
Immunopathology of Chronic Hepatitis B Infection: Role of Innate and Adaptive Immune Response in Disease Progression


Abstract
:1. Introduction
2. Innate and Adaptive Immune Response against HBV Infection
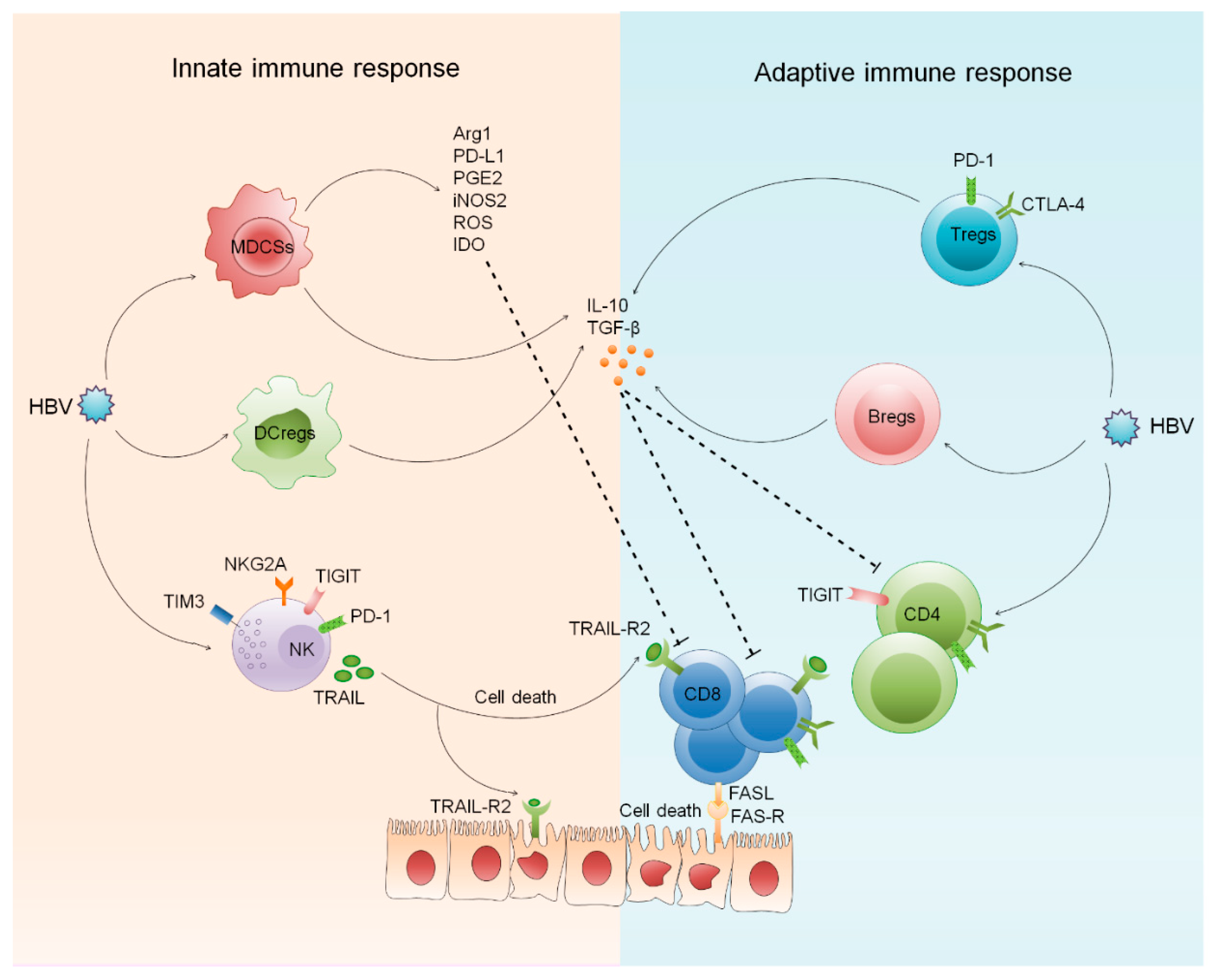
2.1. Role of Innate and Adaptive Regulatory Cells in CHB
2.1.1. Regulatory Dendritic Cells
2.1.2. Myeloid Derived Suppressor Cells
2.1.3. Regulatory T Cells
2.1.4. Regulatory B Cells
2.2. Involvement of Innate and Adaptive Inhibitory Receptors in CHB
2.2.1. Inhibitory Receptors on NK Cells
2.2.2. Inhibitory Receptors on CD4 and CD8 T Cells
2.2.3. Inhibitory Receptors on B Cells
2.3. Involvement of B Cells in CHB
2.4. Innate and Adaptive Immune Cell Derived Inflammatory Mediators in CHB
2.5. Function of Innate and Adaptive Immune Cell Derived Exosomes in CHB
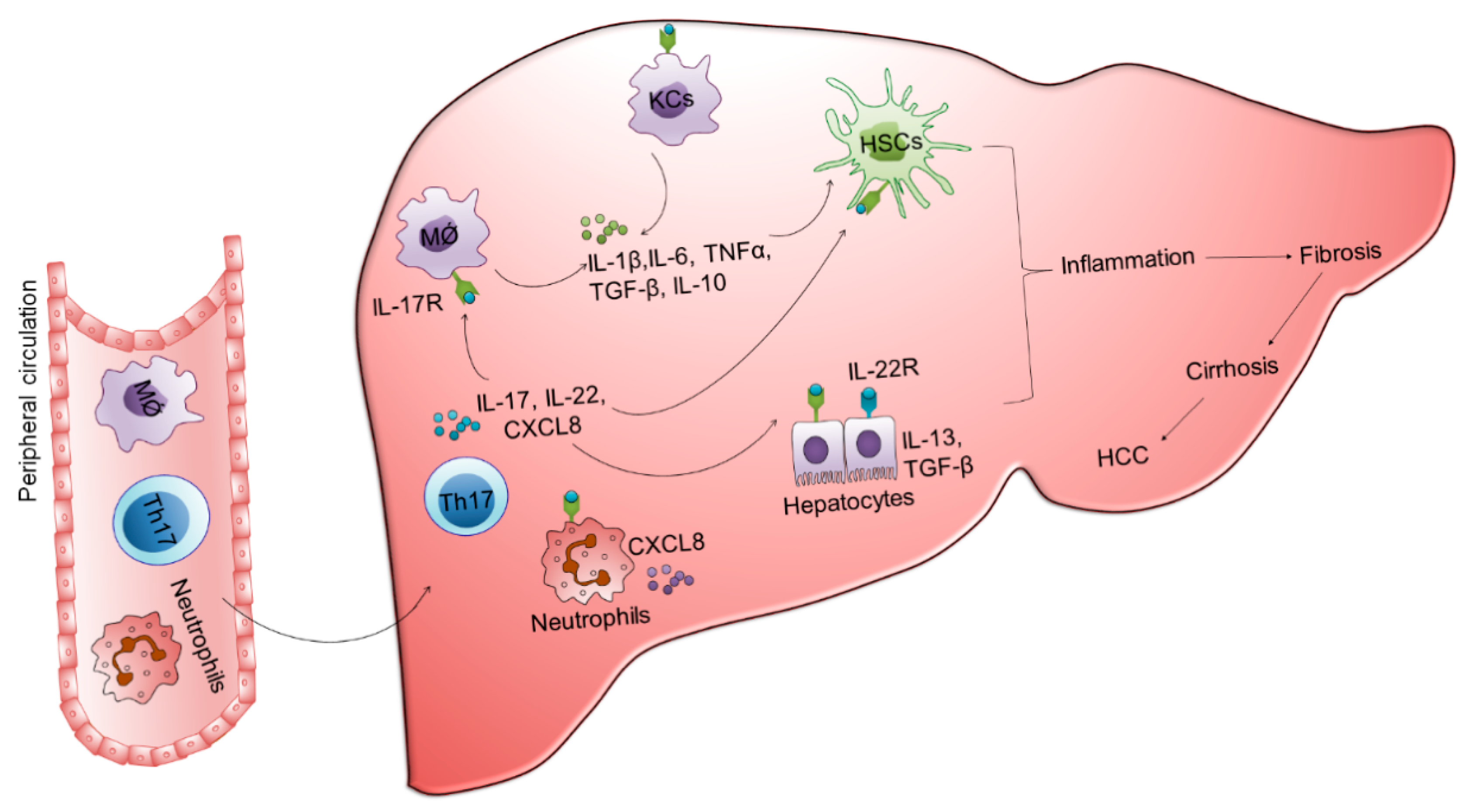
3. Involvement of Innate and Adaptive Immune Cells in the Development of Fibrosis, Cirrhosis, and HCC
3.1. Monocytes/Macrophages
3.2. NK Cells
3.3. CD4 T Cells
3.4. CD8 T Cells
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tang, L.S.Y.; Covert, E.; Wilson, E.; Kottilil, S. Chronic Hepatitis B Infection: A Review. JAMA 2018, 319, 1802–1813. [Google Scholar] [CrossRef]
- Bixler, D.; Zhong, Y.; Ly, K.N.; Moorman, A.C.; Spradling, P.R.; Teshale, E.H.; Rupp, L.B.; Gordon, S.C.; Boscarino, J.A.; Schmidt, M.A.; et al. Mortality Among Patients With Chronic Hepatitis B Infection: The Chronic Hepatitis Cohort Study (CHeCS). Clin. Infect. Dis. 2019, 68, 956–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thimme, R.; Wieland, S.; Steiger, C.; Ghrayeb, J.; Reimann, K.A.; Purcell, R.H.; Chisari, F.V. CD8(+) T cells mediate viral clearance and disease pathogenesis during acute hepatitis B virus infection. J. Virol. 2003, 77, 68–76. [Google Scholar] [CrossRef] [Green Version]
- Acerbi, G.; Montali, I.; Ferrigno, G.D.; Barili, V.; Schivazappa, S.; Alfieri, A.; Laccabue, D.; Loglio, A.; Borghi, M.; Massari, M.; et al. Functional reconstitution of HBV-specific CD8 T cells by in vitro polyphenol treatment in chronic hepatitis B. J. Hepatol. 2021, 74, 783–793. [Google Scholar] [CrossRef]
- Ma, Z.; Cao, Q.; Xiong, Y.; Zhang, E.; Lu, M. Interaction between Hepatitis B Virus and Toll-Like Receptors: Current Status and Potential Therapeutic Use for Chronic Hepatitis B. Vaccines (Basel) 2018, 6, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isogawa, M.; Robek, M.D.; Furuichi, Y.; Chisari, F.V. Toll-like receptor signaling inhibits hepatitis B virus replication in vivo. J. Virol. 2005, 79, 7269–7272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.J.; Zhai, N.C.; Song, H.X.; Yang, Y.; Cui, A.; Li, T.Y.; Tu, Z.K. The Role of Immune Cells in Chronic HBV Infection. J. Clin. Transl. Hepatol. 2015, 3, 277–283. [Google Scholar] [CrossRef]
- Li, Y.; Tang, L.; Guo, L.; Chen, C.; Gu, S.; Zhou, Y.; Ye, G.; Li, X.; Wang, W.; Liao, X.; et al. CXCL13-mediated recruitment of intrahepatic CXCR5(+)CD8(+) T cells favors viral control in chronic HBV infection. J. Hepatol. 2020, 72, 420–430. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Liu, Y.; Yang, X.; Sun, H.; Shu, X.; Zhang, Y.; Cao, H.; Wu, M.; Liu, N.; Zou, Y.; et al. HBV promotes the recruitment of IL-17 secreting T cells via chemokines CCL22 and CCL17. Liver Int. 2020, 40, 1327–1338. [Google Scholar] [CrossRef]
- Zhang, H.; Yan, X.; Yang, C.; Zhan, Q.; Fu, Y.; Luo, H. Intrahepatic T helper 17 cells recruited by hepatitis B virus X antigen-activated hepatic stellate cells exacerbate the progression of chronic hepatitis B virus infection. J. Viral Hepat. 2020, 27, 1138–1149. [Google Scholar] [CrossRef]
- Guidotti, L.G.; Rochford, R.; Chung, J.; Shapiro, M.; Purcell, R.; Chisari, F.V. Viral clearance without destruction of infected cells during acute HBV infection. Science 1999, 284, 825–829. [Google Scholar] [CrossRef]
- Ma, Z.; Zhang, E.; Yang, D.; Lu, M. Contribution of Toll-like receptors to the control of hepatitis B virus infection by initiating antiviral innate responses and promoting specific adaptive immune responses. Cell Mol. Immunol. 2015, 12, 273–282. [Google Scholar] [CrossRef] [Green Version]
- Raihan, R.; Akbar, S.M.F.; Al Mahtab, M.; Khan, M.S.I.; Tabassum, S.; Tee, K.K.; Mohamed, R.B. Increased Proinflammatory Cytokine Production by Chronic Hepatitis B Patients with Mutant Hepatitis B Virus: Plausible Mechanisms Underlying Severe Liver Diseases in These Patients. Viral. Immunol. 2020, 33, 530–534. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.; Chen, Y.; Lu, M. Advances in Targeting the Innate and Adaptive Immune Systems to Cure Chronic Hepatitis B Virus Infection. Front. Immunol. 2019, 10, 3127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertoletti, A.; Kennedy, P.T. The immune tolerant phase of chronic HBV infection: New perspectives on an old concept. Cell Mol. Immunol. 2015, 12, 258–263. [Google Scholar] [CrossRef] [Green Version]
- Fu, W.K.; Cao, J.; Mi, N.N.; Huang, C.F.; Gao, L.; Zhang, J.D.; Yue, P.; Bai, B.; Lin, Y.Y.; Meng, W.B. Cytokines predict virological response in chronic hepatitis B patients receiving peginterferon alfa-2a therapy. World J. Clin. Cases 2020, 8, 2255–2265. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Lian, Y.; Gu, L.; Chen, L.; Li, X.; Zhou, L.; Huang, Y.; Wang, J. Correlations between cytokines produced by T cells and clinical-virological characteristics in untreated chronic hepatitis B patients. BMC Infect. Dis. 2019, 19, 216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, K.C.K.; Joshi, S.S.; Mahoney, D.J.; Mason, A.L.; van Marle, G.; Osiowy, C.; Coffin, C.S. Differences in HBV Replication, APOBEC3 Family Expression, and Inflammatory Cytokine Levels Between Wild-Type HBV and Pre-core (G1896A) or Basal Core Promoter (A1762T/G1764A) Mutants. Front. Microbiol. 2020, 11, 1653. [Google Scholar] [CrossRef]
- Lan, S.; Wu, L.; Wang, X.; Wu, J.; Lin, X.; Wu, W.; Huang, Z. Impact of HBeAg on the maturation and function of dendritic cells. Int. J. Infect. Dis. 2016, 46, 42–48. [Google Scholar] [CrossRef] [Green Version]
- Khanam, A.; Ayithan, N.; Tang, L.; Poonia, B.; Kottilil, S. IL-21-Deficient T Follicular Helper Cells Support B Cell Responses Through IL-27 in Patients With Chronic Hepatitis B. Front. Immunol. 2020, 11, 599648. [Google Scholar] [CrossRef]
- Yang, F.; Yu, X.; Zhou, C.; Mao, R.; Zhu, M.; Zhu, H.; Ma, Z.; Mitra, B.; Zhao, G.; Huang, Y.; et al. Hepatitis B e antigen induces the expansion of monocytic myeloid-derived suppressor cells to dampen T-cell function in chronic hepatitis B virus infection. PLoS Pathog. 2019, 15, e1007690. [Google Scholar] [CrossRef]
- Pal, S.; Nandi, M.; Dey, D.; Chakraborty, B.C.; Shil, A.; Ghosh, S.; Banerjee, S.; Santra, A.; Ahammed, S.K.M.; Chowdhury, A.; et al. Myeloid-derived suppressor cells induce regulatory T cells in chronically HBV infected patients with high levels of hepatitis B surface antigen and persist after antiviral therapy. Aliment Pharmacol. Ther. 2019, 49, 1346–1359. [Google Scholar] [CrossRef]
- Wang, L.; Qiu, J.; Yu, L.; Hu, X.; Zhao, P.; Jiang, Y. Increased numbers of CD5+CD19+CD1dhighIL-10+ Bregs, CD4+Foxp3+ Tregs, CD4+CXCR5+Foxp3+ follicular regulatory T (TFR) cells in CHB or CHC patients. J. Transl. Med. 2014, 12, 251. [Google Scholar] [CrossRef] [Green Version]
- Burton, A.R.; Pallett, L.J.; McCoy, L.E.; Suveizdyte, K.; Amin, O.E.; Swadling, L.; Alberts, E.; Davidson, B.R.; Kennedy, P.T.; Gill, U.S.; et al. Circulating and intrahepatic antiviral B cells are defective in hepatitis B. J. Clin. Investig. 2018, 128, 4588–4603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Zhao, C.; Peng, Q.; Shi, J.; Gu, G. Expression levels of CD28, CTLA-4, PD-1 and TIM-3 as novel indicators of T-cell immune function in patients with chronic hepatitis B virus infection. Biomed. Rep. 2014, 2, 270–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schurich, A.; Khanna, P.; Lopes, A.R.; Han, K.J.; Peppa, D.; Micco, L.; Nebbia, G.; Kennedy, P.T.; Geretti, A.M.; Dusheiko, G.; et al. Role of the coinhibitory receptor cytotoxic T lymphocyte antigen-4 on apoptosis-Prone CD8 T cells in persistent hepatitis B virus infection. Hepatology 2011, 53, 1494–1503. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Dong, Q.; Li, Q.; Li, Y.; Zhao, D.; Sun, J.; Fu, J.; Meng, F.; Lin, H.; Luan, J.; et al. Dysregulated Response of Follicular Helper T Cells to Hepatitis B Surface Antigen Promotes HBV Persistence in Mice and Associates With Outcomes of Patients. Gastroenterology 2018, 154, 2222–2236. [Google Scholar] [CrossRef]
- Li, T.Y.; Yang, Y.; Zhou, G.; Tu, Z.K. Immune suppression in chronic hepatitis B infection associated liver disease: A review. World J. Gastroenterol. 2019, 25, 3527–3537. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Ellis, G.; Pallant, C.; Lopes, A.R.; Khanna, P.; Peppa, D.; Chen, A.; Blair, P.; Dusheiko, G.; Gill, U.; et al. IL-10-producing regulatory B cells in the pathogenesis of chronic hepatitis B virus infection. J. Immunol. 2012, 189, 3925–3935. [Google Scholar] [CrossRef]
- Lucifora, J.; Bonnin, M.; Aillot, L.; Fusil, F.; Maadadi, S.; Dimier, L.; Michelet, M.; Floriot, O.; Ollivier, A.; Rivoire, M.; et al. Direct antiviral properties of TLR ligands against HBV replication in immune-competent hepatocytes. Sci. Rep. 2018, 8, 5390. [Google Scholar] [CrossRef] [Green Version]
- Shurin, G.V.; Ma, Y.; Shurin, M.R. Immunosuppressive mechanisms of regulatory dendritic cells in cancer. Cancer Microenviron. 2013, 6, 159–167. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Park, C.S.; Jang, S.; Kim, J.W.; Kim, S.H.; Song, S.; Kim, K.; Lee, C.K. Tolerogenic dendritic cells are efficiently generated using minocycline and dexamethasone. Sci. Rep. 2017, 7, 15087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubo, M.; Trinschek, B.; Kryczanowsky, F.; Tuettenberg, A.; Steinbrink, K.; Jonuleit, H. Costimulatory molecules on immunogenic versus tolerogenic human dendritic cells. Front. Immunol. 2013, 4, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatipoglu, I.; Ercan, D.; Acilan, C.; Basalp, A.; Durali, D.; Baykal, A.T. Hepatitis B virus e antigen (HBeAg) may have a negative effect on dendritic cell generation. Immunobiology 2014, 219, 944–949. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhou, Z.H.; Sun, X.H.; Zhang, X.; Zhu, X.J.; Jin, S.G.; Gao, Y.T.; Jiang, Y.; Gao, Y.Q. Hepatitis B core antigen upregulates B7-H1 on dendritic cells by activating the AKT/ERK/P38 pathway: A possible mechanism of hepatitis B virus persistence. Lab. Investig. 2016, 96, 1156–1164. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Zhang, X.; Lv, Z.; Gao, L.; Yan, H. Increased Expression of Myeloid-Derived Suppressor Cells in Patients with HBV-Related Hepatocellular Carcinoma. BioMed Res. Int. 2020, 2020, 6527192. [Google Scholar] [CrossRef] [Green Version]
- Fisicaro, P.; Barili, V.; Rossi, M.; Montali, I.; Vecchi, A.; Acerbi, G.; Laccabue, D.; Zecca, A.; Penna, A.; Missale, G.; et al. Pathogenetic Mechanisms of T Cell Dysfunction in Chronic HBV Infection and Related Therapeutic Approaches. Front. Immunol. 2020, 11, 849. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.; Li, J.; Yu, X.; Zhang, D.; Ren, G.; Shi, B.; Wang, C.; Kosinska, A.D.; Wang, S.; Zhou, X.; et al. Polarization of Monocytic Myeloid-Derived Suppressor Cells by Hepatitis B Surface Antigen Is Mediated via ERK/IL-6/STAT3 Signaling Feedback and Restrains the Activation of T Cells in Chronic Hepatitis B Virus Infection. J. Immunol. 2015, 195, 4873–4883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drabczyk-Pluta, M.; Werner, T.; Hoffmann, D.; Leng, Q.; Chen, L.; Dittmer, U.; Zelinskyy, G. Granulocytic myeloid-derived suppressor cells suppress virus-specific CD8(+) T cell responses during acute Friend retrovirus infection. Retrovirology 2017, 14, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Özkan, B.; Lim, H.; Park, S.G. Immunomodulatory Function of Myeloid-Derived Suppressor Cells during B Cell-Mediated Immune Responses. Int. J. Mol. Sci. 2018, 19, 1468. [Google Scholar] [CrossRef] [Green Version]
- Monu, N.R.; Frey, A.B. Myeloid-derived suppressor cells and anti-tumor T cells: A complex relationship. Immunol. Investig. 2012, 41, 595–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gol-Ara, M.; Jadidi-Niaragh, F.; Sadria, R.; Azizi, G.; Mirshafiey, A. The role of different subsets of regulatory T cells in immunopathogenesis of rheumatoid arthritis. Arthritis 2012, 2012, 805875. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, TIM-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef] [Green Version]
- Zeng, D.W.; Dong, J.; Liu, Y.R.; Jiang, J.J.; Zhu, Y.Y. Noninvasive models for assessment of liver fibrosis in patients with chronic hepatitis B virus infection. World J. Gastroenterol. 2016, 22, 6663–6672. [Google Scholar] [CrossRef] [PubMed]
- Stross, L.; Gunther, J.; Gasteiger, G.; Asen, T.; Graf, S.; Aichler, M.; Esposito, I.; Busch, D.H.; Knolle, P.; Sparwasser, T.; et al. Foxp3+ regulatory T cells protect the liver from immune damage and compromise virus control during acute experimental hepatitis B virus infection in mice. Hepatology 2012, 56, 873–883. [Google Scholar] [CrossRef] [PubMed]
- Teng, C.F.; Li, T.C.; Wang, T.; Liao, D.C.; Wen, Y.H.; Wu, T.H.; Wang, J.; Wu, H.C.; Shyu, W.C.; Su, I.J.; et al. Increased infiltration of regulatory T cells in hepatocellular carcinoma of patients with hepatitis B virus pre-S2 mutant. Sci. Rep. 2021, 11, 1136. [Google Scholar] [CrossRef]
- Peng, G.; Li, S.; Wu, W.; Sun, Z.; Chen, Y.; Chen, Z. Circulating CD4+ CD25+ regulatory T cells correlate with chronic hepatitis B infection. Immunology 2008, 123, 57–65. [Google Scholar] [CrossRef]
- Tang, R.; Lei, Z.; Wang, X.; Qi, Q.; He, J.; Liu, D.; Chen, X.; Zhu, J.; Li, Y.; Zhou, S.; et al. Hepatitis B envelope antigen increases Tregs by converting CD4+CD25(-) T cells into CD4(+)CD25(+)Foxp3(+) Tregs. Exp. Ther. Med. 2020, 20, 3679–3686. [Google Scholar] [CrossRef]
- Ma, Q.; Dong, X.; Liu, S.; Zhong, T.; Sun, D.; Zong, L.; Zhao, C.; Lu, Q.; Zhang, M.; Gao, Y.; et al. Hepatitis B e Antigen Induces NKG2A(+) Natural Killer Cell Dysfunction via Regulatory T Cell-Derived Interleukin 10 in Chronic Hepatitis B Virus Infection. Front. Cell Dev. Biol. 2020, 8, 421. [Google Scholar] [CrossRef]
- Vazquez, M.I.; Catalan-Dibene, J.; Zlotnik, A. B cells responses and cytokine production are regulated by their immune microenvironment. Cytokine 2015, 74, 318–326. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Cheng, L.S.; Wu, S.D.; Wang, S.Q.; Li, L.; She, W.M.; Li, J.; Wang, J.Y.; Jiang, W. IL-10-producing regulatory B-cells suppressed effector T-cells but enhanced regulatory T-cells in chronic HBV infection. Clin. Sci. (Lond.) 2016, 130, 907–919. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Yin, W. The Multiple Functions of B Cells in Chronic HBV Infection. Front. Immunol. 2020, 11, 582292. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Liu, Y.; Huang, R.; Jia, B.; Su, R.; Sun, Z.; Tian, C.; Xiong, Y.; Xia, J.; Yan, X.; et al. Characteristics of regulatory B cells in patients with chronic hepatitis B virus infection in different immune phases. Discov. Med. 2017, 23, 295–304. [Google Scholar] [PubMed]
- Chen, Y.; Tian, Z. HBV-Induced Immune Imbalance in the Development of HCC. Front. Immunol. 2019, 10, 2048. [Google Scholar] [CrossRef] [Green Version]
- Karim, M.R.; Wang, Y.F. Phenotypic identification of CD19(+)CD5(+)CD1d(+) regulatory B cells that produce interleukin 10 and transforming growth factor beta1 in human peripheral blood. Arch. Med. Sci. 2019, 15, 1176–1183. [Google Scholar] [CrossRef]
- Gong, Y.; Zhao, C.; Zhao, P.; Wang, M.; Zhou, G.; Han, F.; Cui, Y.; Qian, J.; Zhang, H.; Xiong, H.; et al. Role of IL-10-Producing Regulatory B Cells in Chronic Hepatitis B Virus Infection. Dig. Dis. Sci. 2015, 60, 1308–1314. [Google Scholar] [CrossRef]
- Alatrakchi, N. Bregs in Chronic HBV: Is It Time for Bragging Rights? Dig. Dis. Sci. 2015, 60, 1115–1117. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Xu, J.; Huang, Q.; Huang, M.; Wen, H.; Zhang, C.; Wang, J.; Song, J.; Zheng, M.; Sun, H.; et al. High NKG2A expression contributes to NK cell exhaustion and predicts a poor prognosis of patients with liver cancer. Oncoimmunology 2017, 6, e1264562. [Google Scholar] [CrossRef]
- Marotel, M.; Villard, M.; Drouillard, A.; Tout, I.; Besson, L.; Allatif, O.; Pujol, M.; Rocca, Y.; Ainouze, M.; Roblot, G.; et al. Peripheral natural killer cells in chronic hepatitis B patients display multiple molecular features of T cell exhaustion. Elife 2021, 10. [Google Scholar] [CrossRef]
- Xu, Y.; Wang, Z.; Du, X.; Liu, Y.; Song, X.; Wang, T.; Tan, S.; Liang, X.; Gao, L.; Ma, C. TIM-3 blockade promotes iNKT cell function to inhibit HBV replication. J. Cell Mol. Med. 2018, 22, 3192–3201. [Google Scholar] [CrossRef] [Green Version]
- Schuch, A.; Hoh, A.; Thimme, R. The role of natural killer cells and CD8(+) T cells in hepatitis B virus infection. Front. Immunol. 2014, 5, 258. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.N.; Zhang, N.; Liu, H.H.; Xia, P.; Zhang, C.; Song, J.W.; Fan, X.; Shi, M.; Jin, L.; Zhang, J.Y.; et al. Skewed CD39/CD73/adenosine pathway contributes to B-cell hyperactivation and disease progression in patients with chronic hepatitis B. Gastroenterol. Rep. 2021, 9, 49–58. [Google Scholar] [CrossRef]
- Dong, Y.; Li, X.; Zhang, L.; Zhu, Q.; Chen, C.; Bao, J.; Chen, Y. CD4(+) T cell exhaustion revealed by high PD-1 and LAG-3 expression and the loss of helper T cell function in chronic hepatitis B. BMC Immunol. 2019, 20, 27. [Google Scholar] [CrossRef]
- Nebbia, G.; Peppa, D.; Schurich, A.; Khanna, P.; Singh, H.D.; Cheng, Y.; Rosenberg, W.; Dusheiko, G.; Gilson, R.; ChinAleong, J.; et al. Upregulation of the TIM-3/galectin-9 pathway of T cell exhaustion in chronic hepatitis B virus infection. PLoS ONE 2012, 7, e47648. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.S.; Cox, M.A.; Zajac, A.J. T-cell exhaustion: Characteristics, causes and conversion. Immunology 2010, 129, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Riley, J.L. PD-1 signaling in primary T cells. Immunol. Rev. 2009, 229, 114–125. [Google Scholar] [CrossRef]
- Salimzadeh, L.; Le Bert, N.; Dutertre, C.A.; Gill, U.S.; Newell, E.W.; Frey, C.; Hung, M.; Novikov, N.; Fletcher, S.; Kennedy, P.T.; et al. PD-1 blockade partially recovers dysfunctional virus-specific B cells in chronic hepatitis B infection. J. Clin. Investig. 2018, 128, 4573–4587. [Google Scholar] [CrossRef] [PubMed]
- Peng, G.; Li, S.; Wu, W.; Tan, X.; Chen, Y.; Chen, Z. PD-1 upregulation is associated with HBV-specific T cell dysfunction in chronic hepatitis B patients. Mol. Immunol. 2008, 45, 963–970. [Google Scholar] [CrossRef]
- Fisicaro, P.; Valdatta, C.; Massari, M.; Loggi, E.; Biasini, E.; Sacchelli, L.; Cavallo, M.C.; Silini, E.M.; Andreone, P.; Missale, G.; et al. Antiviral intrahepatic T-cell responses can be restored by blocking programmed death-1 pathway in chronic hepatitis B. Gastroenterology 2010, 138, 682–693. [Google Scholar] [CrossRef]
- Wu, W.; Shi, Y.; Li, S.; Zhang, Y.; Liu, Y.; Wu, Y.; Chen, Z. Blockade of TIM-3 signaling restores the virus-specific CD8(+) T-cell response in patients with chronic hepatitis B. Eur. J. Immunol. 2012, 42, 1180–1191. [Google Scholar] [CrossRef] [PubMed]
- Zong, L.; Peng, H.; Sun, C.; Li, F.; Zheng, M.; Chen, Y.; Wei, H.; Sun, R.; Tian, Z. Breakdown of adaptive immunotolerance induces hepatocellular carcinoma in HBsAg-tg mice. Nat. Commun. 2019, 10, 221. [Google Scholar] [CrossRef] [PubMed]
- Ye, B.; Liu, X.; Li, X.; Kong, H.; Tian, L.; Chen, Y. T-cell exhaustion in chronic hepatitis B infection: Current knowledge and clinical significance. Cell Death Dis. 2015, 6, e1694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tangye, S.G.; Ma, C.S. Regulation of the germinal center and humoral immunity by interleukin-21. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Shi, Y.; Li, J.; Chen, F.; Chen, Z.; Zheng, M. TIM-3 expression on peripheral T cell subsets correlates with disease progression in hepatitis B infection. Virol. J. 2011, 8, 113. [Google Scholar] [CrossRef] [Green Version]
- Heim, K.; Neumann-Haefelin, C.; Thimme, R.; Hofmann, M. Heterogeneity of HBV-Specific CD8(+) T-Cell Failure: Implications for Immunotherapy. Front. Immunol. 2019, 10, 2240. [Google Scholar] [CrossRef]
- Lopes, A.R.; Kellam, P.; Das, A.; Dunn, C.; Kwan, A.; Turner, J.; Peppa, D.; Gilson, R.J.; Gehring, A.; Bertoletti, A.; et al. Bim-mediated deletion of antigen-specific CD8 T cells in patients unable to control HBV infection. J. Clin. Investig. 2008, 118, 1835–1845. [Google Scholar] [CrossRef]
- Poonia, B.; Ayithan, N.; Nandi, M.; Masur, H.; Kottilil, S. HBV induces inhibitory FcRL receptor on B cells and dysregulates B cell-T follicular helper cell axis. Sci. Rep. 2018, 8, 15296. [Google Scholar] [CrossRef]
- Xu, X.; Shang, Q.; Chen, X.; Nie, W.; Zou, Z.; Huang, A.; Meng, M.; Jin, L.; Xu, R.; Zhang, J.Y.; et al. Reversal of B-cell hyperactivation and functional impairment is associated with HBsAg seroconversion in chronic hepatitis B patients. Cell Mol. Immunol. 2015, 12, 309–316. [Google Scholar] [CrossRef]
- Gerlich, W.H. Medical virology of hepatitis B: How it began and where we are now. Virol. J. 2013, 10, 239. [Google Scholar] [CrossRef] [Green Version]
- Le Bert, N.; Salimzadeh, L.; Gill, U.S.; Dutertre, C.A.; Facchetti, F.; Tan, A.; Hung, M.; Novikov, N.; Lampertico, P.; Fletcher, S.P.; et al. Comparative characterization of B cells specific for HBV nucleocapsid and envelope proteins in patients with chronic hepatitis B. J. Hepatol. 2020, 72, 34–44. [Google Scholar] [CrossRef]
- Trepo, C.; Chan, H.L.; Lok, A. Hepatitis B virus infection. Lancet 2014, 384, 2053–2063. [Google Scholar] [CrossRef]
- Neumann-Haefelin, C.; Thimme, R. Entering the spotlight: Hepatitis B surface antigen-specific B cells. J. Clin. Investig. 2018, 128, 4257–4259. [Google Scholar] [CrossRef] [Green Version]
- Seto, W.K.; Chan, T.S.; Hwang, Y.Y.; Wong, D.K.; Fung, J.; Liu, K.S.; Gill, H.; Lam, Y.F.; Lie, A.K.; Lai, C.L.; et al. Hepatitis B reactivation in patients with previous hepatitis B virus exposure undergoing rituximab-containing chemotherapy for lymphoma: A prospective study. J. Clin. Oncol. 2014, 32, 3736–3743. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.; Yu, S.J.; Cho, E.J.; Lee, J.H.; Kim, T.M.; Heo, D.S.; Kim, Y.J.; Yoon, J.H. High titers of anti-HBs prevent rituximab-related viral reactivation in resolved hepatitis B patient with non-Hodgkin’s lymphoma. J. Med. Virol. 2016, 88, 1010–1017. [Google Scholar] [CrossRef] [PubMed]
- Tsutsumi, Y.; Yamamoto, Y.; Ito, S.; Ohigashi, H.; Shiratori, S.; Naruse, H.; Teshima, T. Hepatitis B virus reactivation with a rituximab-containing regimen. World J. Hepatol. 2015, 7, 2344–2351. [Google Scholar] [CrossRef] [PubMed]
- Pape, K.A.; Catron, D.M.; Itano, A.A.; Jenkins, M.K. The humoral immune response is initiated in lymph nodes by B cells that acquire soluble antigen directly in the follicles. Immunity 2007, 26, 491–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batista, F.D.; Harwood, N.E. The who, how and where of antigen presentation to B cells. Nat. Rev. Immunol. 2009, 9, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Khlaiphuengsin, A.; Chuaypen, N.; Hirankarn, N.; Avihingsanon, A.; Crane, M.; Lewin, S.R.; Tangkijvanich, P. Circulating BAFF and CXCL10 levels predict response to pegylated interferon in patients with HBeAg-positive chronic hepatitis B. Asian Pac. J. Allergy Immunol. 2019. [Google Scholar] [CrossRef]
- Khlaiphuengsin, A.; Chuaypen, N.; Pinjaroen, N.; Sirichindakul, B.; Hirankarn, N.; Tangkijvanich, P. Plasma B-cell activating factor levels and polymorphisms in hepatitis B-related hepatocellular carcinoma: Clinical correlation and prognosis. Asian Pac. J. Allergy Immunol. 2019. [Google Scholar] [CrossRef]
- Tan, G.; Song, H.; Xu, F.; Cheng, G. When Hepatitis B Virus Meets Interferons. Front. Microbiol. 2018, 9, 1611. [Google Scholar] [CrossRef] [PubMed]
- Iannacone, M.; Sitia, G.; Ruggeri, Z.M.; Guidotti, L.G. HBV pathogenesis in animal models: Recent advances on the role of platelets. J. Hepatol. 2007, 46, 719–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Chen, Z.; Hu, C.; Qian, F.; Cheng, Y.; Wu, M.; Shi, B.; Chen, J.; Hu, Y.; Yuan, Z. Hepatitis B virus surface antigen selectively inhibits TLR2 ligand-induced IL-12 production in monocytes/macrophages by interfering with JNK activation. J. Immunol. 2013, 190, 5142–5151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Hou, Y.; Meng, S.H.; Yang, B.; Yang, P.; Zhang, H.; Zhu, Y. Abnormal IL-10 levels were related to alanine aminotransferase abnormalities during postpartum in HBeAg positive women with chronic hepatitis B. Medicine (Baltimore) 2019, 98, e17969. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Chen, Y.; Gao, B. Natural killer cells in liver disease. Hepatology 2013, 57, 1654–1662. [Google Scholar] [CrossRef] [PubMed]
- Couper, K.N.; Blount, D.G.; Riley, E.M. IL-10: The master regulator of immunity to infection. J. Immunol. 2008, 180, 5771–5777. [Google Scholar] [CrossRef] [PubMed]
- Lan, T.; Chang, L.; Wu, L.; Yuan, Y.F. IL-6 Plays a Crucial Role in HBV Infection. J. Clin. Transl. Hepatol. 2015, 3, 271–276. [Google Scholar] [CrossRef]
- Li, C.; Deng, M.; Hu, J.; Li, X.; Chen, L.; Ju, Y.; Hao, J.; Meng, S. Chronic inflammation contributes to the development of hepatocellular carcinoma by decreasing miR-122 levels. Oncotarget 2016, 7, 17021–17034. [Google Scholar] [CrossRef]
- Xia, C.; Liu, Y.; Chen, Z.; Zheng, M. Involvement of Interleukin 6 in Hepatitis B Viral Infection. Cell Physiol. Biochem. 2015, 37, 677–686. [Google Scholar] [CrossRef]
- Bouezzedine, F.; Fardel, O.; Gripon, P. Interleukin 6 inhibits HBV entry through NTCP down regulation. Virology 2015, 481, 34–42. [Google Scholar] [CrossRef]
- Le Vee, M.; Lecureur, V.; Stieger, B.; Fardel, O. Regulation of drug transporter expression in human hepatocytes exposed to the proinflammatory cytokines tumor necrosis factor-alpha or interleukin-6. Drug Metab. Dispos. Biol. Fate Chem. 2009, 37, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Guan, S.H.; Zhang, H.; Pan, Y.; Wu, Y.Y.; Wang, A.H.; Sun, B.B. Enhanced levels of interleukin-8 are associated with hepatitis B virus infection and resistance to interferon-alpha therapy. Int. J. Mol. Sci. 2014, 15, 21286–21298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khanam, A.; Trehanpati, N.; Riese, P.; Rastogi, A.; Guzman, C.A.; Sarin, S.K. Blockade of Neutrophil’s Chemokine Receptors CXCR1/2 Abrogate Liver Damage in Acute-on-Chronic Liver Failure. Front. Immunol. 2017, 8, 464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, S.Y.; Lu, D.H.; Guo, X.Y.; Luo, W.; Hu, B.L.; Huang, X.L.; Chen, M.; Wang, J.X.; Ma, S.J.; Yang, X.W.; et al. A deleterious role for Th9/IL-9 in hepatic fibrogenesis. Sci. Rep. 2016, 6, 18694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaseen, M.M.; Abuharfeil, N.M.; Darmani, H.; Daoud, A. Mechanisms of immune suppression by myeloid-derived suppressor cells: The role of interleukin-10 as a key immunoregulatory cytokine. Open Biol. 2020, 10, 200111. [Google Scholar] [CrossRef]
- Schurich, A.; Pallett, L.J.; Lubowiecki, M.; Singh, H.D.; Gill, U.S.; Kennedy, P.T.; Nastouli, E.; Tanwar, S.; Rosenberg, W.; Maini, M.K. The third signal cytokine IL-12 rescues the anti-viral function of exhausted HBV-specific CD8 T cells. PLoS Pathog. 2013, 9, e1003208. [Google Scholar] [CrossRef]
- Schurich, A.; Pallett, L.J.; Jajbhay, D.; Wijngaarden, J.; Otano, I.; Gill, U.S.; Hansi, N.; Kennedy, P.T.; Nastouli, E.; Gilson, R.; et al. Distinct Metabolic Requirements of Exhausted and Functional Virus-Specific CD8 T Cells in the Same Host. Cell Rep. 2016, 16, 1243–1252. [Google Scholar] [CrossRef] [Green Version]
- Wong, S.W.; Ting, Y.W.; Yong, Y.K.; Tan, H.Y.; Barathan, M.; Riazalhosseini, B.; Bee, C.J.; Tee, K.K.; Larsson, M.; Velu, V.; et al. Chronic inflammation involves CCL11 and IL-13 to facilitate the development of liver cirrhosis and fibrosis in chronic hepatitis B virus infection. Scand. J. Clin. Lab Invest. 2021, 81, 147–159. [Google Scholar] [CrossRef]
- Di Scala, M.; Otano, I.; Gil-Farina, I.; Vanrell, L.; Hommel, M.; Olague, C.; Vales, A.; Galarraga, M.; Guembe, L.; Ortiz de Solorzano, C.; et al. Complementary Effects of Interleukin-15 and Alpha Interferon Induce Immunity in Hepatitis B Virus Transgenic Mice. J. Virol. 2016, 90, 8563–8574. [Google Scholar] [CrossRef] [Green Version]
- Du, W.J.; Zhen, J.H.; Zeng, Z.Q.; Zheng, Z.M.; Xu, Y.; Qin, L.Y.; Chen, S.J. Expression of interleukin-17 associated with disease progression and liver fibrosis with hepatitis B virus infection: IL-17 in HBV infection. Diagn Pathol. 2013, 8, 40. [Google Scholar] [CrossRef] [Green Version]
- Shi, M.; Wei, J.; Dong, J.; Meng, W.; Ma, J.; Wang, T.; Wang, N.; Wang, Y. Function of interleukin-17 and -35 in the blood of patients with hepatitis B-related liver cirrhosis. Mol. Med. Rep. 2015, 11, 121–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khanam, A.; Trehanpati, N.; Sarin, S.K. Increased interleukin-23 receptor (IL-23R) expression is associated with disease severity in acute-on-chronic liver failure. Liver Int. 2019, 39, 1062–1070. [Google Scholar] [CrossRef]
- Dai, Z.J.; Liu, X.H.; Wang, M.; Guo, Y.; Zhu, W.; Li, X.; Lin, S.; Tian, T.; Liu, K.; Zheng, Y.; et al. IL-18 polymorphisms contribute to hepatitis B virus-related cirrhosis and hepatocellular carcinoma susceptibility in Chinese population: A case-control study. Oncotarget 2017, 8, 81350–81360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Tang, L.; Hou, J. Role of interleukin-21 in HBV infection: Friend or foe? Cell Mol. Immunol. 2015, 12, 303–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, X.; Gui, H.; King, N.J.; Cole, L.; Wang, H.; Xie, Q.; Bao, S. IL-22 and non-ELR-CXC chemokine expression in chronic hepatitis B virus-infected liver. Immunol. Cell Biol. 2012, 90, 611–619. [Google Scholar] [CrossRef]
- Meng, F.; Wang, K.; Aoyama, T.; Grivennikov, S.I.; Paik, Y.; Scholten, D.; Cong, M.; Iwaisako, K.; Liu, X.; Zhang, M.; et al. Interleukin-17 signaling in inflammatory, Kupffer cells, and hepatic stellate cells exacerbates liver fibrosis in mice. Gastroenterology 2012, 143, 765–776.e763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zang, M.; Li, Y.; He, H.; Ding, H.; Chen, K.; Du, J.; Chen, T.; Wu, Z.; Liu, H.; Wang, D.; et al. IL-23 production of liver inflammatory macrophages to damaged hepatocytes promotes hepatocellular carcinoma development after chronic hepatitis B virus infection. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3759–3770. [Google Scholar] [CrossRef]
- Wang, H.L.; Zhang, H.Y.; Zhai, Z.L.; Zhou, X. The correlation between hepatitis B virus infection and IL-27. Biomed. Mater. Eng. 2012, 22, 187–193. [Google Scholar] [CrossRef]
- Cao, Y.; Zhang, R.; Zhang, W.; Zhu, C.; Yu, Y.; Song, Y.; Wang, Q.; Bai, L.; Liu, Y.; Wu, K.; et al. IL-27, a cytokine, and IFN-lambda1, a type III IFN, are coordinated to regulate virus replication through type I IFN. J. Immunol. 2014, 192, 691–703. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Cai, Y.; Ji, H.; Feng, J.; Ayana, D.A.; Niu, J.; Jiang, Y. Serum IL-33 levels are associated with liver damage in patients with chronic hepatitis B. J. Interferon. Cytokine Res. 2012, 32, 248–253. [Google Scholar] [CrossRef] [Green Version]
- Zhao, P.W.; Shi, X.; Li, C.; Ayana, D.A.; Niu, J.Q.; Feng, J.Y.; Wang, J.; Jiang, Y.F. IL-33 Enhances Humoral Immunity Against Chronic HBV Infection Through Activating CD4(+)CXCR5(+) TFH Cells. J. Interferon. Cytokine Res. 2015, 35, 454–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Liu, X.; Wang, W. IL-35: A Novel Immunomodulator in Hepatitis B Virus-Related Liver Diseases. Front. Cell Dev. Biol. 2021, 9, 614847. [Google Scholar] [CrossRef] [PubMed]
- Shao, X.; Ma, J.; Jia, S.; Yang, L.; Wang, W.; Jin, Z. Interleukin-35 Suppresses Antiviral Immune Response in Chronic Hepatitis B Virus Infection. Front. Cell Infect. Microbiol. 2017, 7, 472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, Z.; Li, L.; Chen, Y.; Wei, H.; Sun, R.; Tian, Z. Interferon-gamma facilitates hepatic antiviral T cell retention for the maintenance of liver-induced systemic tolerance. J. Exp. Med. 2016, 213, 1079–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suri, D.; Schilling, R.; Lopes, A.R.; Mullerova, I.; Colucci, G.; Williams, R.; Naoumov, N.V. Non-cytolytic inhibition of hepatitis B virus replication in human hepatocytes. J. Hepatol. 2001, 35, 790–797. [Google Scholar] [CrossRef]
- Khanam, A.; Trehanpati, N.; Garg, V.; Kumar, C.; Garg, H.; Sharma, B.C.; Sarin, S.K. Altered frequencies of dendritic cells and IFN-gamma-secreting T cells with granulocyte colony-stimulating factor (G-CSF) therapy in acute-on- chronic liver failure. Liver Int. 2014, 34, 505–513. [Google Scholar] [CrossRef]
- Valaydon, Z.; Pellegrini, M.; Thompson, A.; Desmond, P.; Revill, P.; Ebert, G. The role of tumour necrosis factor in hepatitis B infection: Jekyll and Hyde. Clin. Transl. Immunol. 2016, 5, e115. [Google Scholar] [CrossRef] [Green Version]
- Biermer, M.; Puro, R.; Schneider, R.J. Tumor necrosis factor alpha inhibition of hepatitis B virus replication involves disruption of capsid Integrity through activation of NF-kappaB. J. Virol. 2003, 77, 4033–4042. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Xu, Y.; Ma, H.; Wang, B.; Xu, L.; Zhang, H.; Song, X.; Gao, L.; Liang, X.; Ma, C. Hepatitis B virus X protein amplifies TGF-beta promotion on HCC motility through down-regulating PPM1a. Oncotarget 2016, 7, 33125–33135. [Google Scholar] [CrossRef] [Green Version]
- Dong, K.S.; Chen, Y.; Yang, G.; Liao, Z.B.; Zhang, H.W.; Liang, H.F.; Chen, X.P.; Dong, H.H. TGF-beta1 accelerates the hepatitis B virus X-induced malignant transformation of hepatic progenitor cells by upregulating miR-199a-3p. Oncogene 2020, 39, 1807–1820. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Xu, X.; Tao, Y.; Qian, Z.; Yu, Y. Exosomes in hepatocellular carcinoma: A new horizon. Cell Commun. Signal 2019, 17, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.; Huang, A.C.; Zhang, W.; Zhang, G.; Wu, M.; Xu, W.; Yu, Z.; Yang, J.; Wang, B.; Sun, H.; et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature 2018, 560, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Gao, W.; Yao, K.; Ge, J. Roles of Exosomes Derived From Immune Cells in Cardiovascular Diseases. Front. Immunol. 2019, 10, 648. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhang, C.; Zhang, P.; Guo, G.; Jiang, T.; Zhao, X.; Jiang, J.; Huang, X.; Tong, H.; Tian, Y. Serum exosomal microRNAs combined with alpha-fetoprotein as diagnostic markers of hepatocellular carcinoma. Cancer Med. 2018, 7, 1670–1679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, Y.; Kim, M.; Kim, Y.; Jung, H.S.; Jeoung, D. Exosomal MicroRNAs as Mediators of Cellular Interactions Between Cancer Cells and Macrophages. Front. Immunol. 2020, 11, 1167. [Google Scholar] [CrossRef] [PubMed]
- Kouwaki, T.; Fukushima, Y.; Daito, T.; Sanada, T.; Yamamoto, N.; Mifsud, E.J.; Leong, C.R.; Tsukiyama-Kohara, K.; Kohara, M.; Matsumoto, M.; et al. Extracellular Vesicles Including Exosomes Regulate Innate Immune Responses to Hepatitis B Virus Infection. Front. Immunol. 2016, 7, 335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Han, Q.; Hou, Z.; Zhang, C.; Tian, Z.; Zhang, J. Exosomes mediate hepatitis B virus (HBV) transmission and NK-cell dysfunction. Cell Mol. Immunol. 2017, 14, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Qiao, Y.; Li, X.; Chen, J.; Ding, J.; Bai, L.; Shen, F.; Shi, B.; Liu, J.; Peng, L.; et al. Exosomes Exploit the Virus Entry Machinery and Pathway To Transmit Alpha Interferon-Induced Antiviral Activity. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.; Wu, D.; Yan, W.; Wang, Y.; You, J.; Wan, X.; Xi, D.; Luo, X.; Han, M.; Ning, Q. Interferon-Induced Macrophage-Derived Exosomes Mediate Antiviral Activity Against Hepatitis B Virus Through miR-574-5p. J. Infect. Dis. 2021, 223, 686–698. [Google Scholar] [CrossRef]
- Zhang, H.; Xie, Y.; Li, W.; Chibbar, R.; Xiong, S.; Xiang, J. CD4 (+) T cell-released exosomes inhibit CD8(+) cytotoxic T-lymphocyte responses and antitumor immunity. Cell Mol. Immunol. 2011, 8, 23–30. [Google Scholar] [CrossRef]
- Huang, E.; Peng, N.; Xiao, F.; Hu, D.; Wang, X.; Lu, L. The Roles of Immune Cells in the Pathogenesis of Fibrosis. Int. J. Mol. Sci. 2020, 21, 5203. [Google Scholar] [CrossRef] [PubMed]
- Rehermann, B.; Nascimbeni, M. Immunology of hepatitis B virus and hepatitis C virus infection. Nat. Rev. Immunol. 2005, 5, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Maini, M.K.; Boni, C.; Lee, C.K.; Larrubia, J.R.; Reignat, S.; Ogg, G.S.; King, A.S.; Herberg, J.; Gilson, R.; Alisa, A.; et al. The role of virus-specific CD8 (+) cells in liver damage and viral control during persistent hepatitis B virus infection. J. Exp. Med. 2000, 191, 1269–1280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, I.S.; Park, S.H. Immune-mediated Liver Injury in Hepatitis B Virus Infection. Immune. Netw. 2015, 15, 191–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunn, C.; Peppa, D.; Khanna, P.; Nebbia, G.; Jones, M.; Brendish, N.; Lascar, R.M.; Brown, D.; Gilson, R.J.; Tedder, R.J.; et al. Temporal analysis of early immune responses in patients with acute hepatitis B virus infection. Gastroenterology 2009, 137, 1289–1300. [Google Scholar] [CrossRef]
- Dewidar, B.; Meyer, C.; Dooley, S.; Meindl-Beinker, A.N. TGF-beta in Hepatic Stellate Cell Activation and Liver Fibrogenesis-Updated 2019. Cells 2019, 8, 1419. [Google Scholar] [CrossRef] [Green Version]
- Khanam, A.; Saleeb, P.G.; Kottilil, S. Pathophysiology and Treatment Options for Hepatic Fibrosis: Can It Be Completely Cured? Cells 2021, 10, 1097. [Google Scholar] [CrossRef]
- Bility, M.T.; Cheng, L.; Zhang, Z.; Luan, Y.; Li, F.; Chi, L.; Zhang, L.; Tu, Z.; Gao, Y.; Fu, Y.; et al. Hepatitis B virus infection and immunopathogenesis in a humanized mouse model: Induction of human-specific liver fibrosis and M2-like macrophages. PLoS Pathog. 2014, 10, e1004032. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Zhai, N.; Wang, Z.; Song, H.; Yang, Y.; Cui, A.; Li, T.; Wang, G.; Niu, J.; Crispe, I.N.; et al. Regulatory NK cells mediated between immunosuppressive monocytes and dysfunctional T cells in chronic HBV infection. Gut 2018, 67, 2035–2044. [Google Scholar] [CrossRef]
- Zhang, J.Y.; Zou, Z.S.; Huang, A.; Zhang, Z.; Fu, J.L.; Xu, X.S.; Chen, L.M.; Li, B.S.; Wang, F.S. Hyper-activated pro-inflammatory CD16 monocytes correlate with the severity of liver injury and fibrosis in patients with chronic hepatitis B. PLoS ONE 2011, 6, e17484. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Zhao, W.; Cheng, L.; Guo, M.; Li, D.; Li, X.; Tan, Y.; Ma, S.; Li, S.; Yang, Y.; et al. CD137-mediated pathogenesis from chronic hepatitis to hepatocellular carcinoma in hepatitis B virus-transgenic mice. J. Immunol. 2010, 185, 7654–7662. [Google Scholar] [CrossRef] [Green Version]
- Song, G.; Shi, Y.; Zhang, M.; Goswami, S.; Afridi, S.; Meng, L.; Ma, J.; Chen, Y.; Lin, Y.; Zhang, J.; et al. Global immune characterization of HBV/HCV-related hepatocellular carcinoma identifies macrophage and T-cell subsets associated with disease progression. Cell Discov. 2020, 6, 90. [Google Scholar] [CrossRef]
- Fisicaro, P.; Valdatta, C.; Boni, C.; Massari, M.; Mori, C.; Zerbini, A.; Orlandini, A.; Sacchelli, L.; Missale, G.; Ferrari, C. Early kinetics of innate and adaptive immune responses during hepatitis B virus infection. Gut 2009, 58, 974–982. [Google Scholar] [CrossRef] [Green Version]
- Lunemann, S.; Malone, D.F.; Hengst, J.; Port, K.; Grabowski, J.; Deterding, K.; Markova, A.; Bremer, B.; Schlaphoff, V.; Cornberg, M.; et al. Compromised function of natural killer cells in acute and chronic viral hepatitis. J. Infect. Dis. 2014, 209, 1362–1373. [Google Scholar] [CrossRef] [Green Version]
- Boni, C.; Lampertico, P.; Talamona, L.; Giuberti, T.; Invernizzi, F.; Barili, V.; Fisicaro, P.; Rossi, M.; Cavallo, M.C.; Vecchi, A.; et al. Natural killer cell phenotype modulation and natural killer/T-cell interplay in nucleos(t)ide analogue-treated hepatitis e antigen-negative patients with chronic hepatitis B. Hepatology 2015, 62, 1697–1709. [Google Scholar] [CrossRef]
- Okazaki, A.; Hiraga, N.; Imamura, M.; Hayes, C.N.; Tsuge, M.; Takahashi, S.; Aikata, H.; Abe, H.; Miki, D.; Ochi, H.; et al. Severe necroinflammatory reaction caused by natural killer cell-mediated Fas/Fas ligand interaction and dendritic cells in human hepatocyte chimeric mouse. Hepatology 2012, 56, 555–566. [Google Scholar] [CrossRef] [Green Version]
- Zou, Y.; Chen, T.; Han, M.; Wang, H.; Yan, W.; Song, G.; Wu, Z.; Wang, X.; Zhu, C.; Luo, X.; et al. Increased killing of liver NK cells by Fas/Fas ligand and NKG2D/NKG2D ligand contributes to hepatocyte necrosis in virus-induced liver failure. J. Immunol. 2010, 184, 466–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wijaya, R.S.; Read, S.A.; Schibeci, S.; Eslam, M.; Azardaryany, M.K.; El-Khobar, K.; van der Poorten, D.; Lin, R.; Yuen, L.; Lam, V.; et al. KLRG1+ natural killer cells exert a novel antifibrotic function in chronic hepatitis B. J. Hepatol. 2019, 71, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Ge, D.; You, Z. Expression of interleukin-17RC protein in normal human tissues. Int. Arch. Med. 2008, 1, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemmers, A.; Moreno, C.; Gustot, T.; Maréchal, R.; Degré, D.; Demetter, P.; de Nadai, P.; Geerts, A.; Quertinmont, E.; Vercruysse, V.; et al. The interleukin-17 pathway is involved in human alcoholic liver disease. Hepatology 2009, 49, 646–657. [Google Scholar] [CrossRef]
- Paquissi, F.C. Immunity and Fibrogenesis: The Role of Th17/IL-17 Axis in HBV and HCV-induced Chronic Hepatitis and Progression to Cirrhosis. Front. Immunol. 2017, 8, 1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Chen, S.; Xu, K. IL-17 expression is correlated with hepatitis B-related liver diseases and fibrosis. Int. J. Mol. Med. 2011, 27, 385–392. [Google Scholar] [CrossRef] [Green Version]
- Lan, Y.T.; Wang, Z.L.; Tian, P.; Gong, X.N.; Fan, Y.C.; Wang, K. Treg/Th17 imbalance and its clinical significance in patients with hepatitis B-associated liver cirrhosis. Diagn Pathol. 2019, 14, 114. [Google Scholar] [CrossRef] [Green Version]
- Ye, Y.; Xie, X.; Yu, J.; Zhou, L.; Xie, H.; Jiang, G.; Yu, X.; Zhang, W.; Wu, J.; Zheng, S. Involvement of Th17 and Th1 effector responses in patients with Hepatitis B. J. Clin. Immunol. 2010, 30, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.L.; Zhang, T.; Zhao, Q.Y.; Lin, C.S.; Gao, Z.L. Th17 cells over 5.9% at admission indicate poor prognosis in patients with HBV-related acute-on-chronic liver failure. Medicine (Baltimore) 2018, 97, e12656. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Ma, Z.; Xin, G.; Yan, H.; Li, W.; Xu, H.; Hao, C.; Niu, J.; Zhao, P. Th1 and Th2 immune response in chronic hepatitis B patients during a long-term treatment with adefovir dipivoxil. Mediat. Inflamm. 2010, 2010, 143026. [Google Scholar] [CrossRef]
- Jacobson, N.G.; Szabo, S.J.; Weber-Nordt, R.M.; Zhong, Z.; Schreiber, R.D.; Darnell, J.E., Jr.; Murphy, K.M. Interleukin 12 signaling in T helper type 1 (Th1) cells involves tyrosine phosphorylation of signal transducer and activator of transcription (Stat)3 and Stat4. J. Exp. Med. 1995, 181, 1755–1762. [Google Scholar] [CrossRef]
- Athie-Morales, V.; Smits, H.H.; Cantrell, D.A.; Hilkens, C.M. Sustained IL-12 signaling is required for Th1 development. J. Immunol. 2004, 172, 61–69. [Google Scholar] [CrossRef]
- Kanhere, A.; Hertweck, A.; Bhatia, U.; Gokmen, M.R.; Perucha, E.; Jackson, I.; Lord, G.M.; Jenner, R.G. T-bet and GATA3 orchestrate Th1 and Th2 differentiation through lineage-specific targeting of distal regulatory elements. Nat. Commun. 2012, 3, 1268. [Google Scholar] [CrossRef] [Green Version]
- Lugo-Villarino, G.; Maldonado-Lopez, R.; Possemato, R.; Penaranda, C.; Glimcher, L.H. T-bet is required for optimal production of IFN-gamma and antigen-specific T cell activation by dendritic cells. Proc. Natl. Acad. Sci. UCA 2003, 100, 7749–7754. [Google Scholar] [CrossRef] [Green Version]
- Mosmann, T.R.; Sad, S. The expanding universe of T-cell subsets: Th1, Th2 and more. Immunol. Today 1996, 17, 138–146. [Google Scholar] [CrossRef]
- Jiang, R.; Feng, X.; Guo, Y.; Lu, Q.; Hou, J.; Luo, K.; Fu, N. T helper cells in patients with chronic hepatitis B virus infection. Chin. Med. J. (Engl.) 2002, 115, 422–424. [Google Scholar] [PubMed]
- Han, Y.P.; Li, J.; Jiang, L.F.; Xu, Q.Q.; Liu, B.; Dong, L.; Chen, N.; Kong, L.H.; Xie, F.R.; Huang, Z.H. [Hepatitis B e antigen from chronic hepatitis B patients induces Th1/Th2 cytokine imbalance in vitro]. Zhonghua Gan Zang Bing Za Zhi 2013, 21, 584–589. [Google Scholar] [CrossRef] [PubMed]
- Bertoletti, A.; Ferrari, C.; Fiaccadori, F.; Penna, A.; Margolskee, R.; Schlicht, H.J.; Fowler, P.; Guilhot, S.; Chisari, F.V. HLA class I-restricted human cytotoxic T cells recognize endogenously synthesized hepatitis B virus nucleocapsid antigen. Proc. Natl. Acad. Sci. UCA 1991, 88, 10445–10449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamoto, Y.; Kaneko, S.; Fan, H.; Momoi, T.; Tsutsui, H.; Nakanishi, K.; Kobayashi, K.; Suda, T. Prevention of hepatocellular carcinoma development associated with chronic hepatitis by anti-fas ligand antibody therapy. J. Exp. Med. 2002, 196, 1105–1111. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Q.; Xu, J.; Gu, X.; Wu, F.; Deng, J.; Cai, X.; Wang, G.; Li, G.; Chen, Z. Immune checkpoint targeting TIGIT in hepatocellular carcinoma. Am. J. Transl. Res. 2020, 12, 3212–3224. [Google Scholar]
- Xin, H.; Liang, D.; Zhang, M.; Ren, D.; Chen, H.; Zhang, H.; Li, S.; Ding, G.; Zhang, C.; Ding, Z.; et al. The CD68+ macrophages to CD8+ T-cell ratio is associated with clinical outcomes in hepatitis B virus (HBV)-related hepatocellular carcinoma. HPB Off. J. Int. Hepato Pancreato Biliary Assoc. 2020. [Google Scholar] [CrossRef]
- Pacella, I.; Cammarata, I.; Martire, C.; Brancaccio, G.; Gaeta, G.B.; Barnaba, V.; Piconese, S. CD8(+) T cells specific to apoptosis-associated epitopes are expanded in patients with chronic HBV infection and fibrosis. Liver Int. 2021, 41, 470–481. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Cytokines | Functions in CHB Patients | References |
|---|---|---|
| IL-6 | Mediate HBV entry into hepatocytes, induce inflammation and inflammation-driven HCC by the downregulation of miR-122, inhibits HBV replication, inhibits HBV entry through downregulation of HBV-specific receptor Na(+)/taurocholate cotransporting polypeptide (NTCP) | [97,98,99,100,101] |
| IL-8 | Cause resistance to IFN-α therapy, induce inflammation and apoptosis, induce fibrosis, recruits neutrophil | [102,103] |
| IL-9 | Induce inflammation, necrosis, and fibrosis | [104] |
| IL-10 | Inhibit cytokine production, regulate T cell immunity, develop immune tolerance, and persistence HBV infection | [29,96,105] |
| IL-12 | Reverse immune tolerance towards HBV, induce HBV-specific T cell response, reverse mitochondrial defects in HBV-specific CD8 T cells | [106,107] |
| IL-13 | Induce liver fibrosis and cirrhosis | [108] |
| IL-15 | Enhance CD8 T cell response, upregulate PD-1 and PD-L1 | [109] |
| IL-17 | Immune activation, inflammation, induce liver fibrosis | [110,111,112] |
| IL-18 | Increase risk of cirrhosis, enhance IFN-γ release, and improves clearance of virus infected cells | [113] |
| IL-21 | Activate T and B cells, induce IFN-γ secretion and clear HBV antigen, generation of plasmablasts and plasma cells, development of HBV induced liver cirrhosis, and exacerbates liver injury | [20,114] |
| IL-22 | Inhibit liver inflammation and fibrosis, induce fibrosis and HCC | [115,116] |
| IL-23 | Induce inflammation and HCC development | [117] |
| IL-27 | Support plasmablasts and plasma cell generation, enhance HBsAg-specific antibody production, inhibits HBV protein expression and viral capsid associated DNA replication | [20,118,119] |
| IL-33 | Induce liver damage and fibrosis, Activate TFH cells and enhance humoral immunity, suppress HBV replication and HBeAg secretion | [120,121] |
| IL-35 | Development of cirrhosis and HCC, inhibit HBV-specific CD8 T cells proliferation and cytotoxicity, inhibit cytokine induce antiviral immunity | [122,123] |
| IFN-γ | Antiviral immunity, promote CXCL-9 secretion by macrophages, inhibit HBV replication, induce inflammation | [124,125,126] |
| TNF-α | Inhibit HBV replication, provide antiviral immunity, induce inflammation | [127,128] |
| TGF-β | Impair NK cell function, encourage fibrosis and HCC | [129,130] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khanam, A.; Chua, J.V.; Kottilil, S. Immunopathology of Chronic Hepatitis B Infection: Role of Innate and Adaptive Immune Response in Disease Progression. Int. J. Mol. Sci. 2021, 22, 5497. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22115497
Khanam A, Chua JV, Kottilil S. Immunopathology of Chronic Hepatitis B Infection: Role of Innate and Adaptive Immune Response in Disease Progression. International Journal of Molecular Sciences. 2021; 22(11):5497. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22115497
Chicago/Turabian StyleKhanam, Arshi, Joel V. Chua, and Shyam Kottilil. 2021. "Immunopathology of Chronic Hepatitis B Infection: Role of Innate and Adaptive Immune Response in Disease Progression" International Journal of Molecular Sciences 22, no. 11: 5497. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22115497