
NO● Represses the Oxygenation of Arachidonoyl PE by 15LOX/PEBP1: Mechanism and Role in Ferroptosis

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
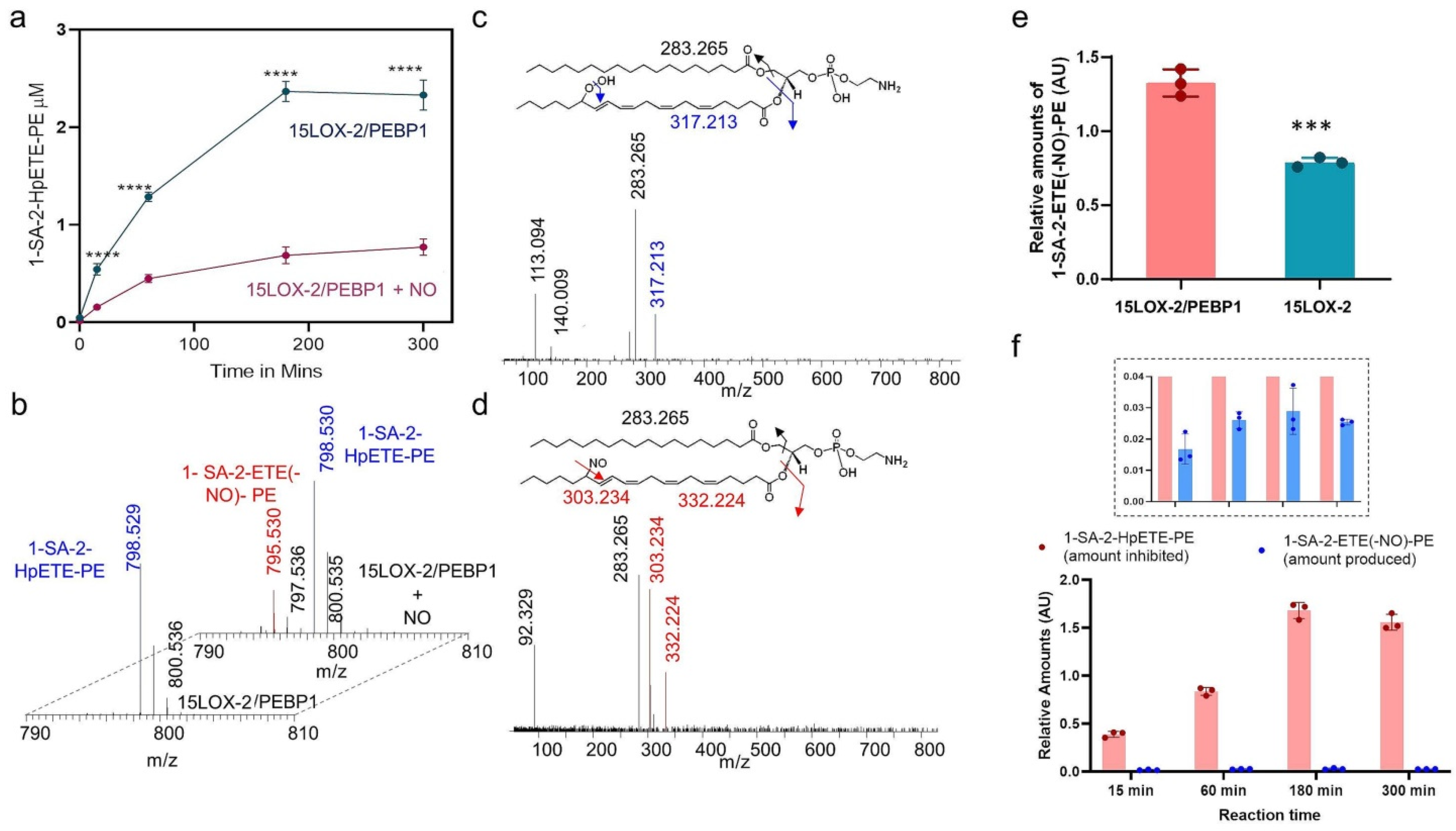
2.1. Lipidomics Analysis of the Effect of NO● on 15LOX-2/PEBP1 Peroxidation Activity
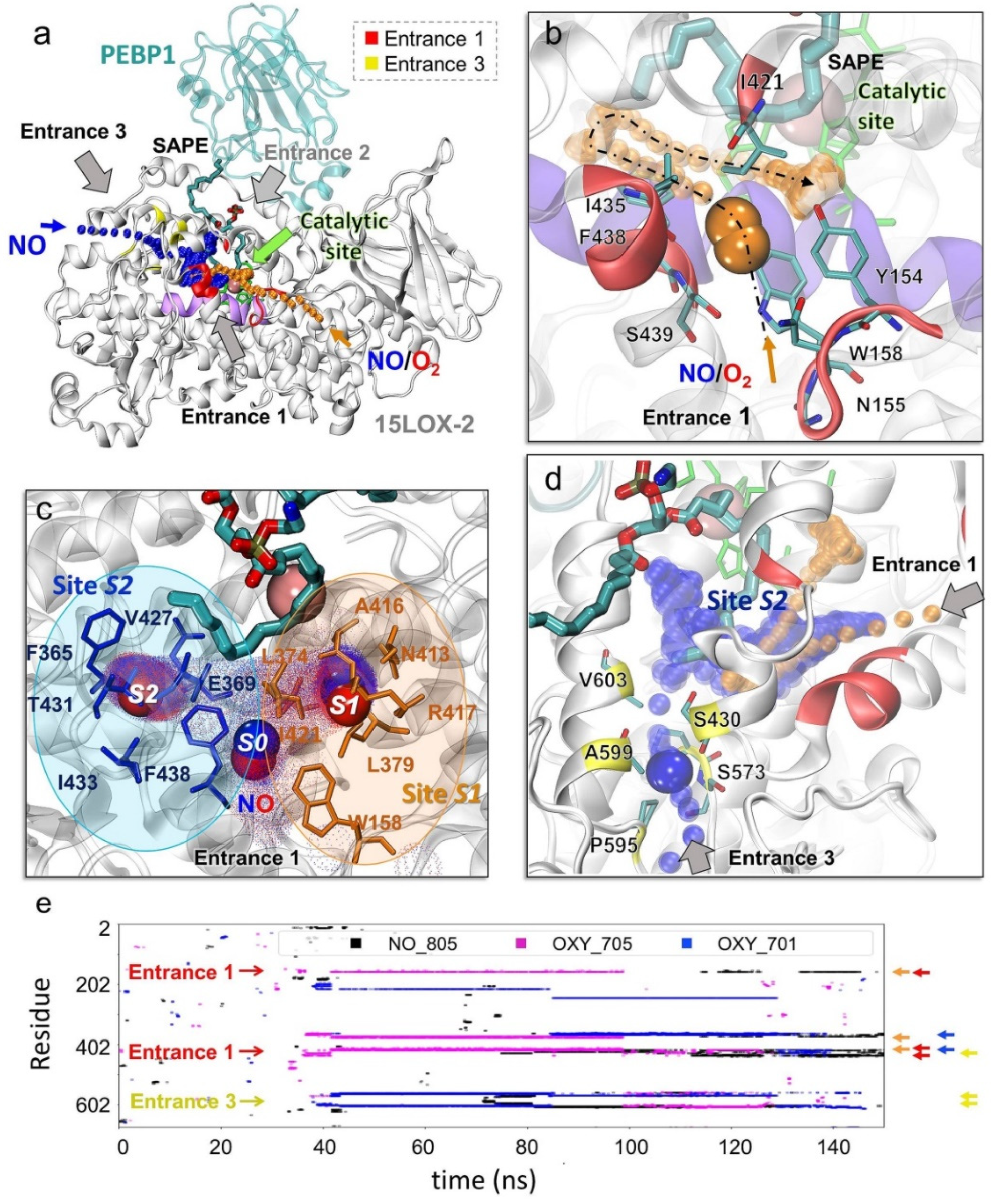
2.2. In silico Analysis of the Effect of NO● on the Structure, Dynamics, and Interactions of 15LOX-2/PEBP1
2.3. Specific Porous Regions on 15LOX-2 Surface Enable Access of O2 and NO● to the Catalytic Cavity
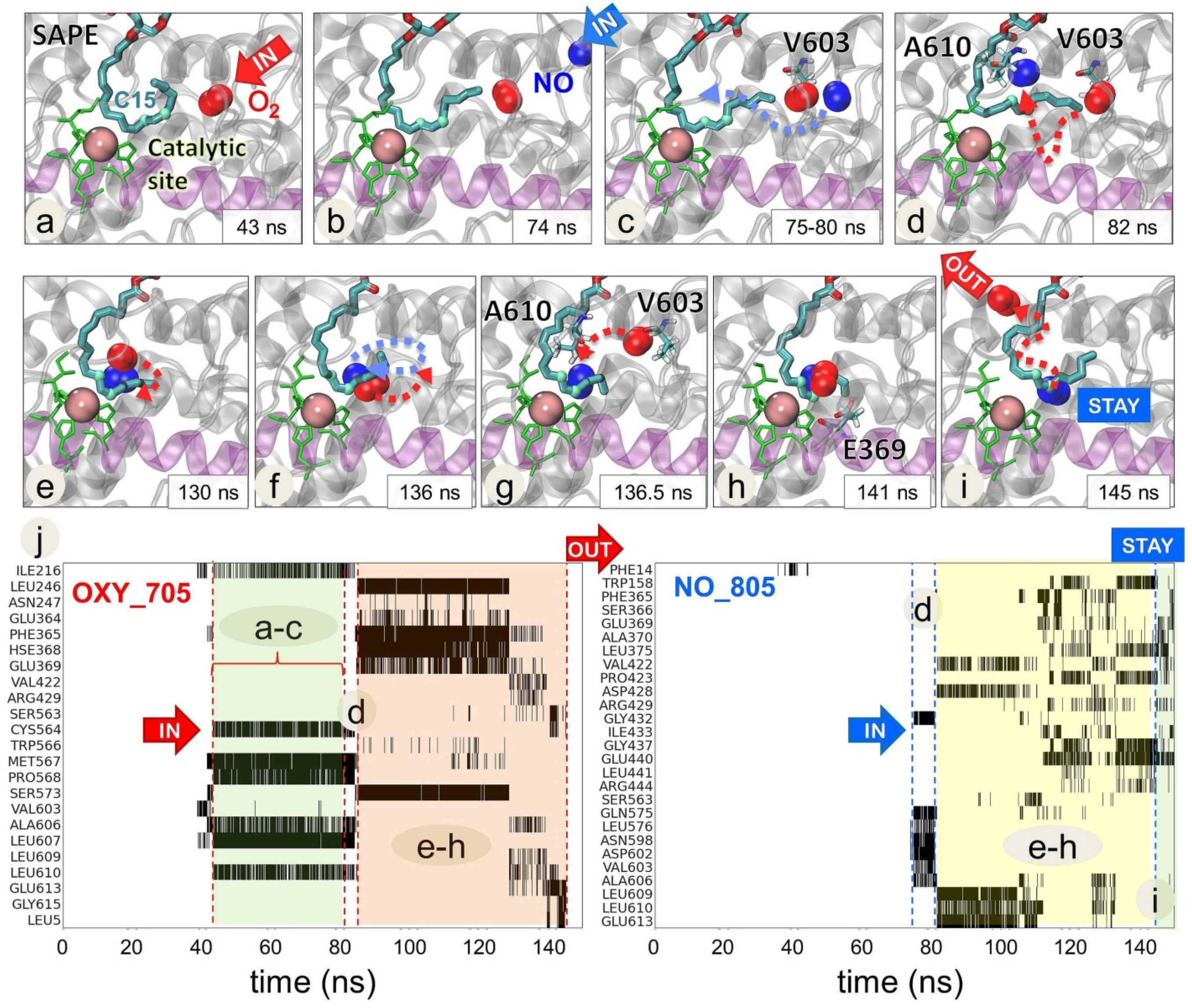
2.4. O2 and NO● Compete for the Catalytic Site
2.5. Selected Residues Stabilize O2 and NO● Near 15LOX-2 Catalytic Site in a Substrate-Dependent Manner
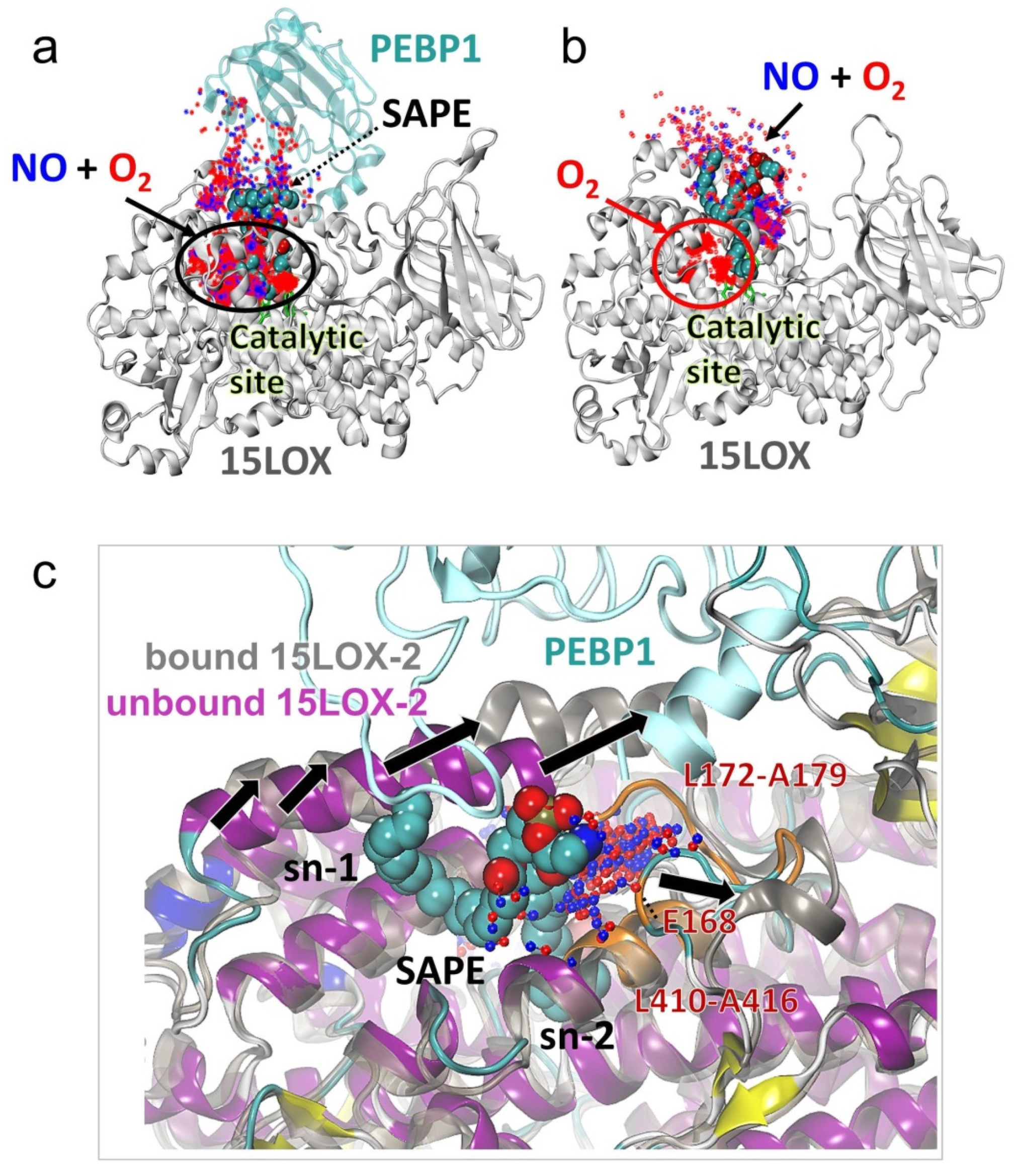
2.6. The Change in 15LOX-2 Structure Upon PEBP1 Binding Renders the Catalytic Site Accessible to Both O2 and NO● Molecules
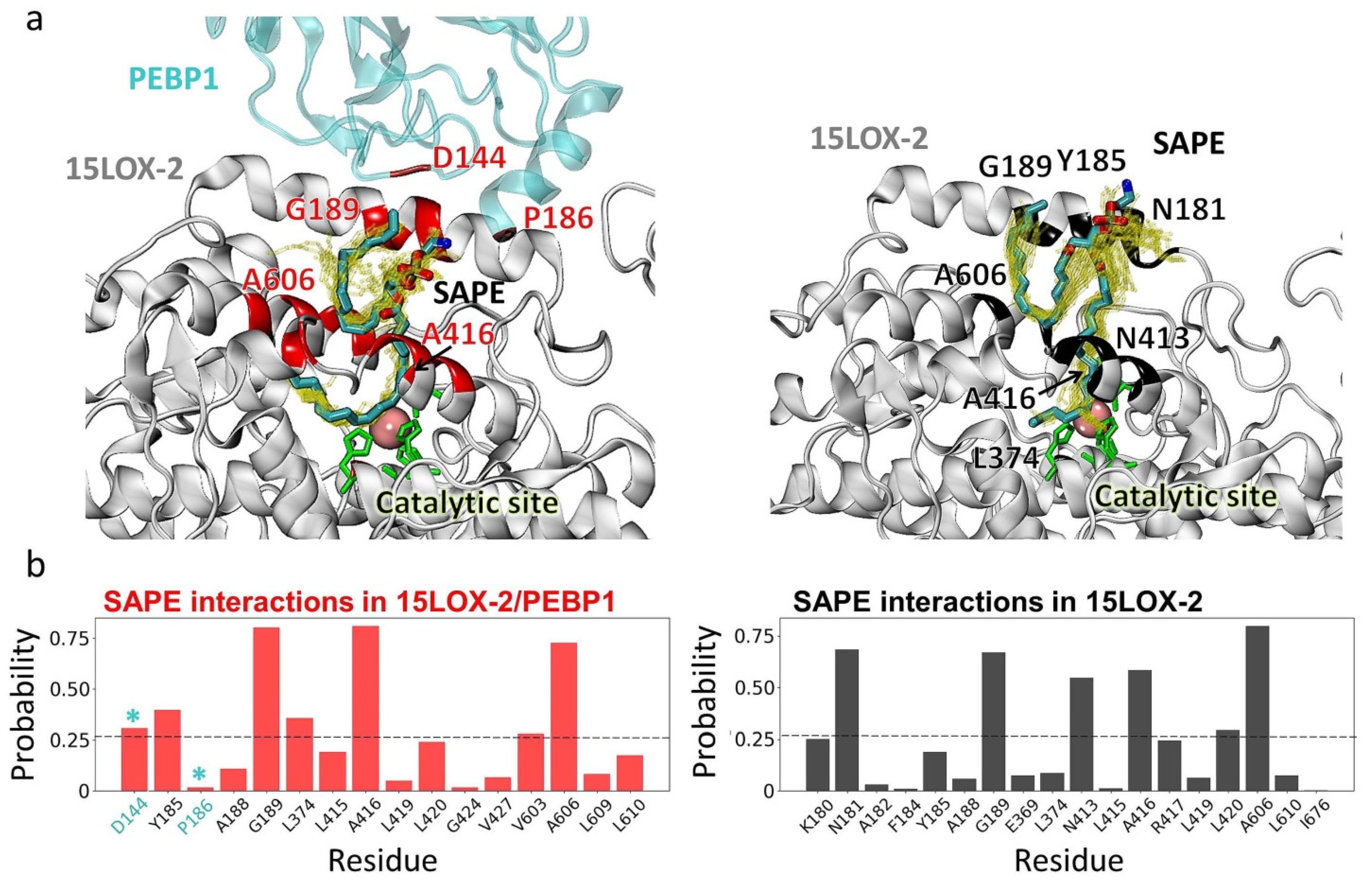
2.7. The Precise Positioning of SAPE for Peroxidation by 15LOX-2 Is Assisted by α2 Residues N181, Y185, and G189, and by E3/S1 Residues A416 and A606
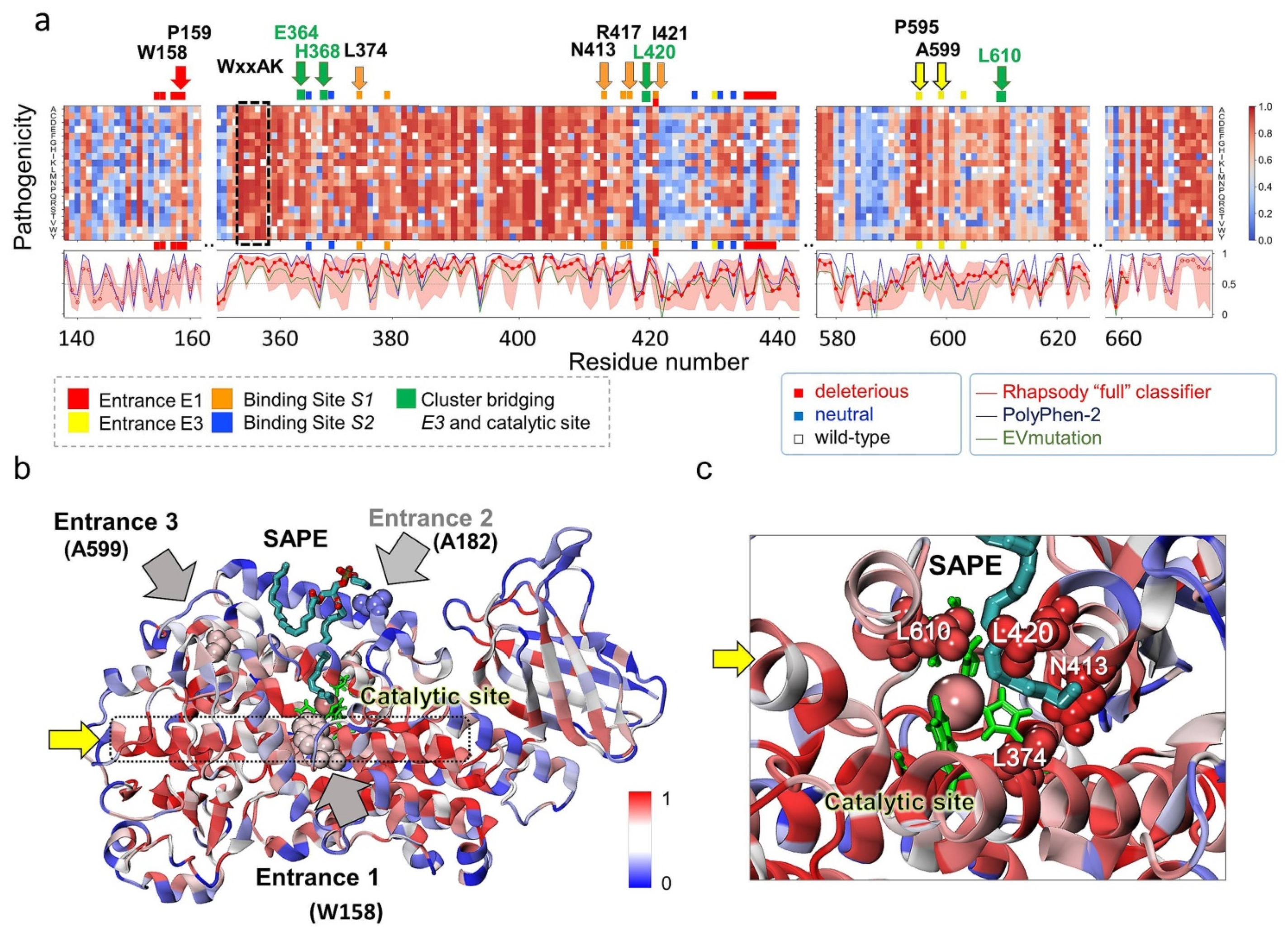
2.8. In Silico Saturation Mutagenesis Analysis Confirms the Critical Role of Selected Residues Amongst Those Identified to Mediate O2/NO● Entry and Translocation to the Catalytic Site
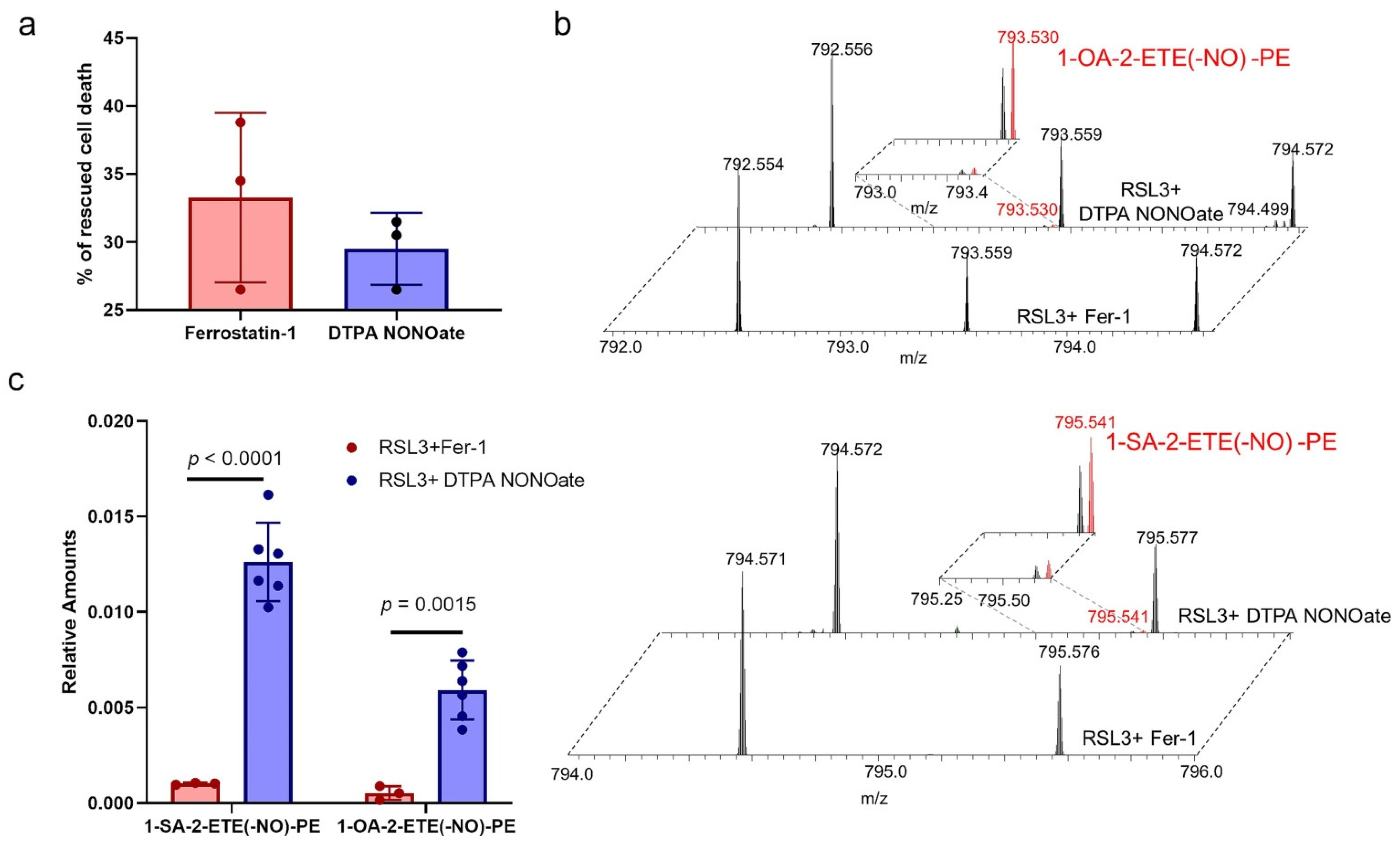
2.9. Identification of Nitrosylated PE Species in Cells Treated with NO● Donors
3. Discussion
4. Materials and Methods
4.1. Molecular Dynamics (MD) Simulations
4.2. Rhapsody and in Silico Saturation Mutagenesis Analysis
4.3. Lipoxygenase Assay
4.4. Cell Culture
4.5. LC–MS Analysis of Phospholipids
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stockwell, B.R.; Angeli, J.P.F.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E. Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, S.E.; Tyurina, Y.Y.; Zhao, J.; Croix, C.M.S.; Dar, H.H.; Mao, G.; Tyurin, V.A.; Anthonymuthu, T.S.; Kapralov, A.A.; Amoscato, A.A. PEBP1 Wardens Ferroptosis by Enabling Lipoxygenase Generation of Lipid Death Signals. Cell 2017, 171, 628–641.e26. [Google Scholar] [CrossRef] [PubMed]
- Anthonymuthu, T.S.; Tyurina, Y.Y.; Sun, W.-Y.; Mikulska-Ruminska, K.; Shrivastava, I.H.; Tyurin, V.A.; Cinemre, F.B.; Dar, H.H.; VanDemark, A.P.; Holman, T.R.; et al. Resolving the paradox of ferroptotic cell death: Ferrostatin-1 binds to 15LOX/PEBP1 complex, suppresses generation of peroxidized ETE-PE, and protects against ferroptosis. Redox Biol. 2020, 38, 101744. [Google Scholar] [CrossRef]
- Anthonymuthu, T.S.; Kenny, E.M.; Shrivastava, I.; Tyurina, Y.Y.; Hier, Z.E.; Ting, H.-C.; Dar, H.H.; Tyurin, V.A.; Nesterova, A.; Amoscato, A.A.; et al. Empowerment of 15-Lipoxygenase Catalytic Competence in Selective Oxidation of Membrane ETE-PE to Ferroptotic Death Signals, HpETE-PE. J. Am. Chem. Soc. 2018, 140, 17835–17839. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.; Shchepinov, M.S.; Pratt, D.A. Resolving the role of lipoxygenases in the initiation and execution of ferroptosis. ACS Cent. Sci. 2018, 4, 387–396. [Google Scholar] [CrossRef]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91. [Google Scholar] [CrossRef]
- Bersuker, K.; Hendricks, J.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Grocin, A.G.; da Silva, T.N.X.; Panzilius, E.; Scheel, C. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef]
- Imai, H. Biological significance of lipid hydroperoxide and its reducing enzyme, phospholipid hydroperoxide glutathione peroxidase, in mammalian cells. Yakugaku Zasshi J. Pharm. Soc. Jpn. 2004, 124, 937–957. [Google Scholar] [CrossRef]
- Björnstedt, M.; Hamberg, M.; Kumar, S.; Xue, J.; Holmgren, A. Human thioredoxin reductase directly reduces lipid hydroperoxides by NADPH and selenocystine strongly stimulates the reaction via catalytically generated selenols. J. Biol. Chem. 1995, 270, 11761–11764. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.-Y.; Tyurin, V.A.; Mikulska-Ruminska, K.; Shrivastava, I.H.; Anthonymuthu, T.S.; Zhai, Y.-J.; Pan, M.-H.; Gong, H.-B.; Lu, D.-H.; Sun, J. Phospholipase iPLA 2 β averts ferroptosis by eliminating a redox lipid death signal. Nat. Chem. Biol. 2021, 17, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Kapralov, A.A.; Yang, Q.; Dar, H.H.; Tyurina, Y.Y.; Anthonymuthu, T.S.; Kim, R.; Croix, C.M.S.; Mikulska-Ruminska, K.; Liu, B.; Shrivastava, I.H.; et al. Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nat. Chem. Biol. 2020, 16, 278–290. [Google Scholar] [CrossRef] [PubMed]
- Hogg, N.; Kalyanaraman, B. Nitric oxide and lipid peroxidation. BBA Bioenerg. 1999, 1411, 378–384. [Google Scholar] [CrossRef]
- Kanner, J.; Harel, S.; Granit, R. Nitric oxide, an inhibitor of lipid oxidation by lipoxygenase, cyclooxygenase and hemoglobin. Lipids 1992, 27, 46. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.; Heydeck, D.; Hofheinz, K.; Roffeis, J.; O’Donnell, V.B.; Kuhn, H.; Walther, M. Molecular enzymology of lipoxygenases. Arch. Biochem. Biophys. 2010, 503, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Keefer, L.K.; Nims, R.W.; Davies, K.M.; Wink, D.A. “NONOates”(1-substituted diazen-1-ium-1, 2-diolates) as nitric oxide donors: Convenient nitric oxide dosage forms. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 1996; Volume 268, pp. 281–293. [Google Scholar]
- Anthonymuthu, T.S.; Kenny, E.M.; Bayır, H. Therapies targeting lipid peroxidation in traumatic brain injury. Brain Res. 2016, 1640, 57–76. [Google Scholar] [CrossRef]
- Wood, I.; Trostchansky, A.; Rubbo, H. Structural considerations on lipoxygenase function, inhibition and crosstalk with nitric oxide pathways. Biochimie 2020, 178, 170–180. [Google Scholar] [CrossRef]
- Kobe, M.J.; Neau, D.B.; Mitchell, C.E.; Bartlett, S.G.; Newcomer, M.E. The structure of human 15-lipoxygenase-2 with a substrate mimic. J. Biol. Chem. 2014, 289, 8562–8569. [Google Scholar] [CrossRef]
- Laskowski, R.A. PDBsum: Summaries and analyses of PDB structures. Nucleic Acids Res. 2001, 29, 221–222. [Google Scholar] [CrossRef]
- Mikulska-Ruminska, K.; Shrivastava, I.; Krieger, J.; Zhang, S.; Li, H.; Bayır, H.; Wenzel, S.E.; VanDemark, A.P.; Kagan, V.E.; Bahar, I. Characterization of Differential Dynamics, Specificity, and Allostery of Lipoxygenase Family Members. J. Chem. Inf. Model. 2019, 59, 2496–2508. [Google Scholar] [CrossRef] [PubMed]
- Ponzoni, L.; Peñaherrera, D.A.; Oltvai, Z.N.; Bahar, I. Rhapsody: Predicting the pathogenicity of human missense variants. Bioinformatics 2020, 36, 3084–3309. [Google Scholar] [CrossRef] [PubMed]
- Ponzoni, L.; Bahar, I. Structural dynamics is a determinant of the functional significance of missense variants. Proc. Natl. Acad. Sci. USA 2018, 115, 4164–4169. [Google Scholar] [CrossRef] [PubMed]
- Hopf, T.A.; Ingraham, J.B.; Poelwijk, F.J.; Scharfe, C.P.; Springer, M.; Sander, C.; Marks, D.S. Mutation effects predicted from sequence co-variation. Nat. Biotechnol. 2017, 35, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7.20.1–7.20.41. [Google Scholar] [CrossRef] [PubMed]
- Maiorino, M.; Conrad, M.; Ursini, F. GPx4, lipid peroxidation, and cell death: Discoveries, rediscoveries, and open issues. Antioxid. Redox Signal. 2018, 29, 61–74. [Google Scholar] [CrossRef]
- Kühn, H.; Barnett, J.; Grunberger, D.; Baecker, P.; Chow, J.; Nguyen, B.; Bursztyn-Pettegrew, H.; Chan, H.; Sigal, E. Overexpression, purification and characterization of human recombinant 15-lipoxygenase. BBA Lipids Lipid Metab. 1993, 1169, 80–89. [Google Scholar] [CrossRef]
- Brown, G.C. Nitric oxide regulates mitochondrial respiration and cell functions by inhibiting cytochrome oxidase. FEBS Lett. 1995, 369, 136–139. [Google Scholar] [CrossRef]
- Jarazo Dietrich, S.; Fass, M.I.; Jacobo, P.V.; Sobarzo, C.M.A.; Lustig, L.; Theas, M.S. Inhibition of NOS-NO system prevents autoimmune orchitis development in rats: Relevance of NO released by testicular macrophages in germ cell apoptosis and testosterone secretion. PLoS ONE 2015, 10, e0128709. [Google Scholar] [CrossRef]
- Stuart, J.A.; Fonseca, J.; Moradi, F.; Cunningham, C.; Seliman, B.; Worsfold, C.R.; Dolan, S.; Abando, J.; Maddalena, L.A. How supraphysiological oxygen levels in standard cell culture affect oxygen-consuming reactions. Oxid. Med. Cell. Longev. 2018, 2018, 1–13. [Google Scholar] [CrossRef]
- Schmidt-Rohr, K. Oxygen is the high-energy molecule powering complex multicellular life: Fundamental corrections to traditional bioenergetics. ACS Omega 2020, 5, 2221–2233. [Google Scholar] [CrossRef] [PubMed]
- Clancy, R.; Leszczynska-Piziak, J.; Abramson, S. Nitric oxide, an endothelial cell relaxation factor, inhibits neutrophil superoxide anion production via a direct action on the NADPH oxidase. J. Clin. Investig. 1992, 90, 1116–1121. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, E.M.; Gonzalez-Cotto, M.; Baseler, W.A.; Davies, L.C.; Ghesquière, B.; Maio, N.; Rice, C.M.; Rouault, T.A.; Cassel, T.; Higashi, R.M. Nitric oxide orchestrates metabolic rewiring in M1 macrophages by targeting aconitase 2 and pyruvate dehydrogenase. Nat. Commun. 2020, 11, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Jaffrey, S.R.; Erdjument-Bromage, H.; Ferris, C.D.; Tempst, P.; Snyder, S.H. Protein S-nitrosylation: A physiological signal for neuronal nitric oxide. Nat. Cell Biol. 2001, 3, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Ischiropoulos, H.; Zhu, L.; Beckman, J.S. Peroxynitrite formation from macrophage-derived nitric oxide. Arch. Biochem. Biophys. 1992, 298, 446–451. [Google Scholar] [CrossRef]
- MacMicking, J.; Xie, Q.-W.; Nathan, C. Nitric oxide and macrophage function. Ann. Rev. Immun. 1997, 15, 323–350. [Google Scholar] [CrossRef]
- Babior, B.; Lambeth, J.; Nauseef, W. The neutrophil NADPH oxidase. Arch. Biochem. Biophys. 2002, 397, 342–344. [Google Scholar] [CrossRef]
- Gordon, S.; Martinez, F.O. Alternative activation of macrophages: Mechanism and functions. Immunity 2010, 32, 593–604. [Google Scholar] [CrossRef]
- Victor, V.M.; Rocha, M.; Esplugues, J.V. Role of free radicals in sepsis: Antioxidant therapy. Curr. Pharm. Des. 2005, 11, 3141–3158. [Google Scholar] [CrossRef]
- Darley-Usmar, V.; Wiseman, H.; Halliwell, B. Nitric oxide and oxygen radicals: A question of balance. FEBS Lett. 1995, 369, 131–135. [Google Scholar] [CrossRef]
- Swindle, E.J.; Metcalfe, D.D. The role of reactive oxygen species and nitric oxide in mast cell-dependent inflammatory processes. Immun. Rev. 2007, 217, 186–205. [Google Scholar] [CrossRef]
- Horn, T.; Kakularam, K.R.; Anton, M.; Richter, C.; Reddanna, P.; Kuhn, H. Functional characterization of genetic enzyme variations in human lipoxygenases. Redox Biol. 2013, 1, 566–577. [Google Scholar] [CrossRef] [PubMed]
- Hofheinz, K.; Kakularam, K.R.; Adel, S.; Anton, M.; Polymarasetty, A.; Reddanna, P.; Kuhn, H.; Horn, T. Conversion of pro-inflammatory murine Alox5 into an anti-inflammatory 15S-lipoxygenating enzyme by multiple mutations of sequence determinants. Arch. Biochem. Biophys. 2013, 530, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.S.; Funk, C.D. Structure-function properties of human platelet 12-lipoxygenase: Chimeric enzyme and in vitro mutagenesis studies. FASEB J. 1993, 7, 694–701. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.D.; Liu, X.; Kantrow, S.P.; Lancaster, J.R. The biological lifetime of nitric oxide: Implications for the perivascular dynamics of NO and O2. Proc. Natl. Acad. Sci. USA 2001, 98, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Perilla, J.R.; Schulten, K. Physical properties of the HIV-1 capsid from all-atom molecular dynamics simulations. Nat. Commun. 2017, 8, 1–10. [Google Scholar] [CrossRef]
- Mikulska-Ruminska, K.; Kulik, A.; Benadiba, C.; Bahar, I.; Dietler, G.; Nowak, W. Nanomechanics of multidomain neuronal cell adhesion protein contactin revealed by single molecule AFM and SMD. Sci. Rep. 2017, 7, 8852. [Google Scholar] [CrossRef]
- Cheng, Z.; Lan, Y.; Guo, J.; Ma, D.; Jiang, S.; Lai, Q.; Zhou, Z.; Peplowski, L. Computational Design of Nitrile Hydratase from Pseudonocardia thermophila JCM3095 for Improved Thermostability. Molecules 2020, 25, 4806. [Google Scholar] [CrossRef]
- Banfield, M.J.; Barker, J.J.; Perry, A.C.; Brady, R.L. Function from structure? The crystal structure of human phosphatidylethanolamine-binding protein suggests a role in membrane signal transduction. Structure 1998, 6, 1245–1254. [Google Scholar] [CrossRef]
- Phillips, J.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.; Kale, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed]
- MacKerell, A., Jr.; Bashford, D.; Bellott, M.; Dunbrack, R., Jr.; Evanseck, J.; Field, M.; Fischer, S.; Gao, J.; Guo, H.; Ha, S. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.; Chandrasekhar, J.; Madura, J.; Impey, R.; Klein, M. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926. [Google Scholar] [CrossRef]
- Koes, D.R.; Baumgartner, M.P.; Camacho, C.J. Lessons learned in empirical scoring with smina from the CSAR 2011 benchmarking exercise. J. Chem. Inf. Mod. 2013, 53, 1893–1904. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Montgomery, J., Jr.; Vreven, T.; Kudin, K.; Burant, J. Gaussian 03, Revision B. 05; Gaussian Inc.: Pittsburgh, PA, USA, 2003; p. 12478. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Bakan, A.; Dutta, A.; Mao, W.; Liu, Y.; Chennubhotla, C.; Lezon, T.R.; Bahar, I. Evol and ProDy for bridging protein sequence evolution and structural dynamics. Bioinformatics 2014, 30, 2681–2683. [Google Scholar] [CrossRef]
- Bakan, A.; Meireles, L.M.; Bahar, I. ProDy: Protein dynamics inferred from theory and experiments. Bioinformatics 2011, 27, 1575–1577. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Krieger, J.M.; Zhang, Y.; Kaya, C.; Kaynak, B.; Mikulska-Ruminska, K.; Doruker, P.; Li, H.; Bahar, I. ProDy 2.0: Increased Scale and Scope after 10 Years of Protein Dynamics Modelling with Python. Bioinformatics 2021, btab187. [Google Scholar] [CrossRef] [PubMed]
- Chovancova, E.; Pavelka, A.; Benes, P.; Strnad, O.; Brezovsky, J.; Kozlikova, B.; Gora, A.; Sustr, V.; Klvana, M.; Medek, P. CAVER 3.0: A tool for the analysis of transport pathways in dynamic protein structures. PLoS Comput. Biol. 2012, 8, e1002708. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Jameson, J.B., II; Kenyon, V.; Holman, T.R. A high-throughput mass spectrometric assay for discovery of human lipoxygenase inhibitors and allosteric effectors. Anal. Biochem. 2015, 476, 45–50. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mikulska-Ruminska, K.; Anthonymuthu, T.S.; Levkina, A.; Shrivastava, I.H.; Kapralov, A.A.; Bayır, H.; Kagan, V.E.; Bahar, I. NO● Represses the Oxygenation of Arachidonoyl PE by 15LOX/PEBP1: Mechanism and Role in Ferroptosis. Int. J. Mol. Sci. 2021, 22, 5253. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22105253
Mikulska-Ruminska K, Anthonymuthu TS, Levkina A, Shrivastava IH, Kapralov AA, Bayır H, Kagan VE, Bahar I. NO● Represses the Oxygenation of Arachidonoyl PE by 15LOX/PEBP1: Mechanism and Role in Ferroptosis. International Journal of Molecular Sciences. 2021; 22(10):5253. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22105253
Chicago/Turabian StyleMikulska-Ruminska, Karolina, Tamil S. Anthonymuthu, Anastasia Levkina, Indira H. Shrivastava, Alexandr A. Kapralov, Hülya Bayır, Valerian E. Kagan, and Ivet Bahar. 2021. "NO● Represses the Oxygenation of Arachidonoyl PE by 15LOX/PEBP1: Mechanism and Role in Ferroptosis" International Journal of Molecular Sciences 22, no. 10: 5253. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22105253