A.C.; Jessup, J.M.; vanTuinen, P.; Ledbetter, D.H.; Barker, D.F.; Nakamura, Y.; et al. Chromosome 17 deletions and p53 gene mutations in colorectal carcinomas. Science 1989, 244, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Fearon, E.R.; Hamilton, S.R.; Kern, S.E.; Preisinger, A.C.; Leppert, M.; Nakamura, Y.; White, R.; Smits, A.M.; Bos, J.L. Genetic alterations during colorectal-tumor development. N. Engl. J. Med. 1988, 319, 525–532. [Google Scholar] [CrossRef] [Green Version]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Boland, C.R. Evolution of the nomenclature for the hereditary colorectal cancer syndromes. Fam. Cancer 2005, 4, 211–218. [Google Scholar] [CrossRef]
- Boland, C.R.; Lynch, H.T. The history of Lynch syndrome. Fam. Cancer 2013, 12, 145–157. [Google Scholar] [CrossRef] [Green Version]
- Ionov, Y.; Peinado, M.A.; Malkhosyan, S.; Shibata, D.; Perucho, M. Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature 1993, 363, 558–561. [Google Scholar] [CrossRef] [PubMed]
- Aaltonen, L.A.; Peltomaki, P.; Leach, F.S.; Sistonen, P.; Pylkkanen, L.; Mecklin, J.P.; Jarvinen, H.; Powell, S.M.; Jen, J.; Hamilton, S.R.; et al. Clues to the pathogenesis of familial colorectal cancer. Science 1993, 260, 812–816. [Google Scholar] [CrossRef] [PubMed]
- Thibodeau, S.N.; Bren, G.; Schaid, D. Microsatellite instability in cancer of the proximal colon. Science 1993, 260, 816–819. [Google Scholar] [CrossRef] [PubMed]
- Peltomaki, P. Update on Lynch syndrome genomics. Fam. Cancer 2016, 15, 385–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kane, M.F.; Loda, M.; Gaida, G.M.; Lipman, J.; Mishra, R.; Goldman, H.; Jessup, J.M.; Kolodner, R. Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res. 1997, 57, 808–811. [Google Scholar]
- Cunningham, J.M.; Christensen, E.R.; Tester, D.J.; Kim, C.Y.; Roche, P.C.; Burgart, L.J.; Thibodeau, S.N. Hypermethylation of the hMLH1 promoter in colon cancer with microsatellite instability. Cancer Res. 1998, 58, 3455–3460. [Google Scholar] [PubMed]
- Herman, J.G.; Umar, A.; Polyak, K.; Graff, J.R.; Ahuja, N.; Issa, J.P.; Markowitz, S.; Willson, J.K.; Hamilton, S.R.; Kinzler, K.W.; et al. Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc. Natl. Acad. Sci. USA 1998, 95, 6870–6875. [Google Scholar] [CrossRef] [Green Version]
- Aaltonen, L.A.; Salovaara, R.; Kristo, P.; Canzian, F.; Hemminki, A.; Peltomaki, P.; Chadwick, R.B.; Kaariainen, H.; Eskelinen, M.; Jarvinen, H.; et al. Incidence of hereditary nonpolyposis colorectal cancer and the feasibility of molecular screening for the disease. N. Engl. J. Med. 1998, 338, 1481–1487. [Google Scholar] [CrossRef]
- Giardiello, F.M.; Allen, J.I.; Axilbund, J.E.; Boland, C.R.; Burke, C.A.; Burt, R.W.; Church, J.M.; Dominitz, J.A.; Johnson, D.A.; Kaltenbach, T.; et al. Guidelines on genetic evaluation and management of Lynch syndrome: A consensus statement by the US Multi-Society Task Force on colorectal cancer. Gastroenterology 2014, 147, 502–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinzler, K.W.; Vogelstein, B. Lessons from hereditary colorectal cancer. Cell 1996, 87, 159–170. [Google Scholar] [CrossRef] [Green Version]
- Sankila, R.; Aaltonen, L.A.; Jarvinen, H.J.; Mecklin, J.P. Better survival rates in patients with MLH1-associated hereditary colorectal cancer. Gastroenterology 1996, 110, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Gryfe, R.; Kim, H.; Hsieh, E.T.; Aronson, M.D.; Holowaty, E.J.; Bull, S.B.; Redston, M.; Gallinger, S. Tumor microsatellite instability and clinical outcome in young patients with colorectal cancer. N. Engl. J. Med. 2000, 342, 69–77. [Google Scholar] [CrossRef]
- Malesci, A.; Laghi, L.; Bianchi, P.; Delconte, G.; Randolph, A.; Torri, V.; Carnaghi, C.; Doci, R.; Rosati, R.; Montorsi, M.; et al. Reduced likelihood of metastases in patients with microsatellite-unstable colorectal cancer. Clin. Cancer Res. 2007, 13, 3831–3839. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, T.; Wu, T.T.; Catalano, P.J.; Ueki, T.; Satriano, R.; Haller, D.G.; Benson, A.B., 3rd; Hamilton, S.R. Molecular predictors of survival after adjuvant chemotherapy for colon cancer. N. Engl. J. Med. 2001, 344, 1196–1206. [Google Scholar] [CrossRef] [Green Version]
- Laghi, L.; Malesci, A. Microsatellite instability and therapeutic consequences in colorectal cancer. Dig. Dis. 2012, 30, 304–309. [Google Scholar] [CrossRef]
- Sargent, D.J.; Marsoni, S.; Monges, G.; Thibodeau, S.N.; Labianca, R.; Hamilton, S.R.; French, A.J.; Kabat, B.; Foster, N.R.; Torri, V.; et al. Defective mismatch repair as a predictive marker for lack of efficacy of fluorouracil-based adjuvant therapy in colon cancer. J. Clin. Oncol. 2010, 28, 3219–3226. [Google Scholar] [CrossRef] [Green Version]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [Green Version]
- Kuismanen, S.A.; Holmberg, M.T.; Salovaara, R.; Schweizer, P.; Aaltonen, L.A.; de La Chapelle, A.; Nystrom-Lahti, M.; Peltomaki, P. Epigenetic phenotypes distinguish microsatellite-stable and -unstable colorectal cancers. Proc. Natl. Acad. Sci. USA 1999, 96, 12661–12666. [Google Scholar] [CrossRef] [Green Version]
- Ahuja, N.; Mohan, A.L.; Li, Q.; Stolker, J.M.; Herman, J.G.; Hamilton, S.R.; Baylin, S.B.; Issa, J.P. Association between CpG island methylation and microsatellite instability in colorectal cancer. Cancer Res. 1997, 57, 3370–3374. [Google Scholar]
- Toyota, M.; Ahuja, N.; Ohe-Toyota, M.; Herman, J.G.; Baylin, S.B.; Issa, J.P. CpG island methylator phenotype in colorectal cancer. Proc. Natl. Acad. Sci. USA 1999, 96, 8681–8686. [Google Scholar] [CrossRef] [Green Version]
- Kambara, T.; Simms, L.A.; Whitehall, V.L.; Spring, K.J.; Wynter, C.V.; Walsh, M.D.; Barker, M.A.; Arnold, S.; McGivern, A.; Matsubara, N.; et al. BRAF mutation is associated with DNA methylation in serrated polyps and cancers of the colorectum. GUT 2004, 53, 1137–1144. [Google Scholar] [CrossRef] [Green Version]
- Esteller, M.; Sparks, A.; Toyota, M.; Sanchez-Cespedes, M.; Capella, G.; Peinado, M.A.; Gonzalez, S.; Tarafa, G.; Sidransky, D.; Meltzer, S.J.; et al. Analysis of adenomatous polyposis coli promoter hypermethylation in human cancer. Cancer Res. 2000, 60, 4366–4371. [Google Scholar]
- Nosho, K.; Irahara, N.; Shima, K.; Kure, S.; Kirkner, G.J.; Schernhammer, E.S.; Hazra, A.; Hunter, D.J.; Quackenbush, J.; Spiegelman, D.; et al. Comprehensive biostatistical analysis of CpG island methylator phenotype in colorectal cancer using a large population-based sample. PLoS ONE 2008, 3, e3698. [Google Scholar] [CrossRef]
- Ogino, S.; Goel, A. Molecular classification and correlates in colorectal cancer. J. Mol. Diagn 2008, 10, 13–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogino, S.; Chan, A.T.; Fuchs, C.S.; Giovannucci, E. Molecular pathological epidemiology of colorectal neoplasia: An emerging transdisciplinary and interdisciplinary field. GUT 2011, 60, 397–411. [Google Scholar] [CrossRef]
- Miranda, E.; Bianchi, P.; Destro, A.; Morenghi, E.; Malesci, A.; Santoro, A.; Laghi, L.; Roncalli, M. Genetic and epigenetic alterations in primary colorectal cancers and related lymph node and liver metastases. Cancer 2013, 119, 266–276. [Google Scholar] [CrossRef]
- Sjoblom, T.; Jones, S.; Wood, L.D.; Parsons, D.W.; Lin, J.; Barber, T.D.; Mandelker, D.; Leary, R.J.; Ptak, J.; Silliman, N.; et al. The consensus coding sequences of human breast and colorectal cancers. Science 2006, 314, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Wood, L.D.; Parsons, D.W.; Jones, S.; Lin, J.; Sjoblom, T.; Leary, R.J.; Shen, D.; Boca, S.M.; Barber, T.; Ptak, J.; et al. The genomic landscapes of human breast and colorectal cancers. Science 2007, 318, 1108–1113. [Google Scholar] [CrossRef] [Green Version]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reynies, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef]
- Isella, C.; Terrasi, A.; Bellomo, S.E.; Petti, C.; Galatola, G.; Muratore, A.; Mellano, A.; Senetta, R.; Cassenti, A.; Sonetto, C.; et al. Stromal contribution to the colorectal cancer transcriptome. Nat. Genet. 2015, 47, 312–319. [Google Scholar] [CrossRef]
- Isella, C.; Brundu, F.; Bellomo, S.E.; Galimi, F.; Zanella, E.; Porporato, R.; Petti, C.; Fiori, A.; Orzan, F.; Senetta, R.; et al. Selective analysis of cancer-cell intrinsic transcriptional traits defines novel clinically relevant subtypes of colorectal cancer. Nat. Commun. 2017, 8, 15107. [Google Scholar] [CrossRef]
- Celesti, G.; Di Caro, G.; Bianchi, P.; Grizzi, F.; Basso, G.; Marchesi, F.; Doni, A.; Marra, G.; Roncalli, M.; Mantovani, A.; et al. Presence of Twist1-positive neoplastic cells in the stroma of chromosome-unstable colorectal tumors. Gastroenterology 2013, 145, 647–657.e615. [Google Scholar] [CrossRef]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pages, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Caro, G.; Marchesi, F.; Laghi, L.; Grizzi, F. Immune cells: Plastic players along colorectal cancer progression. J. Cell Mol. Med. 2013, 17, 1088–1095. [Google Scholar] [CrossRef] [PubMed]
- Grizzi, F.; Bianchi, P.; Malesci, A.; Laghi, L. Prognostic value of innate and adaptive immunity in colorectal cancer. World J. Gastroenterol. 2013, 19, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Angelova, M.; Mlecnik, B.; Vasaturo, A.; Bindea, G.; Fredriksen, T.; Lafontaine, L.; Buttard, B.; Morgand, E.; Bruni, D.; Jouret-Mourin, A.; et al. Evolution of Metastases in Space and Time under Immune Selection. Cell 2018, 175, 751–765.e716. [Google Scholar] [CrossRef] [Green Version]
- Laghi, L.; Bianchi, P.; Miranda, E.; Balladore, E.; Pacetti, V.; Grizzi, F.; Allavena, P.; Torri, V.; Repici, A.; Santoro, A.; et al. CD3+ cells at the invasive margin of deeply invading (pT3-T4) colorectal cancer and risk of post-surgical metastasis: A longitudinal study. Lancet Oncol. 2009, 10, 877–884. [Google Scholar] [CrossRef]
- Galon, J.; Angell, H.K.; Bedognetti, D.; Marincola, F.M. The continuum of cancer immunosurveillance: Prognostic, predictive, and mechanistic signatures. Immunity 2013, 39, 11–26. [Google Scholar] [CrossRef] [Green Version]
- Fridman, W.H.; Pages, F.; Sautes-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef]
- Galon, J.; Pages, F.; Marincola, F.M.; Thurin, M.; Trinchieri, G.; Fox, B.A.; Gajewski, T.F.; Ascierto, P.A. The immune score as a new possible approach for the classification of cancer. J. Transl Med. 2012, 10, 1. [Google Scholar] [CrossRef]
- Galon, J.; Mlecnik, B.; Bindea, G.; Angell, H.K.; Berger, A.; Lagorce, C.; Lugli, A.; Zlobec, I.; Hartmann, A.; Bifulco, C.; et al. Towards the introduction of the ‘Immunoscore’ in the classification of malignant tumours. J. Pathol. 2014, 232, 199–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [Green Version]
- Vesely, M.D.; Kershaw, M.H.; Schreiber, R.D.; Smyth, M.J. Natural innate and adaptive immunity to cancer. Annu. Rev. Immunol. 2011, 29, 235–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Zitvogel, L.; Palucka, A.K. Neutralizing tumor-promoting chronic inflammation: A magic bullet? Science 2013, 339, 286–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef]
- Zhang, L.; Li, Z.; Skrzypczynska, K.M.; Fang, Q.; Zhang, W.; O’Brien, S.A.; He, Y.; Wang, L.; Zhang, Q.; Kim, A.; et al. Single-Cell Analyses Inform Mechanisms of Myeloid-Targeted Therapies in Colon Cancer. Cell 2020, 181, 442–459.e429. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fridman, W.H.; Zitvogel, L.; Sautes-Fridman, C.; Kroemer, G. The immune contexture in cancer prognosis and treatment. Nat. Rev. Clin. Oncol. 2017, 14, 717–734. [Google Scholar] [CrossRef]
- Mikucki, M.E.; Fisher, D.T.; Matsuzaki, J.; Skitzki, J.J.; Gaulin, N.B.; Muhitch, J.B.; Ku, A.W.; Frelinger, J.G.; Odunsi, K.; Gajewski, T.F.; et al. Non-redundant requirement for CXCR3 signalling during tumoricidal T-cell trafficking across tumour vascular checkpoints. Nat. Commun. 2015, 6, 7458. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [Green Version]
- Bain, C.C.; Bravo-Blas, A.; Scott, C.L.; Perdiguero, E.G.; Geissmann, F.; Henri, S.; Malissen, B.; Osborne, L.C.; Artis, D.; Mowat, A.M. Constant replenishment from circulating monocytes maintains the macrophage pool in the intestine of adult mice. Nat. Immunol. 2014, 15, 929–937. [Google Scholar] [CrossRef] [Green Version]
- Eaden, J.A.; Abrams, K.R.; Mayberry, J.F. The risk of colorectal cancer in ulcerative colitis: A meta-analysis. Gut 2001, 48, 526–535. [Google Scholar] [CrossRef] [Green Version]
- Grivennikov, S.I.; Wang, K.; Mucida, D.; Stewart, C.A.; Schnabl, B.; Jauch, D.; Taniguchi, K.; Yu, G.Y.; Osterreicher, C.H.; Hung, K.E.; et al. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature 2012, 491, 254–258. [Google Scholar] [CrossRef] [Green Version]
- Shaked, H.; Hofseth, L.J.; Chumanevich, A.; Chumanevich, A.A.; Wang, J.; Wang, Y.; Taniguchi, K.; Guma, M.; Shenouda, S.; Clevers, H.; et al. Chronic epithelial NF-kappaB activation accelerates APC loss and intestinal tumor initiation through iNOS up-regulation. Proc. Natl. Acad. Sci. USA 2012, 109, 14007–14012. [Google Scholar] [CrossRef] [Green Version]
- Dmitrieva-Posocco, O.; Dzutsev, A.; Posocco, D.F.; Hou, V.; Yuan, W.; Thovarai, V.; Mufazalov, I.A.; Gunzer, M.; Shilovskiy, I.P.; Khaitov, M.R.; et al. Cell-Type-Specific Responses to Interleukin-1 Control Microbial Invasion and Tumor-Elicited Inflammation in Colorectal Cancer. Immunity 2019, 50, 166–180.e167. [Google Scholar] [CrossRef] [Green Version]
- West, N.R.; McCuaig, S.; Franchini, F.; Powrie, F. Emerging cytokine networks in colorectal cancer. Nat. Rev. Immunol. 2015, 15, 615–629. [Google Scholar] [CrossRef] [PubMed]
- Lasry, A.; Zinger, A.; Ben-Neriah, Y. Inflammatory networks underlying colorectal cancer. Nat. Immunol. 2016, 17, 230–240. [Google Scholar] [CrossRef] [PubMed]
- Mager, L.F.; Wasmer, M.H.; Rau, T.T.; Krebs, P. Cytokine-Induced Modulation of Colorectal Cancer. Front. Oncol. 2016, 6, 96. [Google Scholar] [CrossRef]
- Trinchieri, G. Cancer and inflammation: An old intuition with rapidly evolving new concepts. Annu. Rev. Immunol. 2012, 30, 677–706. [Google Scholar] [CrossRef]
- McCuaig, S.; Barras, D.; Mann, E.H.; Friedrich, M.; Bullers, S.J.; Janney, A.; Garner, L.C.; Domingo, E.; Koelzer, V.H.; Delorenzi, M.; et al. The Interleukin 22 Pathway Interacts with Mutant KRAS to Promote Poor Prognosis in Colon Cancer. Clin. Cancer Res. 2020, 26, 4313–4325. [Google Scholar] [CrossRef] [PubMed]
- Mlecnik, B.; Bindea, G.; Angell, H.K.; Sasso, M.S.; Obenauf, A.C.; Fredriksen, T.; Lafontaine, L.; Bilocq, A.M.; Kirilovsky, A.; Tosolini, M.; et al. Functional network pipeline reveals genetic determinants associated with in situ lymphocyte proliferation and survival of cancer patients. Sci. Transl. Med. 2014, 6, 228ra237. [Google Scholar] [CrossRef]
- Calon, A.; Lonardo, E.; Berenguer-Llergo, A.; Espinet, E.; Hernando-Momblona, X.; Iglesias, M.; Sevillano, M.; Palomo-Ponce, S.; Tauriello, D.V.; Byrom, D.; et al. Stromal gene expression defines poor-prognosis subtypes in colorectal cancer. Nat. Genet. 2015, 47, 320–329. [Google Scholar] [CrossRef] [Green Version]
- Di Caro, G.; Carvello, M.; Pesce, S.; Erreni, M.; Marchesi, F.; Todoric, J.; Sacchi, M.; Montorsi, M.; Allavena, P.; Spinelli, A. Circulating Inflammatory Mediators as Potential Prognostic Markers of Human Colorectal Cancer. PLoS ONE 2016, 11, e0148186. [Google Scholar] [CrossRef] [Green Version]
- Gunawardene, A.; Dennett, E.; Larsen, P. Prognostic value of multiple cytokine analysis in colorectal cancer: A systematic review. J. Gastrointest. Oncol. 2019, 10, 134–143. [Google Scholar] [CrossRef]
- Boakye, D.; Jansen, L.; Schottker, B.; Jansen, E.; Schneider, M.; Halama, N.; Gao, X.; Chang-Claude, J.; Hoffmeister, M.; Brenner, H. Blood markers of oxidative stress are strongly associated with poorer prognosis in colorectal cancer patients. Int. J. Cancer 2020, 147, 2373–2386. [Google Scholar] [CrossRef] [Green Version]
- Mlecnik, B.; Bindea, G.; Angell, H.K.; Maby, P.; Angelova, M.; Tougeron, D.; Church, S.E.; Lafontaine, L.; Fischer, M.; Fredriksen, T.; et al. Integrative Analyses of Colorectal Cancer Show Immunoscore Is a Stronger Predictor of Patient Survival Than Microsatellite Instability. Immunity 2016, 44, 698–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Yu, X.; Zheng, L.; Zhang, Y.; Li, Y.; Fang, Q.; Gao, R.; Kang, B.; Zhang, Q.; Huang, J.Y.; et al. Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature 2018, 564, 268–272. [Google Scholar] [CrossRef] [PubMed]
- Salama, P.; Phillips, M.; Grieu, F.; Morris, M.; Zeps, N.; Joseph, D.; Platell, C.; Iacopetta, B. Tumor-infiltrating FOXP3+ T regulatory cells show strong prognostic significance in colorectal cancer. J. Clin. Oncol. 2009, 27, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, K.; Tanaka, K.; Homma, Y.; Nojiri, K.; Kumamoto, T.; Takeda, K.; Endo, I. Low infiltration of peritumoral regulatory T cells predicts worse outcome following resection of colorectal liver metastases. Ann. Surg. Oncol. 2015, 22, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Cavalleri, T.; Bianchi, P.; Basso, G.; Celesti, G.; Grizzi, F.; Bossi, P.; Greco, L.; Pitrone, C.; Valtorta, E.; Mauri, G.; et al. Combined Low Densities of FoxP3(+) and CD3(+) Tumor-Infiltrating Lymphocytes Identify Stage II Colorectal Cancer at High Risk of Progression. Cancer Immunol. Res. 2019, 7, 751–758. [Google Scholar] [CrossRef] [Green Version]
- Whiteside, T.L. What are regulatory T cells (Treg) regulating in cancer and why? Semin Cancer Biol. 2012, 22, 327–334. [Google Scholar] [CrossRef] [Green Version]
- Coppola, D.; Nebozhyn, M.; Khalil, F.; Dai, H.; Yeatman, T.; Loboda, A.; Mule, J.J. Unique ectopic lymph node-like structures present in human primary colorectal carcinoma are identified by immune gene array profiling. Am. J. Pathol. 2011, 179, 37–45. [Google Scholar] [CrossRef]
- Di Caro, G.; Bergomas, F.; Grizzi, F.; Doni, A.; Bianchi, P.; Malesci, A.; Laghi, L.; Allavena, P.; Mantovani, A.; Marchesi, F. Occurrence of tertiary lymphoid tissue is associated with T-cell infiltration and predicts better prognosis in early-stage colorectal cancers. Clin. Cancer Res. 2014, 20, 2147–2158. [Google Scholar] [CrossRef] [Green Version]
- Sautes-Fridman, C.; Petitprez, F.; Calderaro, J.; Fridman, W.H. Tertiary lymphoid structures in the era of cancer immunotherapy. Nat. Rev. Cancer 2019, 19, 307–325. [Google Scholar] [CrossRef] [PubMed]
- Castino, G.F.; Cortese, N.; Capretti, G.; Serio, S.; Di Caro, G.; Mineri, R.; Magrini, E.; Grizzi, F.; Cappello, P.; Novelli, F.; et al. Spatial distribution of B cells predicts prognosis in human pancreatic adenocarcinoma. Oncoimmunology 2016, 5, e1085147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shalapour, S.; Font-Burgada, J.; Di Caro, G.; Zhong, Z.; Sanchez-Lopez, E.; Dhar, D.; Willimsky, G.; Ammirante, M.; Strasner, A.; Hansel, D.E.; et al. Immunosuppressive plasma cells impede T-cell-dependent immunogenic chemotherapy. Nature 2015, 521, 94–98. [Google Scholar] [CrossRef]
- Biswas, S.K.; Allavena, P.; Mantovani, A. Tumor-associated macrophages: Functional diversity, clinical significance, and open questions. Semin Immunopathol. 2013, 35, 585–600. [Google Scholar] [CrossRef] [PubMed]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeNardo, D.G.; Ruffell, B. Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 2019, 19, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Yang, C.; Wang, S.; Shi, D.; Wei, C.; Song, J.; Lin, X.; Dou, R.; Bai, J.; Xiang, Z.; et al. Wnt5a-induced M2 polarization of tumor-associated macrophages via IL-10 promotes colorectal cancer progression. Cell Commun. Signal. 2020, 18, 51. [Google Scholar] [CrossRef] [Green Version]
- Forssell, J.; Oberg, A.; Henriksson, M.L.; Stenling, R.; Jung, A.; Palmqvist, R. High macrophage infiltration along the tumor front correlates with improved survival in colon cancer. Clin. Cancer Res. 2007, 13, 1472–1479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Q.; Peng, R.Q.; Wu, X.J.; Xia, Q.; Hou, J.H.; Ding, Y.; Zhou, Q.M.; Zhang, X.; Pang, Z.Z.; Wan, D.S.; et al. The density of macrophages in the invasive front is inversely correlated to liver metastasis in colon cancer. J. Transl Med. 2010, 8, 13. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.W.; Liu, L.; Gong, C.Y.; Shi, H.S.; Zeng, Y.H.; Wang, X.Z.; Zhao, Y.W.; Wei, Y.Q. Prognostic significance of tumor-associated macrophages in solid tumor: A meta-analysis of the literature. PLoS ONE 2012, 7, e50946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malesci, A.; Bianchi, P.; Celesti, G.; Basso, G.; Marchesi, F.; Grizzi, F.; Di Caro, G.; Cavalleri, T.; Rimassa, L.; Palmqvist, R.; et al. Tumor-associated macrophages and response to 5-fluorouracil adjuvant therapy in stage III colorectal cancer. Oncoimmunology 2017, 6, e1342918. [Google Scholar] [CrossRef]
- Jass, J.R.; Do, K.A.; Simms, L.A.; Iino, H.; Wynter, C.; Pillay, S.P.; Searle, J.; Radford-Smith, G.; Young, J.; Leggett, B. Morphology of sporadic colorectal cancer with DNA replication errors. Gut 1998, 42, 673–679. [Google Scholar] [CrossRef]
- Rodriguez-Bigas, M.A.; Boland, C.R.; Hamilton, S.R.; Henson, D.E.; Jass, J.R.; Khan, P.M.; Lynch, H.; Perucho, M.; Smyrk, T.; Sobin, L.; et al. A National Cancer Institute Workshop on Hereditary Nonpolyposis Colorectal Cancer Syndrome: Meeting highlights and Bethesda guidelines. J. Natl. Cancer Inst. 1997, 89, 1758–1762. [Google Scholar] [CrossRef] [Green Version]
- Perucho, M. Microsatellite instability: The mutator that mutates the other mutator. Nat. Med. 1996, 2, 630–631. [Google Scholar] [CrossRef] [PubMed]
- Malkhosyan, S.; Rampino, N.; Yamamoto, H.; Perucho, M. Frameshift mutator mutations. Nature 1996, 382, 499–500. [Google Scholar] [CrossRef]
- Perucho, M. Tumors with microsatellite instability: Many mutations, targets and paradoxes. Oncogene 2003, 22, 2223–2225. [Google Scholar] [CrossRef] [Green Version]
- Schwitalle, Y.; Kloor, M.; Eiermann, S.; Linnebacher, M.; Kienle, P.; Knaebel, H.P.; Tariverdian, M.; Benner, A.; von Knebel Doeberitz, M. Immune response against frameshift-induced neopeptides in HNPCC patients and healthy HNPCC mutation carriers. Gastroenterology 2008, 134, 988–997. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Jen, J.; Vogelstein, B.; Hamilton, S.R. Clinical and pathological characteristics of sporadic colorectal carcinomas with DNA replication errors in microsatellite sequences. Am. J. Pathol. 1994, 145, 148–156. [Google Scholar]
- Domingo, E.; Freeman-Mills, L.; Rayner, E.; Glaire, M.; Briggs, S.; Vermeulen, L.; Fessler, E.; Medema, J.P.; Boot, A.; Morreau, H.; et al. Somatic POLE proofreading domain mutation, immune response, and prognosis in colorectal cancer: A retrospective, pooled biomarker study. Lancet Gastroenterol. Hepatol. 2016, 1, 207–216. [Google Scholar] [CrossRef] [Green Version]
- Mo, S.; Ma, X.; Li, Y.; Zhang, L.; Hou, T.; Han-Zhang, H.; Qian, J.; Cai, S.; Huang, D.; Peng, J. Somatic POLE exonuclease domain mutations elicit enhanced intratumoral immune responses in stage II colorectal cancer. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef]
- Vayrynen, J.P.; Haruki, K.; Lau, M.C.; Vayrynen, S.A.; Zhong, R.; Dias Costa, A.; Borowsky, J.; Zhao, M.; Fujiyoshi, K.; Arima, K.; et al. The Prognostic Role of Macrophage Polarization in the Colorectal Cancer Microenvironment. Cancer Immunol. Res. 2021, 9, 8–19. [Google Scholar] [CrossRef]
- Laghi, L.; Negri, F.; Gaiani, F.; Cavalleri, T.; Grizzi, F.; De’ Angelis, G.L.; Malesci, A. Prognostic and Predictive Cross-Roads of Microsatellite Instability and Immune Response to Colon Cancer. Int. J. Mol. Sci. 2020, 21, 9680. [Google Scholar] [CrossRef]
- Giannakis, M.; Mu, X.J.; Shukla, S.A.; Qian, Z.R.; Cohen, O.; Nishihara, R.; Bahl, S.; Cao, Y.; Amin-Mansour, A.; Yamauchi, M.; et al. Genomic Correlates of Immune-Cell Infiltrates in Colorectal Carcinoma. Cell Rep. 2016, 17, 1206. [Google Scholar] [CrossRef] [PubMed]
- Jussila, A.; Virta, L.J.; Pukkala, E.; Farkkila, M.A. Malignancies in patients with inflammatory bowel disease: A nationwide register study in Finland. Scand. J. Gastroenterol. 2013, 48, 1405–1413. [Google Scholar] [CrossRef] [PubMed]
- Jess, T.; Horvath-Puho, E.; Fallingborg, J.; Rasmussen, H.H.; Jacobsen, B.A. Cancer risk in inflammatory bowel disease according to patient phenotype and treatment: A Danish population-based cohort study. Am. J. Gastroenterol. 2013, 108, 1869–1876. [Google Scholar] [CrossRef]
- Kappelman, M.D.; Farkas, D.K.; Long, M.D.; Erichsen, R.; Sandler, R.S.; Sorensen, H.T.; Baron, J.A. Risk of cancer in patients with inflammatory bowel diseases: A nationwide population-based cohort study with 30 years of follow-up evaluation. Clin. Gastroenterol. Hepatol. 2014, 12, 265–273.e261. [Google Scholar] [CrossRef] [Green Version]
- Peneau, A.; Savoye, G.; Turck, D.; Dauchet, L.; Fumery, M.; Salleron, J.; Lerebours, E.; Ligier, K.; Vasseur, F.; Dupas, J.L.; et al. Mortality and cancer in pediatric-onset inflammatory bowel disease: A population-based study. Am. J. Gastroenterol. 2013, 108, 1647–1653. [Google Scholar] [CrossRef]
- Duricova, D.; Fumery, M.; Annese, V.; Lakatos, P.L.; Peyrin-Biroulet, L.; Gower-Rousseau, C. The natural history of Crohn’s disease in children: A review of population-based studies. Eur. J. Gastroenterol. Hepatol. 2017, 29, 125–134. [Google Scholar] [CrossRef]
- Iannone, A.; Ruospo, M.; Wong, G.; Principi, M.; Barone, M.; Strippoli, G.F.M.; Di Leo, A. Chromoendoscopy for Surveillance in Ulcerative Colitis and Crohn’s Disease: A Systematic Review of Randomized Trials. Clin. Gastroenterol. Hepatol. 2017, 15, 1684–1697.e1611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutgens, M.W.; van Oijen, M.G.; van der Heijden, G.J.; Vleggaar, F.P.; Siersema, P.D.; Oldenburg, B. Declining risk of colorectal cancer in inflammatory bowel disease: An updated meta-analysis of population-based cohort studies. Inflamm Bowel. Dis. 2013, 19, 789–799. [Google Scholar] [CrossRef]
- Choi, C.R.; Bakir, I.A.; Hart, A.L.; Graham, T.A. Clonal evolution of colorectal cancer in IBD. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 218–229. [Google Scholar] [CrossRef]
- Bopanna, S.; Roy, M.; Das, P.; Dattagupta, S.; Sreenivas, V.; Mouli, V.P.; Kedia, S.; Dhingra, R.; Pradhan, R.; Kumar, N.S.; et al. Role of random biopsies in surveillance of dysplasia in ulcerative colitis patients with high risk of colorectal cancer. Intest. Res. 2016, 14, 264–269. [Google Scholar] [CrossRef] [Green Version]
- Dulai, P.S.; Sandborn, W.J.; Gupta, S. Colorectal Cancer and Dysplasia in Inflammatory Bowel Disease: A Review of Disease Epidemiology, Pathophysiology, and Management. Cancer Prev. Res. 2016, 9, 887–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fornaro, R.; Caratto, M.; Caratto, E.; Caristo, G.; Fornaro, F.; Giovinazzo, D.; Sticchi, C.; Casaccia, M.; Andorno, E. Colorectal Cancer in Patients With Inflammatory Bowel Disease: The Need for a Real Surveillance Program. Clin. Colorectal. Cancer 2016, 15, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Jewel Samadder, N.; Valentine, J.F.; Guthery, S.; Singh, H.; Bernstein, C.N.; Wan, Y.; Wong, J.; Boucher, K.; Pappas, L.; Rowe, K.; et al. Colorectal Cancer in Inflammatory Bowel Diseases: A Population-Based Study in Utah. Dig. Dis. Sci. 2017, 62, 2126–2132. [Google Scholar] [CrossRef] [PubMed]
- Hata, K.; Kishikawa, J.; Anzai, H.; Shinagawa, T.; Kazama, S.; Ishii, H.; Nozawa, H.; Kawai, K.; Kiyomatsu, T.; Tanaka, J.; et al. Surveillance colonoscopy for colitis-associated dysplasia and cancer in ulcerative colitis patients. Dig. Endosc. 2016, 28, 260–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Y.D.; Al Bandar, M.H.; Dulskas, A.; Cho, M.S.; Hur, H.; Min, B.S.; Lee, K.Y.; Kim, N.K. Prognosis of ulcerative colitis colorectal cancer vs. sporadic colorectal cancer: Propensity score matching analysis. BMC Surg. 2017, 17, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sengupta, N.; Yee, E.; Feuerstein, J.D. Colorectal Cancer Screening in Inflammatory Bowel Disease. Dig. Dis. Sci. 2016, 61, 980–989. [Google Scholar] [CrossRef] [PubMed]
- Annese, V.; Beaugerie, L.; Egan, L.; Biancone, L.; Bolling, C.; Brandts, C.; Dierickx, D.; Dummer, R.; Fiorino, G.; Gornet, J.M.; et al. European Evidence-based Consensus: Inflammatory Bowel Disease and Malignancies. J. Crohns Colitis 2015, 9, 945–965. [Google Scholar] [CrossRef] [Green Version]
- Fumery, M.; Pineton de Chambrun, G.; Stefanescu, C.; Buisson, A.; Bressenot, A.; Beaugerie, L.; Amiot, A.; Altwegg, R.; Savoye, G.; Abitbol, V.; et al. Detection of Dysplasia or Cancer in 3.5% of Patients With Inflammatory Bowel Disease and Colonic Strictures. Clin. Gastroenterol. Hepatol. 2015, 13, 1770–1775. [Google Scholar] [CrossRef]
- Sonnenberg, A.; Genta, R.M. Epithelial Dysplasia and Cancer in IBD Strictures. J. Crohns Colitis 2015, 9, 769–775. [Google Scholar] [CrossRef] [Green Version]
- Popp, C.; Nichita, L.; Voiosu, T.; Bastian, A.; Cioplea, M.; Micu, G.; Pop, G.; Sticlaru, L.; Bengus, A.; Voiosu, A.; et al. Expression Profile of p53 and p21 in Large Bowel Mucosa as Biomarkers of Inflammatory-Related Carcinogenesis in Ulcerative Colitis. Dis. Markers 2016, 2016, 3625279. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, M.; Matsumoto, S. Gut microbiota and colorectal cancer. Genes Environ. 2016, 38, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanauchi, O.; Mitsuyama, K.; Andoh, A. The new prophylactic strategy for colon cancer in inflammatory bowel disease by modulating microbiota. Scand. J. Gastroenterol. 2013, 48, 387–400. [Google Scholar] [CrossRef] [PubMed]
- Messaritakis, I.; Koulouridi, A.; Sfakianaki, M.; Vogiatzoglou, K.; Gouvas, N.; Athanasakis, E.; Tsiaoussis, J.; Xynos, E.; Mavroudis, D.; Tzardi, M.; et al. The Role of Vitamin D Receptor Gene Polymorphisms in Colorectal Cancer Risk. Cancers 2020, 12, 1379. [Google Scholar] [CrossRef] [PubMed]
- Markowska, A.; Antoszczak, M.; Kojs, Z.; Bednarek, W.; Markowska, J.; Huczynski, A. Role of vitamin D3 in selected malignant neoplasms. Nutrition 2020, 79–80, 110964. [Google Scholar] [CrossRef]
- Sadeghi, H.; Nazemalhosseini-Mojarad, E.; Sahebi, U.; Fazeli, E.; Azizi-Tabesh, G.; Yassaee, V.R.; Savabkar, S.; Asadzadeh Aghdaei, H.; Zali, M.R.; Mirfakhraie, R. Novel long noncoding RNAs upregulation may have synergistic effects on the CYP24A1 and PFDN4 biomarker role in human colorectal cancer. J. Cell Physiol. 2021, 236, 2051–2057. [Google Scholar] [CrossRef]
- Sadeghi, H.; Heiat, M. A novel circular RNA hsa_circ_0060927 may serve as a potential diagnostic biomarker for human colorectal cancer. Mol. Biol Rep. 2020, 47, 6649–6655. [Google Scholar] [CrossRef]
- Flores, O.; Wang, Z.; Knudsen, K.E.; Burnstein, K.L. Nuclear targeting of cyclin-dependent kinase 2 reveals essential roles of cyclin-dependent kinase 2 localization and cyclin E in vitamin D-mediated growth inhibition. Endocrinology 2010, 151, 896–908. [Google Scholar] [CrossRef] [Green Version]
- Jensen, S.S.; Madsen, M.W.; Lukas, J.; Binderup, L.; Bartek, J. Inhibitory effects of 1alpha,25-dihydroxyvitamin D(3) on the G(1)-S phase-controlling machinery. Mol. Endocrinol. 2001, 15, 1370–1380. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Qin, X.; Liu, M.; Wang, X. The burgeoning role of cytochrome P450-mediated vitamin D metabolites against colorectal cancer. Pharmacol. Res. 2018, 133, 9–20. [Google Scholar] [CrossRef]
- Feldman, D.; Krishnan, A.V.; Swami, S.; Giovannucci, E.; Feldman, B.J. The role of vitamin D in reducing cancer risk and progression. Nat. Rev. Cancer 2014, 14, 342–357. [Google Scholar] [CrossRef]
- Kure, S.; Nosho, K.; Baba, Y.; Irahara, N.; Shima, K.; Ng, K.; Meyerhardt, J.A.; Giovannucci, E.L.; Fuchs, C.S.; Ogino, S. Vitamin D receptor expression is associated with PIK3CA and KRAS mutations in colorectal cancer. Cancer Epidemiol. Biomarkers Prev. 2009, 18, 2765–2772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mocellin, S. Vitamin D and cancer: Deciphering the truth. Biochim Biophys Acta 2011, 1816, 172–178. [Google Scholar] [CrossRef]
- Gandini, S.; Boniol, M.; Haukka, J.; Byrnes, G.; Cox, B.; Sneyd, M.J.; Mullie, P.; Autier, P. Meta-analysis of observational studies of serum 25-hydroxyvitamin D levels and colorectal, breast and prostate cancer and colorectal adenoma. Int. J. Cancer 2011, 128, 1414–1424. [Google Scholar] [CrossRef] [PubMed]
- Pendas-Franco, N.; Garcia, J.M.; Pena, C.; Valle, N.; Palmer, H.G.; Heinaniemi, M.; Carlberg, C.; Jimenez, B.; Bonilla, F.; Munoz, A.; et al. DICKKOPF-4 is induced by TCF/beta-catenin and upregulated in human colon cancer, promotes tumour cell invasion and angiogenesis and is repressed by 1alpha,25-dihydroxyvitamin D3. Oncogene 2008, 27, 4467–4477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meeker, S.; Seamons, A.; Maggio-Price, L.; Paik, J. Protective links between vitamin D, inflammatory bowel disease and colon cancer. World J. Gastroenterol. 2016, 22, 933–948. [Google Scholar] [CrossRef]
- Shintani, T.; Rosli, S.N.Z.; Takatsu, F.; Choon, Y.F.; Hayashido, Y.; Toratani, S.; Usui, E.; Okamoto, T. Eldecalcitol (ED-71), an analog of 1alpha,25-dihydroxyvitamin D3 as a potential anti-cancer agent for oral squamous cell carcinomas. J. Steroid Biochem. Mol. Biol. 2016, 164, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Bandera Merchan, B.; Morcillo, S.; Martin-Nunez, G.; Tinahones, F.J.; Macias-Gonzalez, M. The role of vitamin D and VDR in carcinogenesis: Through epidemiology and basic sciences. J. Steroid Biochem. Mol. Biol. 2017, 167, 203–218. [Google Scholar] [CrossRef]
- Wesselink, E.; Balvers, M.; Bours, M.J.L.; de Wilt, J.H.W.; Witkamp, R.F.; van Baar, H.; Geijsen, A.; van Halteren, H.; Keulen, E.T.P.; Kok, D.E.; et al. The association between circulating levels of vitamin D and inflammatory markers in the first 2 years after colorectal cancer diagnosis. Therap Adv. Gastroenterol. 2020, 13, 1756284820923922. [Google Scholar] [CrossRef]
- Krishnan, A.V.; Feldman, D. Mechanisms of the anti-cancer and anti-inflammatory actions of vitamin D. Annu. Rev. Pharmacol. Toxicol. 2011, 51, 311–336. [Google Scholar] [CrossRef] [Green Version]
- Karin, M.; Lin, A. NF-kappaB at the crossroads of life and death. Nat. Immunol. 2002, 3, 221–227. [Google Scholar] [CrossRef]
- Al-Jaderi, Z.; Maghazachi, A.A. Effects of vitamin D3, calcipotriol and FTY720 on the expression of surface molecules and cytolytic activities of human natural killer cells and dendritic cells. Toxins 2013, 5, 1932–1947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theodoratou, E.; Farrington, S.M.; Tenesa, A.; McNeill, G.; Cetnarskyj, R.; Barnetson, R.A.; Porteous, M.E.; Dunlop, M.G.; Campbell, H. Modification of the inverse association between dietary vitamin D intake and colorectal cancer risk by a FokI variant supports a chemoprotective action of Vitamin D intake mediated through VDR binding. Int. J. Cancer 2008, 123, 2170–2179. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; He, B.; Pan, Y.; Deng, Q.; Sun, H.; Li, R.; Gao, T.; Song, G.; Wang, S. Systematic review and meta-analysis on vitamin D receptor polymorphisms and cancer risk. Tumour Biol. 2014, 35, 4153–4169. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Fang, Y.J.; Feng, X.L.; Abulimiti, A.; Huang, C.Y.; Luo, H.; Zhang, C.X. Interactions Between Vitamin D and Calcium Intake, Vitamin D Receptor Genetic Polymorphisms, and Colorectal Cancer Risk. Dig. Dis. Sci. 2020. [Google Scholar] [CrossRef]
- Yu, K.; Yang, J.; Jiang, Y.; Song, R.; Lu, Q. Vitamin D receptor BsmI polymorphism and colorectal cancer risk: An updated analysis. Asian Pac. J. Cancer Prev. 2014, 15, 4801–4807. [Google Scholar] [CrossRef] [Green Version]
- Slattery, M.L.; Wolff, R.K.; Curtin, K.; Fitzpatrick, F.; Herrick, J.; Potter, J.D.; Caan, B.J.; Samowitz, W.S. Colon tumor mutations and epigenetic changes associated with genetic polymorphism: Insight into disease pathways. Mutat Res. 2009, 660, 12–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keum, N.; Giovannucci, E. Vitamin D supplements and cancer incidence and mortality: A meta-analysis. Br. J. Cancer 2014, 111, 976–980. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.; Feskanich, D.; Fuchs, C.S.; Willett, W.C.; Hollis, B.W.; Giovannucci, E.L. A nested case control study of plasma 25-hydroxyvitamin D concentrations and risk of colorectal cancer. J. Natl. Cancer Inst. 2007, 99, 1120–1129. [Google Scholar] [CrossRef] [Green Version]
- Jenab, M.; Bueno-de-Mesquita, H.B.; Ferrari, P.; van Duijnhoven, F.J.; Norat, T.; Pischon, T.; Jansen, E.H.; Slimani, N.; Byrnes, G.; Rinaldi, S.; et al. Association between pre-diagnostic circulating vitamin D concentration and risk of colorectal cancer in European populations:a nested case-control study. BMJ 2010, 340, b5500. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.E.; Li, H.; Chan, A.T.; Hollis, B.W.; Lee, I.M.; Stampfer, M.J.; Wu, K.; Giovannucci, E.; Ma, J. Circulating levels of vitamin D and colon and rectal cancer: The Physicians’ Health Study and a meta-analysis of prospective studies. Cancer Prev. Res. 2011, 4, 735–743. [Google Scholar] [CrossRef] [Green Version]
- Yin, L.; Grandi, N.; Raum, E.; Haug, U.; Arndt, V.; Brenner, H. Meta-analysis: Longitudinal studies of serum vitamin D and colorectal cancer risk. Aliment. Pharmacol. Ther. 2009, 30, 113–125. [Google Scholar] [CrossRef]
- Li, M.; Chen, P.; Li, J.; Chu, R.; Xie, D.; Wang, H. Review: The impacts of circulating 25-hydroxyvitamin D levels on cancer patient outcomes: A systematic review and meta-analysis. J. Clin. Endocrinol. Metab. 2014, 99, 2327–2336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, C.; Song, M.; Zhang, Y.; Wolpin, B.M.; Meyerhardt, J.A.; Ogino, S.; Hollis, B.W.; Chan, A.T.; Fuchs, C.S.; Wu, K.; et al. Prediagnostic Circulating Concentrations of Vitamin D Binding Protein and Survival among Patients with Colorectal Cancer. Cancer Epidemiol. Biomarkers Prev. 2020, 29, 2323–2331. [Google Scholar] [CrossRef] [PubMed]
- Vaughan-Shaw, P.G.; Buijs, L.F.; Blackmur, J.P.; Theodoratou, E.; Zgaga, L.; Din, F.V.N.; Farrington, S.M.; Dunlop, M.G. The effect of vitamin D supplementation on survival in patients with colorectal cancer: Systematic review and meta-analysis of randomised controlled trials. Br. J. Cancer 2020, 123, 1705–1712. [Google Scholar] [CrossRef] [PubMed]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Ion Channels in Cancer: Are Cancer Hallmarks Oncochannelopathies? Physiol. Rev. 2018, 98, 559–621. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.J.; Kim, S.Y.; Lee, S.; Jeon, J.H.; Matsui, H.; Kwon, Y.K.; Kim, S.J.; So, I. The role of transient receptor potential channel blockers in human gastric cancer cell viability. Can. J. Physiol. Pharmacol. 2012, 90, 175–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, B.; Zhao, R.; Bai, B.; Wu, Y.; Xu, Y.; Lu, S.; Fang, Y.; Wang, Z.; Maswikiti, E.P.; Zhou, X.; et al. Identification of key tumorigenesisrelated genes and their microRNAs in colon cancer. Oncol. Rep. 2018, 40, 3551–3560. [Google Scholar] [CrossRef]
- Wang, C.Y.; Lai, M.D.; Phan, N.N.; Sun, Z.; Lin, Y.C. Meta-Analysis of Public Microarray Datasets Reveals Voltage-Gated Calcium Gene Signatures in Clinical Cancer Patients. PLoS ONE 2015, 10, e0125766. [Google Scholar] [CrossRef] [Green Version]
- Villalobos, C.; Sobradillo, D.; Hernandez-Morales, M.; Nunez, L. Calcium remodeling in colorectal cancer. Biochim Biophys Acta Mol. Cell Res. 2017, 1864, 843–849. [Google Scholar] [CrossRef]
- Lipkin, M.; Newmark, H. Effect of added dietary calcium on colonic epithelial-cell proliferation in subjects at high risk for familial colonic cancer. N. Engl. J. Med. 1985, 313, 1381–1384. [Google Scholar] [CrossRef]
- Garland, C.; Shekelle, R.B.; Barrett-Connor, E.; Criqui, M.H.; Rossof, A.H.; Paul, O. Dietary vitamin D and calcium and risk of colorectal cancer: A 19-year prospective study in men. Lancet 1985, 1, 307–309. [Google Scholar] [CrossRef]
- Bonovas, S.; Fiorino, G.; Lytras, T.; Malesci, A.; Danese, S. Calcium supplementation for the prevention of colorectal adenomas: A systematic review and meta-analysis of randomized controlled trials. World J. Gastroenterol. 2016, 22, 4594–4603. [Google Scholar] [CrossRef] [PubMed]
- Newmark, H.L.; Wargovich, M.J.; Bruce, W.R. Colon cancer and dietary fat, phosphate, and calcium: A hypothesis. J. Natl. Cancer Inst. 1984, 72, 1323–1325. [Google Scholar] [PubMed]
- Pence, B.C. Role of calcium in colon cancer prevention: Experimental and clinical studies. Mutat Res. 1993, 290, 87–95. [Google Scholar] [CrossRef]
- Wang, W.; Yu, S.; Huang, S.; Deng, R.; Ding, Y.; Wu, Y.; Li, X.; Wang, A.; Wang, S.; Chen, W.; et al. A Complex Role for Calcium Signaling in Colorectal Cancer Development and Progression. Mol. Cancer Res. 2019, 17, 2145–2153. [Google Scholar] [CrossRef] [Green Version]
- Yashiro, M. Ulcerative colitis-associated colorectal cancer. World J. Gastroenterol. 2014, 20, 16389–16397. [Google Scholar] [CrossRef]
- Velayos, F.S.; Terdiman, J.P.; Walsh, J.M. Effect of 5-aminosalicylate use on colorectal cancer and dysplasia risk: A systematic review and metaanalysis of observational studies. Am. J. Gastroenterol. 2005, 100, 1345–1353. [Google Scholar] [CrossRef]
- Nguyen, G.C.; Gulamhusein, A.; Bernstein, C.N. 5-aminosalicylic acid is not protective against colorectal cancer in inflammatory bowel disease: A meta-analysis of non-referral populations. Am. J. Gastroenterol. 2012, 107, 1298–1304. [Google Scholar] [CrossRef] [PubMed]
- Cahill, C.; Gordon, P.H.; Petrucci, A.; Boutros, M. Small bowel adenocarcinoma and Crohn’s disease: Any further ahead than 50 years ago? World J. Gastroenterol 2014, 20, 11486–11495. [Google Scholar] [CrossRef]
- Gong, J.; Zhu, L.; Guo, Z.; Li, Y.; Zhu, W.; Li, N.; Li, J. Use of thiopurines and risk of colorectal neoplasia in patients with inflammatory bowel diseases: A meta-analysis. PLoS ONE 2013, 8, e81487. [Google Scholar] [CrossRef]
- Arber, N.; Levin, B. Chemoprevention of colorectal neoplasia: The potential for personalized medicine. Gastroenterology 2008, 134, 1224–1237. [Google Scholar] [CrossRef] [Green Version]
- Kune, G.A.; Kune, S.; Watson, L.F. Colorectal cancer risk, chronic illnesses, operations, and medications: Case control results from the Melbourne Colorectal Cancer Study. Cancer Res. 1988, 48, 4399–4404. [Google Scholar] [CrossRef] [Green Version]
- Giovannucci, E.; Egan, K.M.; Hunter, D.J.; Stampfer, M.J.; Colditz, G.A.; Willett, W.C.; Speizer, F.E. Aspirin and the risk of colorectal cancer in women. N. Engl. J. Med. 1995, 333, 609–614. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.T.; Giovannucci, E.L.; Meyerhardt, J.A.; Schernhammer, E.S.; Wu, K.; Fuchs, C.S. Aspirin dose and duration of use and risk of colorectal cancer in men. Gastroenterology 2008, 134, 21–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinakaran, D.; Sergi, C.M. Co-ingestion of aspirin and acetaminophen promoting fulminant liver failure: A critical review of Reye syndrome in the current perspective at the dawn of the 21st century. Clin. Exp. Pharmacol. Physiol. 2018, 45, 117–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, A.T.; Ogino, S.; Fuchs, C.S. Aspirin and the risk of colorectal cancer in relation to the expression of COX-2. N. Engl. J. Med. 2007, 356, 2131–2142. [Google Scholar] [CrossRef]
- Steinbach, G.; Lynch, P.M.; Phillips, R.K.; Wallace, M.H.; Hawk, E.; Gordon, G.B.; Wakabayashi, N.; Saunders, B.; Shen, Y.; Fujimura, T.; et al. The effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenomatous polyposis. N. Engl. J. Med. 2000, 342, 1946–1952. [Google Scholar] [CrossRef] [PubMed]
- Burn, J.; Sheth, H.; Elliott, F.; Reed, L.; Macrae, F.; Mecklin, J.P.; Moslein, G.; McRonald, F.E.; Bertario, L.; Evans, D.G.; et al. Cancer prevention with aspirin in hereditary colorectal cancer (Lynch syndrome), 10-year follow-up and registry-based 20-year data in the CAPP2 study: A double-blind, randomised, placebo-controlled trial. Lancet 2020, 395, 1855–1863. [Google Scholar] [CrossRef]
- Alexander, J.L.; Scott, A.J.; Pouncey, A.L.; Marchesi, J.; Kinross, J.; Teare, J. Colorectal carcinogenesis: An archetype of gut microbiota-host interaction. Ecancermedicalscience 2018, 12, 865. [Google Scholar] [CrossRef] [Green Version]
- Gholizadeh, P.; Eslami, H.; Kafil, H.S. Carcinogenesis mechanisms of Fusobacterium nucleatum. Biomed. Pharmacother. 2017, 89, 918–925. [Google Scholar] [CrossRef]
- Kostic, A.D.; Chun, E.; Robertson, L.; Glickman, J.N.; Gallini, C.A.; Michaud, M.; Clancy, T.E.; Chung, D.C.; Lochhead, P.; Hold, G.L.; et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe 2013, 14, 207–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakatsu, G.; Li, X.; Zhou, H.; Sheng, J.; Wong, S.H.; Wu, W.K.; Ng, S.C.; Tsoi, H.; Dong, Y.; Zhang, N.; et al. Gut mucosal microbiome across stages of colorectal carcinogenesis. Nat. Commun. 2015, 6, 8727. [Google Scholar] [CrossRef]
- Castellarin, M.; Warren, R.L.; Freeman, J.D.; Dreolini, L.; Krzywinski, M.; Strauss, J.; Barnes, R.; Watson, P.; Allen-Vercoe, E.; Moore, R.A.; et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 2012, 22, 299–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kostic, A.D.; Gevers, D.; Pedamallu, C.S.; Michaud, M.; Duke, F.; Earl, A.M.; Ojesina, A.I.; Jung, J.; Bass, A.J.; Tabernero, J.; et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 2012, 22, 292–298. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Chen, J.; Yao, H.; Hu, H. Fusobacterium and Colorectal Cancer. Front. Oncol. 2018, 8, 371. [Google Scholar] [CrossRef] [PubMed]
- Gur, C.; Ibrahim, Y.; Isaacson, B.; Yamin, R.; Abed, J.; Gamliel, M.; Enk, J.; Bar-On, Y.; Stanietsky-Kaynan, N.; Coppenhagen-Glazer, S.; et al. Binding of the Fap2 protein of Fusobacterium nucleatum to human inhibitory receptor TIGIT protects tumors from immune cell attack. Immunity 2015, 42, 344–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubinstein, M.R.; Baik, J.E.; Lagana, S.M.; Han, R.P.; Raab, W.J.; Sahoo, D.; Dalerba, P.; Wang, T.C.; Han, Y.W. Fusobacterium nucleatum promotes colorectal cancer by inducing Wnt/beta-catenin modulator Annexin A1. EMBO Rep. 2019, 20. [Google Scholar] [CrossRef]
- Yan, X.; Liu, L.; Li, H.; Qin, H.; Sun, Z. Clinical significance of Fusobacterium nucleatum, epithelial-mesenchymal transition, and cancer stem cell markers in stage III/IV colorectal cancer patients. Onco Targets Ther. 2017, 10, 5031–5046. [Google Scholar] [CrossRef] [Green Version]
- Ma, C.T.; Luo, H.S.; Gao, F.; Tang, Q.C.; Chen, W. Fusobacterium nucleatum promotes the progression of colorectal cancer by interacting with E-cadherin. Oncol. Lett. 2018, 16, 2606–2612. [Google Scholar] [CrossRef] [Green Version]
- Bullman, S.; Pedamallu, C.S.; Sicinska, E.; Clancy, T.E.; Zhang, X.; Cai, D.; Neuberg, D.; Huang, K.; Guevara, F.; Nelson, T.; et al. Analysis of Fusobacterium persistence and antibiotic response in colorectal cancer. Science 2017, 358, 1443–1448. [Google Scholar] [CrossRef] [Green Version]
- Yu, T.; Guo, F.; Yu, Y.; Sun, T.; Ma, D.; Han, J.; Qian, Y.; Kryczek, I.; Sun, D.; Nagarsheth, N.; et al. Fusobacterium nucleatum Promotes Chemoresistance to Colorectal Cancer by Modulating Autophagy. Cell 2017, 170, 548–563.e516. [Google Scholar] [CrossRef] [Green Version]
- Mima, K.; Nishihara, R.; Qian, Z.R.; Cao, Y.; Sukawa, Y.; Nowak, J.A.; Yang, J.; Dou, R.; Masugi, Y.; Song, M.; et al. Fusobacterium nucleatum in colorectal carcinoma tissue and patient prognosis. Gut 2016, 65, 1973–1980. [Google Scholar] [CrossRef] [Green Version]
- Hamada, T.; Zhang, X.; Mima, K.; Bullman, S.; Sukawa, Y.; Nowak, J.A.; Kosumi, K.; Masugi, Y.; Twombly, T.S.; Cao, Y.; et al. Fusobacterium nucleatum in Colorectal Cancer Relates to Immune Response Differentially by Tumor Microsatellite Instability Status. Cancer Immunol. Res. 2018, 6, 1327–1336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tahara, T.; Yamamoto, E.; Suzuki, H.; Maruyama, R.; Chung, W.; Garriga, J.; Jelinek, J.; Yamano, H.O.; Sugai, T.; An, B.; et al. Fusobacterium in colonic flora and molecular features of colorectal carcinoma. Cancer Res. 2014, 74, 1311–1318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, M.; Kanno, S.; Nosho, K.; Sukawa, Y.; Mitsuhashi, K.; Kurihara, H.; Igarashi, H.; Takahashi, T.; Tachibana, M.; Takahashi, H.; et al. Association of Fusobacterium nucleatum with clinical and molecular features in colorectal serrated pathway. Int. J. Cancer 2015, 137, 1258–1268. [Google Scholar] [CrossRef]
- De Angelis, G.L.; Bottarelli, L.; Azzoni, C.; De Angelis, N.; Leandro, G.; Di Mario, F.; Gaiani, F.; Negri, F. Microsatellite instability in colorectal cancer. Acta Biomed. 2018, 89, 97–101. [Google Scholar] [CrossRef]
- Serna, G.; Ruiz-Pace, F.; Hernando, J.; Alonso, L.; Fasani, R.; Landolfi, S.; Comas, R.; Jimenez, J.; Elez, E.; Bullman, S.; et al. Fusobacterium nucleatum persistence and risk of recurrence after preoperative treatment in locally advanced rectal cancer. Ann. Oncol. 2020, 31, 1366–1375. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.; Shen, L.; Shi, W.; Xia, F.; Zhang, H.; Wang, Y.; Zhang, J.; Wang, Y.; Sun, X.; Zhang, Z.; et al. Gut Microbiome Components Predict Response to Neoadjuvant Chemoradiotherapy in Patients with Locally Advanced Rectal Cancer: A Prospective, Longitudinal Study. Clin. Cancer Res. 2021, 27, 1329–1340. [Google Scholar] [CrossRef]
- Kattan, M.W.; Hess, K.R.; Amin, M.B.; Lu, Y.; Moons, K.G.; Gershenwald, J.E.; Gimotty, P.A.; Guinney, J.H.; Halabi, S.; Lazar, A.J.; et al. American Joint Committee on Cancer acceptance criteria for inclusion of risk models for individualized prognosis in the practice of precision medicine. CA Cancer J. Clin. 2016, 66, 370–374. [Google Scholar] [CrossRef] [Green Version]
- Asare, E.A.; Washington, M.K.; Gress, D.M.; Gershenwald, J.E.; Greene, F.L. Improving the quality of cancer staging. CA Cancer J. Clin. 2015, 65, 261–263. [Google Scholar] [CrossRef]
- Reilly, B.M.; Evans, A.T. Translating clinical research into clinical practice: Impact of using prediction rules to make decisions. Ann. Intern. Med. 2006, 144, 201–209. [Google Scholar] [CrossRef]
- Gervasoni, J.E., Jr.; Sbayi, S.; Cady, B. Role of lymphadenectomy in surgical treatment of solid tumors: An update on the clinical data. Ann. Surg. Oncol. 2007, 14, 2443–2462. [Google Scholar] [CrossRef]
- Cady, B. Lymph node metastases. Indicators, but not governors of survival. Arch. Surg. 1984, 119, 1067–1072. [Google Scholar] [CrossRef] [PubMed]
- Gill, S.; Loprinzi, C.L.; Sargent, D.J.; Thome, S.D.; Alberts, S.R.; Haller, D.G.; Benedetti, J.; Francini, G.; Shepherd, L.E.; Francois Seitz, J.; et al. Pooled analysis of fluorouracil-based adjuvant therapy for stage II and III colon cancer: Who benefits and by how much? J. Clin. Oncol. 2004, 22, 1797–1806. [Google Scholar] [CrossRef]
- Gunderson, L.L.; Jessup, J.M.; Sargent, D.J.; Greene, F.L.; Stewart, A. Revised tumor and node categorization for rectal cancer based on surveillance, epidemiology, and end results and rectal pooled analysis outcomes. J. Clin. Oncol. 2010, 28, 256–263. [Google Scholar] [CrossRef]
- Sobrero, A.F.; Puccini, A.; Shi, Q.; Grothey, A.; Andre, T.; Shields, A.F.; Souglakos, I.; Yoshino, T.; Iveson, T.; Ceppi, M.; et al. A new prognostic and predictive tool for shared decision making in stage III colon cancer. Eur. J. Cancer 2020, 138, 182–188. [Google Scholar] [CrossRef]
- Weinberg, R.A. Mechanisms of malignant progression. Carcinogenesis 2008, 29, 1092–1095. [Google Scholar] [CrossRef]
- Campbell, P.J.; Yachida, S.; Mudie, L.J.; Stephens, P.J.; Pleasance, E.D.; Stebbings, L.A.; Morsberger, L.A.; Latimer, C.; McLaren, S.; Lin, M.L.; et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature 2010, 467, 1109–1113. [Google Scholar] [CrossRef] [Green Version]
- Yachida, S.; Jones, S.; Bozic, I.; Antal, T.; Leary, R.; Fu, B.; Kamiyama, M.; Hruban, R.H.; Eshleman, J.R.; Nowak, M.A.; et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 2010, 467, 1114–1117. [Google Scholar] [CrossRef] [Green Version]
- Coghlin, C.; Murray, G.I. Current and emerging concepts in tumour metastasis. J. Pathol. 2010, 222, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.A. Parallel progression of primary tumours and metastases. Nat. Rev. Cancer 2009, 9, 302–312. [Google Scholar] [CrossRef]
- Gerhardt, H.; Semb, H. Pericytes: Gatekeepers in tumour cell metastasis? J. Mol. Med. 2008, 86, 135–144. [Google Scholar] [CrossRef]
- Liu, W.; Laitinen, S.; Khan, S.; Vihinen, M.; Kowalski, J.; Yu, G.; Chen, L.; Ewing, C.M.; Eisenberger, M.A.; Carducci, M.A.; et al. Copy number analysis indicates monoclonal origin of lethal metastatic prostate cancer. Nat. Med. 2009, 15, 559–565. [Google Scholar] [CrossRef] [Green Version]
- McPherson, A.; Roth, A.; Laks, E.; Masud, T.; Bashashati, A.; Zhang, A.W.; Ha, G.; Biele, J.; Yap, D.; Wan, A.; et al. Divergent modes of clonal spread and intraperitoneal mixing in high-grade serous ovarian cancer. Nat. Genet. 2016, 48, 758–767. [Google Scholar] [CrossRef]
- Gibson, W.J.; Hoivik, E.A.; Halle, M.K.; Taylor-Weiner, A.; Cherniack, A.D.; Berg, A.; Holst, F.; Zack, T.I.; Werner, H.M.; Staby, K.M.; et al. The genomic landscape and evolution of endometrial carcinoma progression and abdominopelvic metastasis. Nat. Genet. 2016, 48, 848–855. [Google Scholar] [CrossRef]
- Reiter, J.G.; Makohon-Moore, A.P.; Gerold, J.M.; Bozic, I.; Chatterjee, K.; Iacobuzio-Donahue, C.A.; Vogelstein, B.; Nowak, M.A. Reconstructing metastatic seeding patterns of human cancers. Nat. Commun. 2017, 8, 14114. [Google Scholar] [CrossRef] [Green Version]
- Haffner, M.C.; Mosbruger, T.; Esopi, D.M.; Fedor, H.; Heaphy, C.M.; Walker, D.A.; Adejola, N.; Gurel, M.; Hicks, J.; Meeker, A.K.; et al. Tracking the clonal origin of lethal prostate cancer. J. Clin. Investig. 2013, 123, 4918–4922. [Google Scholar] [CrossRef] [Green Version]
- Naxerova, K.; Reiter, J.G.; Brachtel, E.; Lennerz, J.K.; van de Wetering, M.; Rowan, A.; Cai, T.; Clevers, H.; Swanton, C.; Nowak, M.A.; et al. Origins of lymphatic and distant metastases in human colorectal cancer. Science 2017, 357, 55–60. [Google Scholar] [CrossRef] [Green Version]
- Talmadge, J.E.; Fidler, I.J. AACR centennial series: The biology of cancer metastasis: Historical perspective. Cancer Res. 2010, 70, 5649–5669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montagut, C.; Dalmases, A.; Bellosillo, B.; Crespo, M.; Pairet, S.; Iglesias, M.; Salido, M.; Gallen, M.; Marsters, S.; Tsai, S.P.; et al. Identification of a mutation in the extracellular domain of the Epidermal Growth Factor Receptor conferring cetuximab resistance in colorectal cancer. Nat. Med. 2012, 18, 221–223. [Google Scholar] [CrossRef]
- Parsons, B.L.; Myers, M.B. KRAS mutant tumor subpopulations can subvert durable responses to personalized cancer treatments. Per. Med. 2013, 10, 191–199. [Google Scholar] [CrossRef] [Green Version]
- Coppe, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Munoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef]
- Floor, S.L.; Dumont, J.E.; Maenhaut, C.; Raspe, E. Hallmarks of cancer: Of all cancer cells, all the time? Trends Mol. Med. 2012, 18, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Fidler, I.J.; Jessup, J.M.; Fogler, W.E.; Staerkel, R.; Mazumder, A. Activation of tumoricidal properties in peripheral blood monocytes of patients with colorectal carcinoma. Cancer Res. 1986, 46, 994–998. [Google Scholar]
- Marusyk, A.; Almendro, V.; Polyak, K. Intra-tumour heterogeneity: A looking glass for cancer? Nat. Rev. Cancer 2012, 12, 323–334. [Google Scholar] [CrossRef]
- Hunter, K.W.; Amin, R.; Deasy, S.; Ha, N.H.; Wakefield, L. Genetic insights into the morass of metastatic heterogeneity. Nat. Rev. Cancer 2018, 18, 211–223. [Google Scholar] [CrossRef] [Green Version]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef]
- Boral, D.; Vishnoi, M.; Liu, H.N.; Yin, W.; Sprouse, M.L.; Scamardo, A.; Hong, D.S.; Tan, T.Z.; Thiery, J.P.; Chang, J.C.; et al. Molecular characterization of breast cancer CTCs associated with brain metastasis. Nat. Commun. 2017, 8, 196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naxerova, K.; Jain, R.K. Using tumour phylogenetics to identify the roots of metastasis in humans. Nat. Rev. Clin. Oncol. 2015, 12, 258–272. [Google Scholar] [CrossRef]
- Robinson, D.R.; Wu, Y.M.; Lonigro, R.J.; Vats, P.; Cobain, E.; Everett, J.; Cao, X.; Rabban, E.; Kumar-Sinha, C.; Raymond, V.; et al. Integrative clinical genomics of metastatic cancer. Nature 2017, 548, 297–303. [Google Scholar] [CrossRef]
- Fidler, I.J.; Nicolson, G.L. Fate of recirculating B16 melanoma metastatic variant cells in parabiotic syngeneic recipients. J. Natl. Cancer Inst. 1977, 58, 1867–1872. [Google Scholar] [CrossRef] [PubMed]
- Welch, D.R. Tumor Heterogeneity—A ‘Contemporary Concept’ Founded on Historical Insights and Predictions. Cancer Res. 2016, 76, 4–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciardiello, F.; Arnold, D.; Casali, P.G.; Cervantes, A.; Douillard, J.Y.; Eggermont, A.; Eniu, A.; McGregor, K.; Peters, S.; Piccart, M.; et al. Delivering precision medicine in oncology today and in future-the promise and challenges of personalised cancer medicine: A position paper by the European Society for Medical Oncology (ESMO). Ann. Oncol. 2014, 25, 1673–1678. [Google Scholar] [CrossRef]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.M.; Zhao, X.; Christensen, J.; et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef]
- Amado, R.G.; Wolf, M.; Peeters, M.; Van Cutsem, E.; Siena, S.; Freeman, D.J.; Juan, T.; Sikorski, R.; Suggs, S.; Radinsky, R.; et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J. Clin. Oncol. 2008, 26, 1626–1634. [Google Scholar] [CrossRef]
- Bardelli, A.; Corso, S.; Bertotti, A.; Hobor, S.; Valtorta, E.; Siravegna, G.; Sartore-Bianchi, A.; Scala, E.; Cassingena, A.; Zecchin, D.; et al. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov. 2013, 3, 658–673. [Google Scholar] [CrossRef] [Green Version]
- Inno, A.; Di Salvatore, M.; Cenci, T.; Martini, M.; Orlandi, A.; Strippoli, A.; Ferrara, A.M.; Bagala, C.; Cassano, A.; Larocca, L.M.; et al. Is there a role for IGF1R and c-MET pathways in resistance to cetuximab in metastatic colorectal cancer? Clin. Colorectal. Cancer 2011, 10, 325–332. [Google Scholar] [CrossRef]
- Karadima, M.L.; Saetta, A.A.; Chatziandreou, I.; Lazaris, A.C.; Patsouris, E.; Tsavaris, N. The Prognostic Influence of BRAF Mutation and other Molecular, Clinical and Laboratory Parameters in Stage IV Colorectal Cancer. Pathol. Oncol. Res. 2016, 22, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Rowland, A.; Dias, M.M.; Wiese, M.D.; Kichenadasse, G.; McKinnon, R.A.; Karapetis, C.S.; Sorich, M.J. Meta-analysis comparing the efficacy of anti-EGFR monoclonal antibody therapy between KRAS G13D and other KRAS mutant metastatic colorectal cancer tumours. Eur. J. Cancer 2016, 55, 122–130. [Google Scholar] [CrossRef]
- Turke, A.B.; Zejnullahu, K.; Wu, Y.L.; Song, Y.; Dias-Santagata, D.; Lifshits, E.; Toschi, L.; Rogers, A.; Mok, T.; Sequist, L.; et al. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell 2010, 17, 77–88. [Google Scholar] [CrossRef] [Green Version]
- Yonesaka, K.; Zejnullahu, K.; Okamoto, I.; Satoh, T.; Cappuzzo, F.; Souglakos, J.; Ercan, D.; Rogers, A.; Roncalli, M.; Takeda, M.; et al. Activation of ERBB2 signaling causes resistance to the EGFR-directed therapeutic antibody cetuximab. Sci. Transl. Med. 2011, 3, 99ra86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knijn, N.; Mekenkamp, L.J.; Klomp, M.; Vink-Borger, M.E.; Tol, J.; Teerenstra, S.; Meijer, J.W.; Tebar, M.; Riemersma, S.; van Krieken, J.H.; et al. KRAS mutation analysis: A comparison between primary tumours and matched liver metastases in 305 colorectal cancer patients. Br. J. Cancer 2011, 104, 1020–1026. [Google Scholar] [CrossRef] [Green Version]
- Misale, S.; Di Nicolantonio, F.; Sartore-Bianchi, A.; Siena, S.; Bardelli, A. Resistance to anti-EGFR therapy in colorectal cancer: From heterogeneity to convergent evolution. Cancer Discov. 2014, 4, 1269–1280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz, L.A., Jr.; Williams, R.T.; Wu, J.; Kinde, I.; Hecht, J.R.; Berlin, J.; Allen, B.; Bozic, I.; Reiter, J.G.; Nowak, M.A.; et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature 2012, 486, 537–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misale, S.; Yaeger, R.; Hobor, S.; Scala, E.; Janakiraman, M.; Liska, D.; Valtorta, E.; Schiavo, R.; Buscarino, M.; Siravegna, G.; et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature 2012, 486, 532–536. [Google Scholar] [CrossRef] [Green Version]
- Siravegna, G.; Mussolin, B.; Buscarino, M.; Corti, G.; Cassingena, A.; Crisafulli, G.; Ponzetti, A.; Cremolini, C.; Amatu, A.; Lauricella, C.; et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat. Med. 2015, 21, 827. [Google Scholar] [CrossRef] [PubMed]
- Arena, S.; Bellosillo, B.; Siravegna, G.; Martinez, A.; Canadas, I.; Lazzari, L.; Ferruz, N.; Russo, M.; Misale, S.; Gonzalez, I.; et al. Emergence of Multiple EGFR Extracellular Mutations during Cetuximab Treatment in Colorectal Cancer. Clin. Cancer Res. 2015, 21, 2157–2166. [Google Scholar] [CrossRef] [Green Version]
- Hata, A.; Katakami, N.; Fujita, S.; Takatori, K.; Horai, A.; Kitajima, N.; Terashima, K. Panitumumab rechallenge in chemorefractory patients with metastatic colorectal cancer. J. Gastrointest Cancer 2013, 44, 456–459. [Google Scholar] [CrossRef]
- Russo, M.; Crisafulli, G.; Sogari, A.; Reilly, N.M.; Arena, S.; Lamba, S.; Bartolini, A.; Amodio, V.; Magri, A.; Novara, L.; et al. Adaptive mutability of colorectal cancers in response to targeted therapies. Science 2019, 366, 1473–1480. [Google Scholar] [CrossRef]
- Troiani, T.; Martinelli, E.; Napolitano, S.; Vitagliano, D.; Ciuffreda, L.P.; Costantino, S.; Morgillo, F.; Capasso, A.; Sforza, V.; Nappi, A.; et al. Increased TGF-alpha as a mechanism of acquired resistance to the anti-EGFR inhibitor cetuximab through EGFR-MET interaction and activation of MET signaling in colon cancer cells. Clin. Cancer Res. 2013, 19, 6751–6765. [Google Scholar] [CrossRef] [Green Version]
- Troiani, T.; Napolitano, S.; Vitagliano, D.; Morgillo, F.; Capasso, A.; Sforza, V.; Nappi, A.; Ciardiello, D.; Ciardiello, F.; Martinelli, E. Primary and acquired resistance of colorectal cancer cells to anti-EGFR antibodies converge on MEK/ERK pathway activation and can be overcome by combined MEK/EGFR inhibition. Clin. Cancer Res. 2014, 20, 3775–3786. [Google Scholar] [CrossRef] [Green Version]
- Russo, M.; Di Nicolantonio, F.; Bardelli, A. Climbing RAS, the everest of oncogenes. Cancer Discov. 2014, 4, 19–21. [Google Scholar] [CrossRef] [Green Version]
- Ciardiello, F.; Bianco, R.; Caputo, R.; Caputo, R.; Damiano, V.; Troiani, T.; Melisi, D.; De Vita, F.; De Placido, S.; Bianco, A.R.; et al. Antitumor activity of ZD6474, a vascular endothelial growth factor receptor tyrosine kinase inhibitor, in human cancer cells with acquired resistance to antiepidermal growth factor receptor therapy. Clin. Cancer Res. 2004, 10, 784–793. [Google Scholar] [CrossRef] [Green Version]
- Santini, D.; Vincenzi, B.; Addeo, R.; Garufi, C.; Masi, G.; Scartozzi, M.; Mancuso, A.; Frezza, A.M.; Venditti, O.; Imperatori, M.; et al. Cetuximab rechallenge in metastatic colorectal cancer patients: How to come away from acquired resistance? Ann. Oncol. 2012, 23, 2313–2318. [Google Scholar] [CrossRef]
- Yancopoulos, G.D.; Davis, S.; Gale, N.W.; Rudge, J.S.; Wiegand, S.J.; Holash, J. Vascular-specific growth factors and blood vessel formation. Nature 2000, 407, 242–248. [Google Scholar] [CrossRef]
- Grothey, A.; Van Cutsem, E.; Sobrero, A.; Siena, S.; Falcone, A.; Ychou, M.; Humblet, Y.; Bouche, O.; Mineur, L.; Barone, C.; et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): An international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013, 381, 303–312. [Google Scholar] [CrossRef]
- Wilhelm, S.M.; Dumas, J.; Adnane, L.; Lynch, M.; Carter, C.A.; Schutz, G.; Thierauch, K.H.; Zopf, D. Regorafenib (BAY 73-4506): A new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int. J. Cancer 2011, 129, 245–255. [Google Scholar] [CrossRef]
- Tsai, J.H.; Lee, W.M. Tie2 in tumor endothelial signaling and survival: Implications for antiangiogenic therapy. Mol. Cancer Res. 2009, 7, 300–310. [Google Scholar] [CrossRef] [Green Version]
- Schmieder, R.; Hoffmann, J.; Becker, M.; Bhargava, A.; Muller, T.; Kahmann, N.; Ellinghaus, P.; Adams, R.; Rosenthal, A.; Thierauch, K.H.; et al. Regorafenib (BAY 73-4506): Antitumor and antimetastatic activities in preclinical models of colorectal cancer. Int. J. Cancer 2014, 135, 1487–1496. [Google Scholar] [CrossRef]
- Napolitano, S.; Martini, G.; Rinaldi, B.; Martinelli, E.; Donniacuo, M.; Berrino, L.; Vitagliano, D.; Morgillo, F.; Barra, G.; De Palma, R.; et al. Primary and Acquired Resistance of Colorectal Cancer to Anti-EGFR Monoclonal Antibody Can Be Overcome by Combined Treatment of Regorafenib with Cetuximab. Clin. Cancer Res. 2015, 21, 2975–2983. [Google Scholar] [CrossRef] [Green Version]
- Shitara, K.; Yamanaka, T.; Denda, T.; Tsuji, Y.; Shinozaki, K.; Komatsu, Y.; Kobayashi, Y.; Furuse, J.; Okuda, H.; Asayama, M.; et al. REVERCE: A randomized phase II study of regorafenib followed by cetuximab versus the reverse sequence for previously treated metastatic colorectal cancer patients. Ann. Oncol. 2019, 30, 259–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takegawa, N.; Yonesaka, K.; Sakai, K.; Ueda, H.; Watanabe, S.; Nonagase, Y.; Okuno, T.; Takeda, M.; Maenishi, O.; Tsurutani, J.; et al. HER2 genomic amplification in circulating tumor DNA from patients with cetuximab-resistant colorectal cancer. Oncotarget 2016, 7, 3453–3460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietrantonio, F.; Vernieri, C.; Siravegna, G.; Mennitto, A.; Berenato, R.; Perrone, F.; Gloghini, A.; Tamborini, E.; Lonardi, S.; Morano, F.; et al. Heterogeneity of Acquired Resistance to Anti-EGFR Monoclonal Antibodies in Patients with Metastatic Colorectal Cancer. Clin. Cancer Res. 2017, 23, 2414–2422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, K.H.; Cunningham, D.; Werner, B.; Vlachogiannis, G.; Spiteri, I.; Heide, T.; Mateos, J.F.; Vatsiou, A.; Lampis, A.; Damavandi, M.D.; et al. Longitudinal Liquid Biopsy and Mathematical Modeling of Clonal Evolution Forecast Time to Treatment Failure in the PROSPECT-C Phase II Colorectal Cancer Clinical Trial. Cancer Discov. 2018, 8, 1270–1285. [Google Scholar] [CrossRef] [Green Version]
- Beaver, J.A.; Jelovac, D.; Balukrishna, S.; Cochran, R.; Croessmann, S.; Zabransky, D.J.; Wong, H.Y.; Toro, P.V.; Cidado, J.; Blair, B.G.; et al. Detection of cancer DNA in plasma of patients with early-stage breast cancer. Clin. Cancer Res. 2014, 20, 2643–2650. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| DNA | mRNA | ||
|---|---|---|---|
| Type of Gene Damage | Methylation | Gene Expression Patterns | |
| Microsatellite instability (MSI) = mismatch repair (MMR) deficient | CpG island methylator (CIMP+) | Consensus molecular subtype (CMS) 1 CRC intrinsic subtype (CRIS)-A/B | |
| Microsatellite stable (MSS) = MMR proficient = chromosomal instability (CIN) | Mostly CIMP- | CMS2, canonical | CRIS-C |
| CMS3, metabolic | CRIS-D | ||
| CMS4, mesenchymal | CRIS-E/B | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gaiani, F.; Marchesi, F.; Negri, F.; Greco, L.; Malesci, A.; de’Angelis, G.L.; Laghi, L. Heterogeneity of Colorectal Cancer Progression: Molecular Gas and Brakes. Int. J. Mol. Sci. 2021, 22, 5246. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22105246
Gaiani F, Marchesi F, Negri F, Greco L, Malesci A, de’Angelis GL, Laghi L. Heterogeneity of Colorectal Cancer Progression: Molecular Gas and Brakes. International Journal of Molecular Sciences. 2021; 22(10):5246. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22105246
Chicago/Turabian StyleGaiani, Federica, Federica Marchesi, Francesca Negri, Luana Greco, Alberto Malesci, Gian Luigi de’Angelis, and Luigi Laghi. 2021. "Heterogeneity of Colorectal Cancer Progression: Molecular Gas and Brakes" International Journal of Molecular Sciences 22, no. 10: 5246. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22105246