
Urolithin and Reduced Urolithin Derivatives as Potent Inhibitors of Tyrosinase and Melanogenesis: Importance of the 4-Substituted Resorcinol Moiety

 , , ,
, , ,
Abstract
:
1. Introduction
2. Results and Discussion
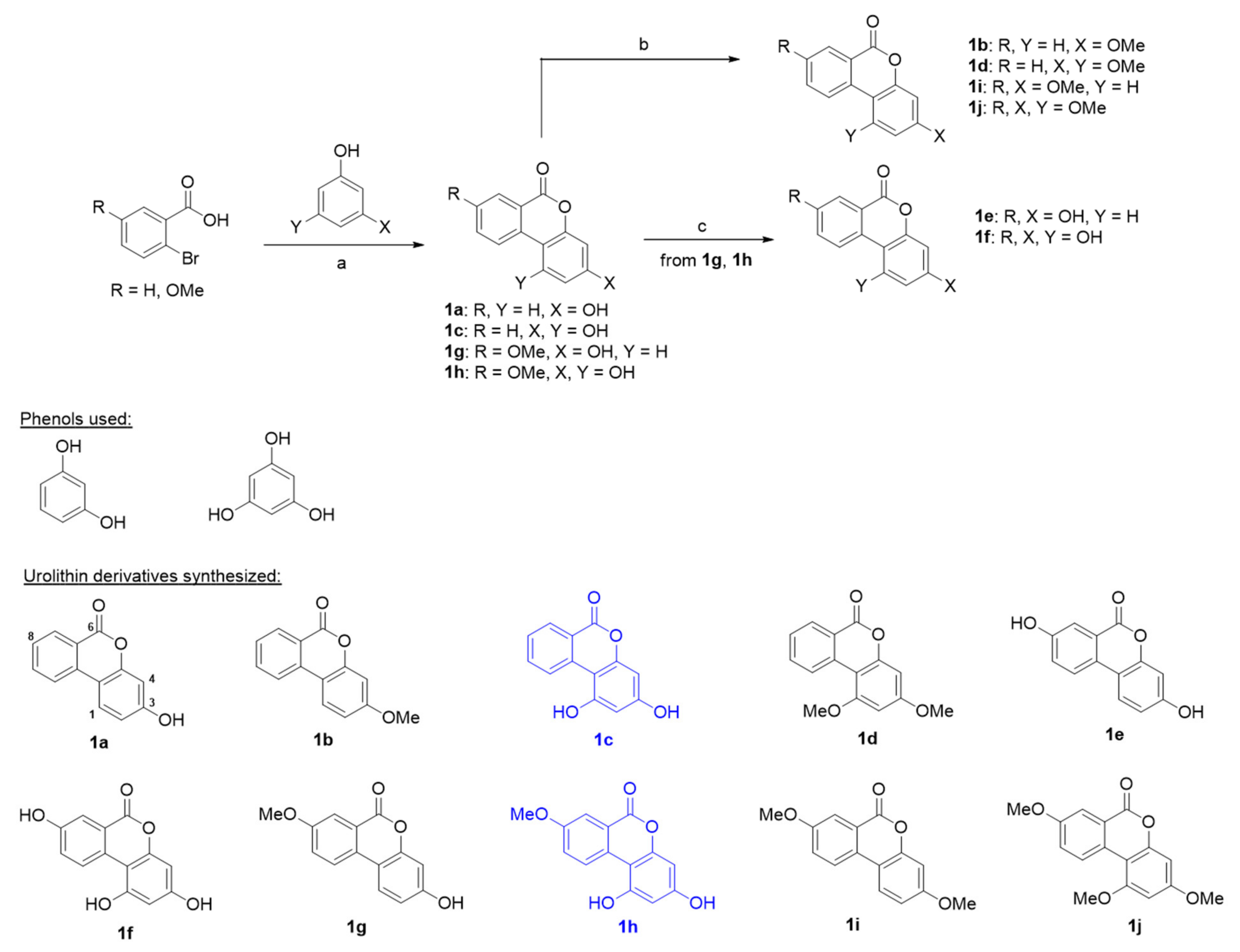
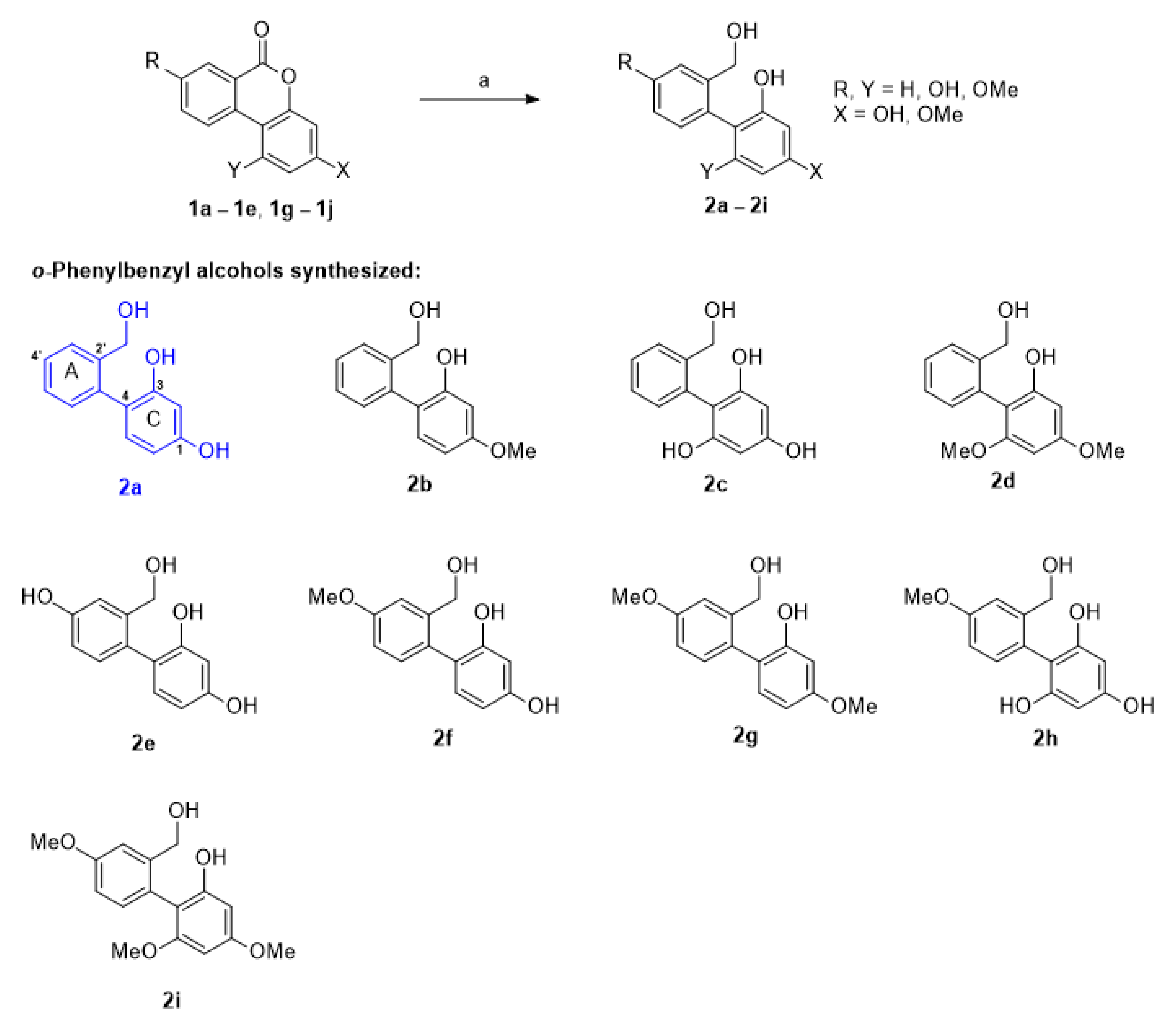
2.1. Chemistry
2.2. Mushroom Tyrosinase-Inhibitory Activities of Urolithin Derivatives
2.3. Mushroom Tyrosinase-Inhibitory Activities of Reduced Urolithin Derivatives
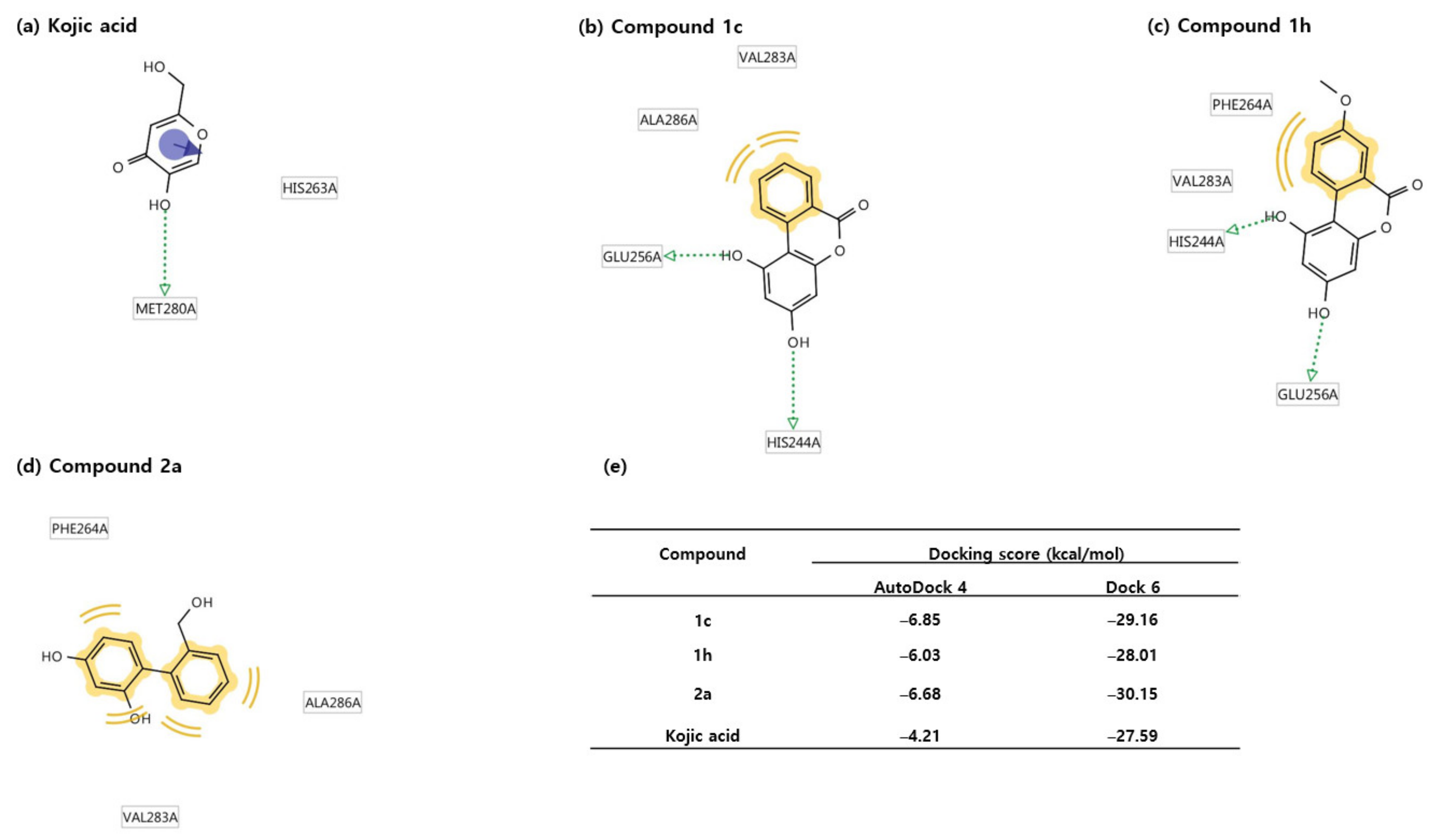
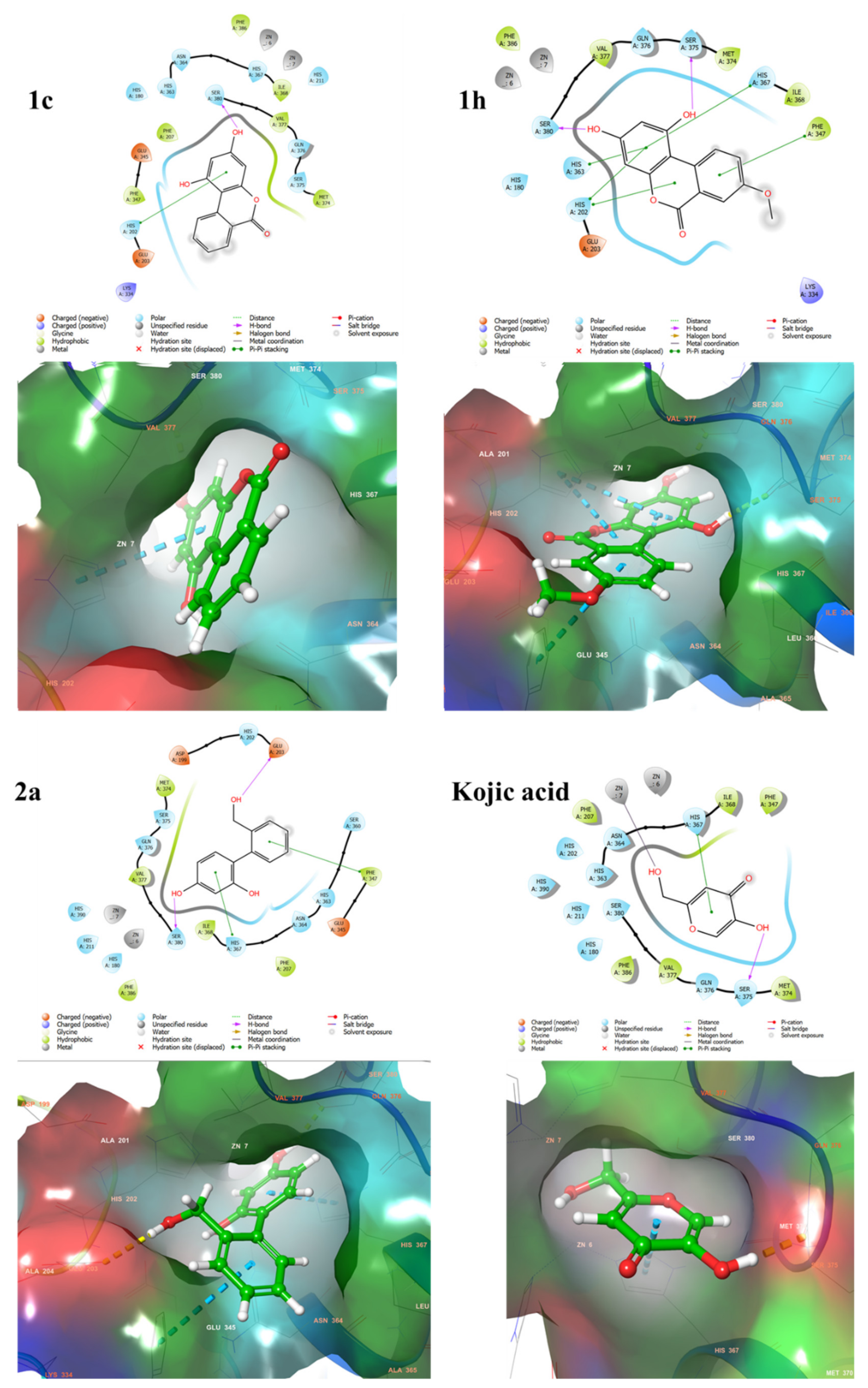
2.4. Docking Studies Using Mushroom Tyrosinase Crystal Structure
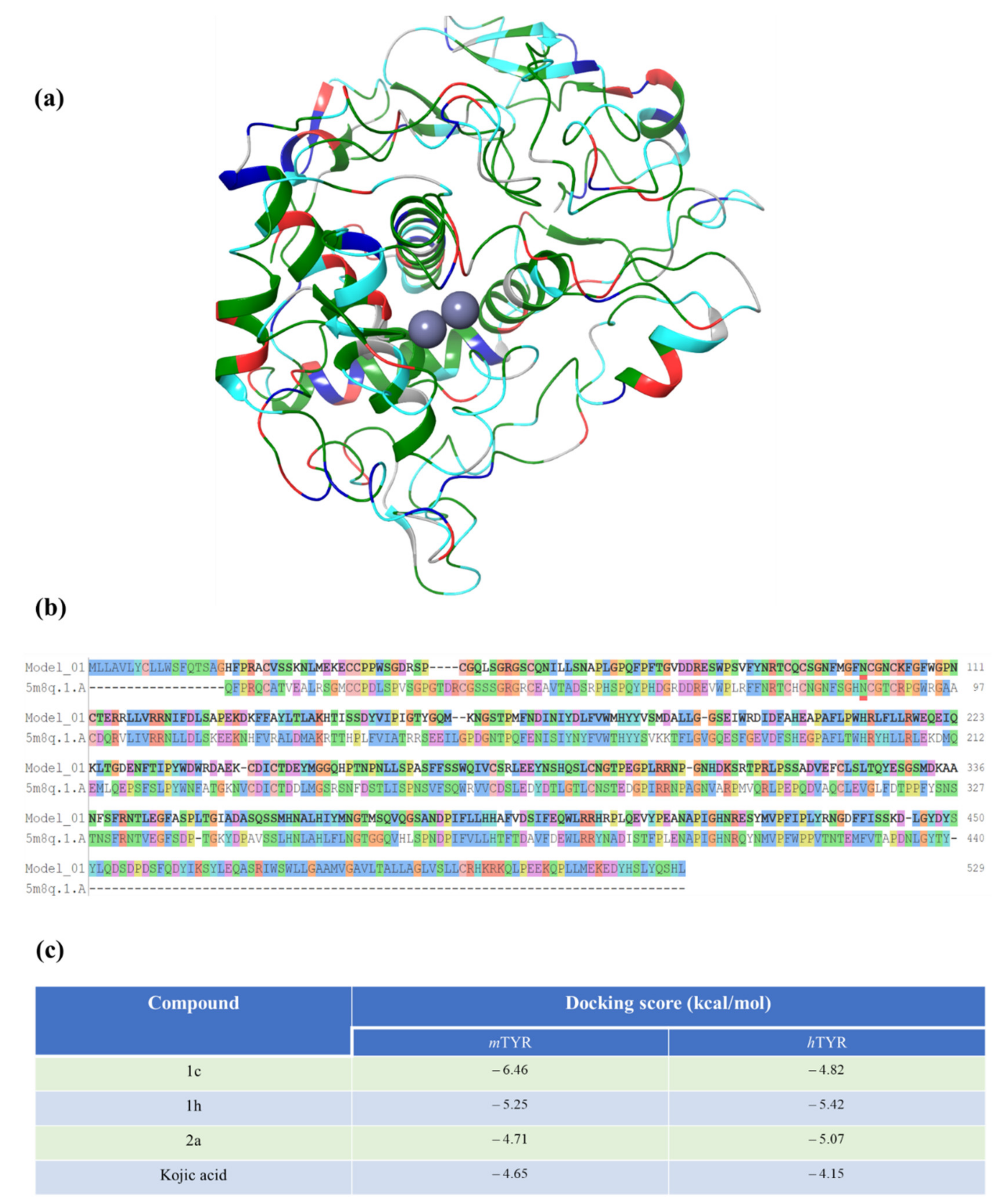
2.5. Docking Studies Using Human Tyrosinase Homology Model
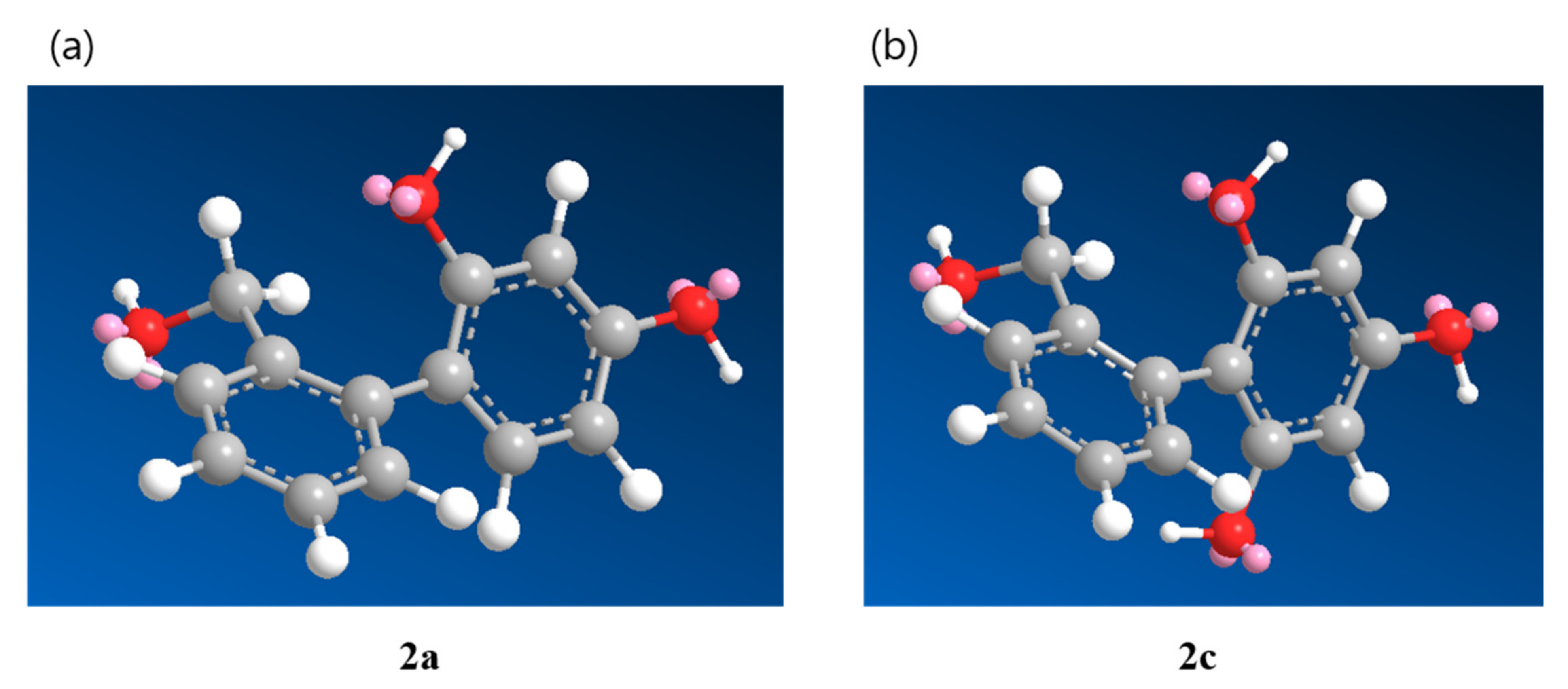
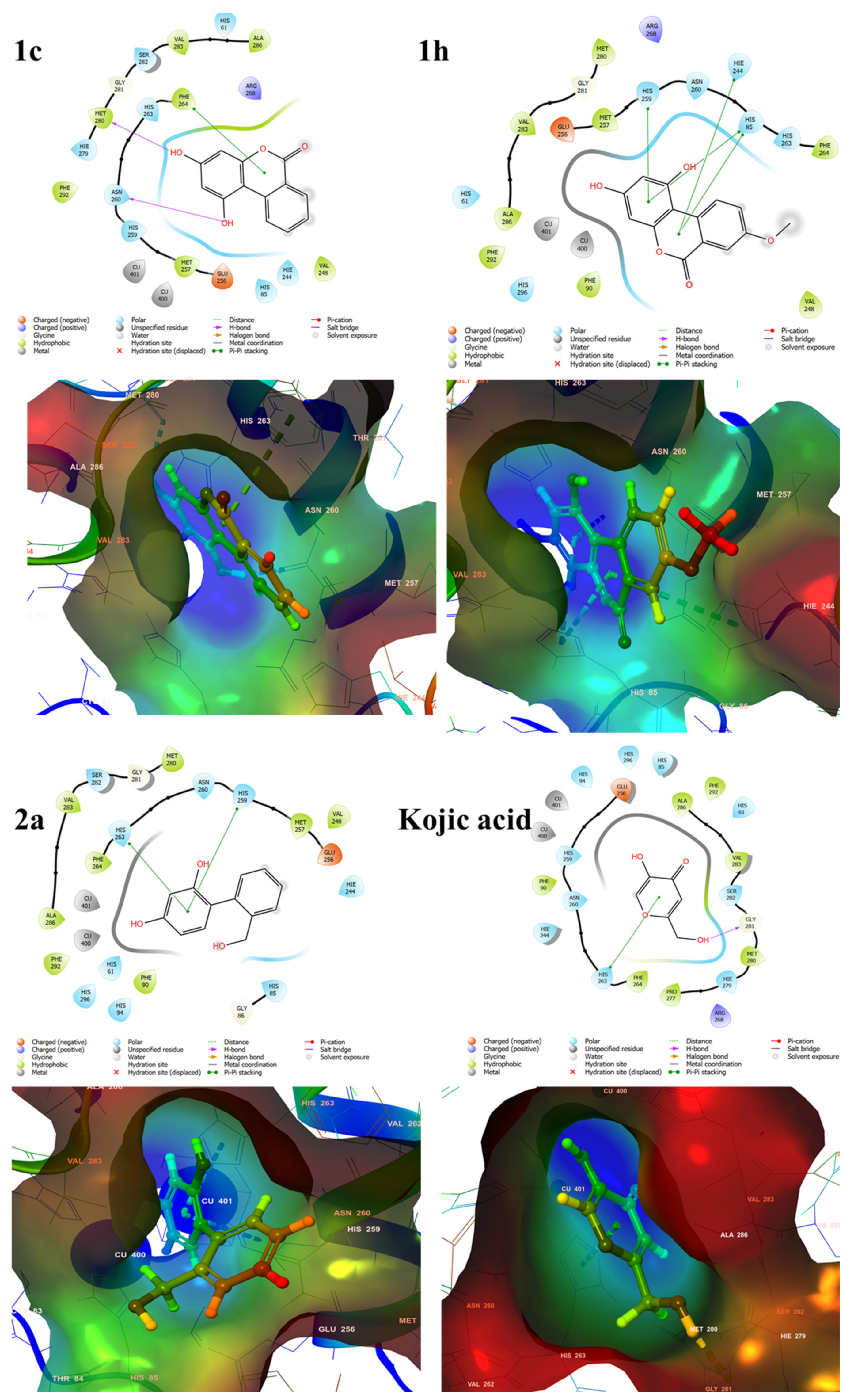
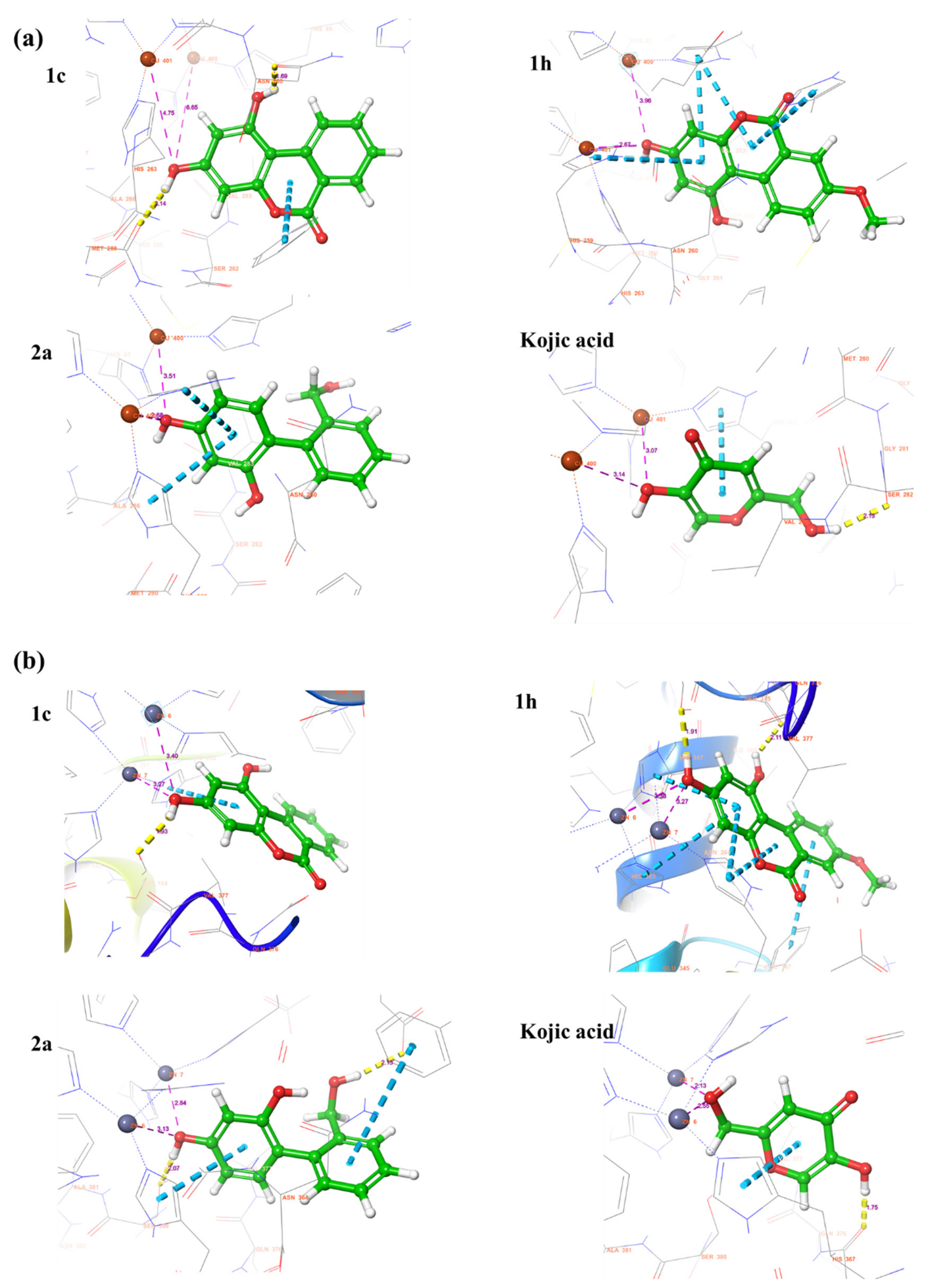
2.6. Docking Score and Binding Mode of Compounds 1c, 1h, 2a and Kojic Acid at the Active Site of Human Tyrosinase Homology Model
2.7. Binding Analysis of 1c, 1h, 2a and Kojic acid in a Human Tyrosinase Homology model and Mushroom Tyrosinase Enzyme
2.8. IC50 Evaluation of the Urolithin and Reduced Urolithin Derivatives
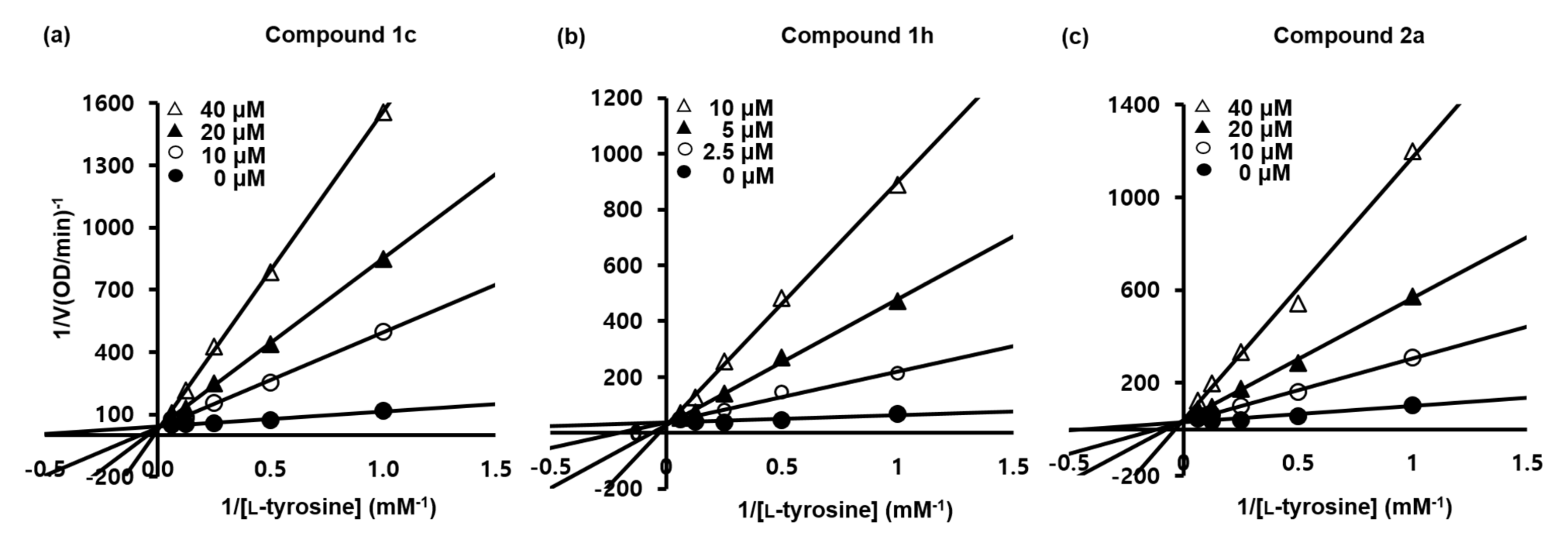
2.9. Kinetic Analyses of the Urolithin and Reduced Urolithin Derivatives
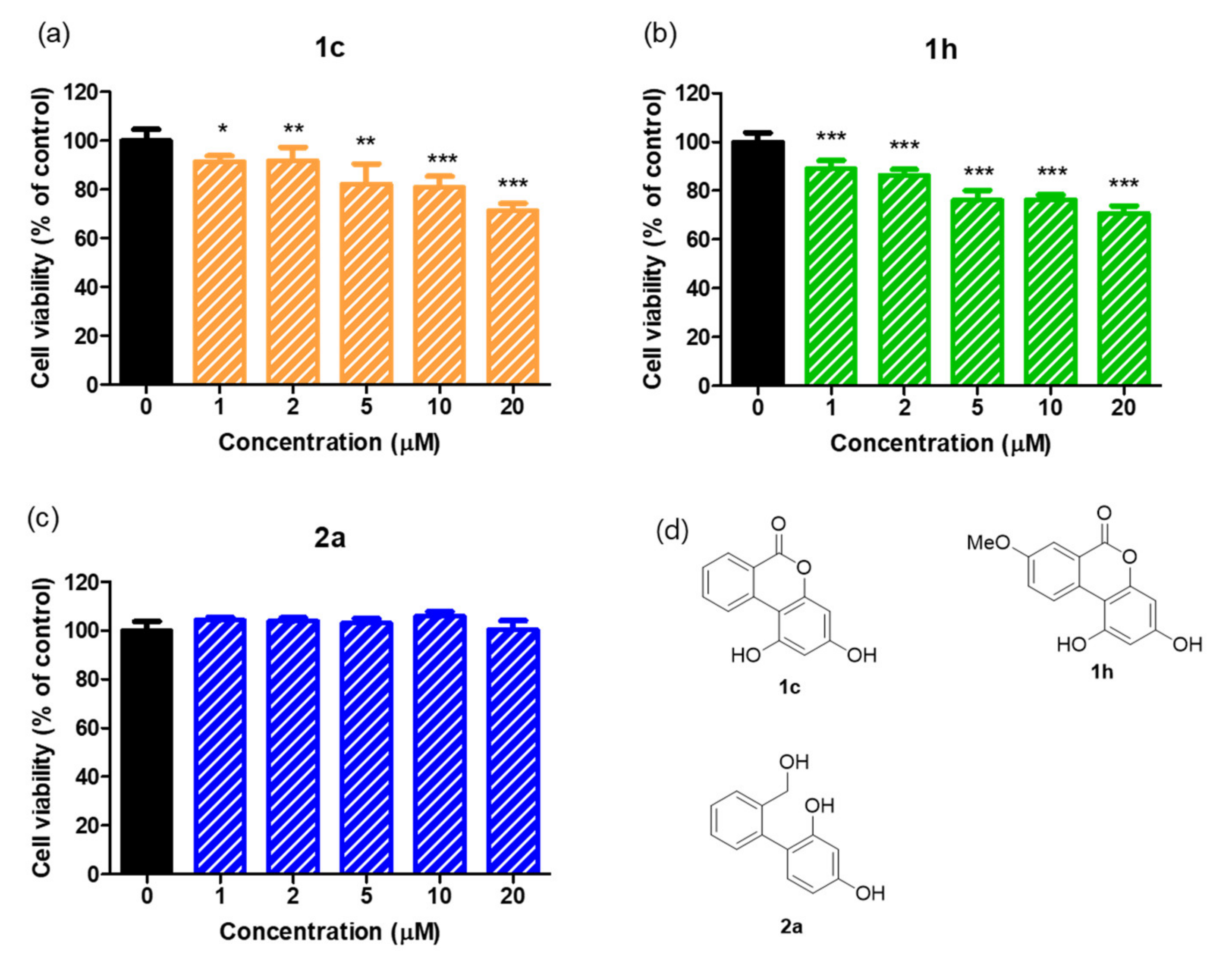
2.10. Cell Viabilities of 1c and 1h and of 2a in B16F10 Melanoma Cells
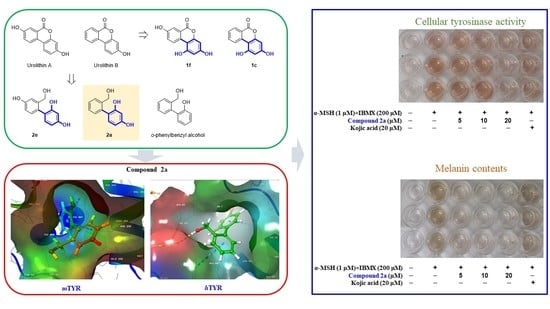
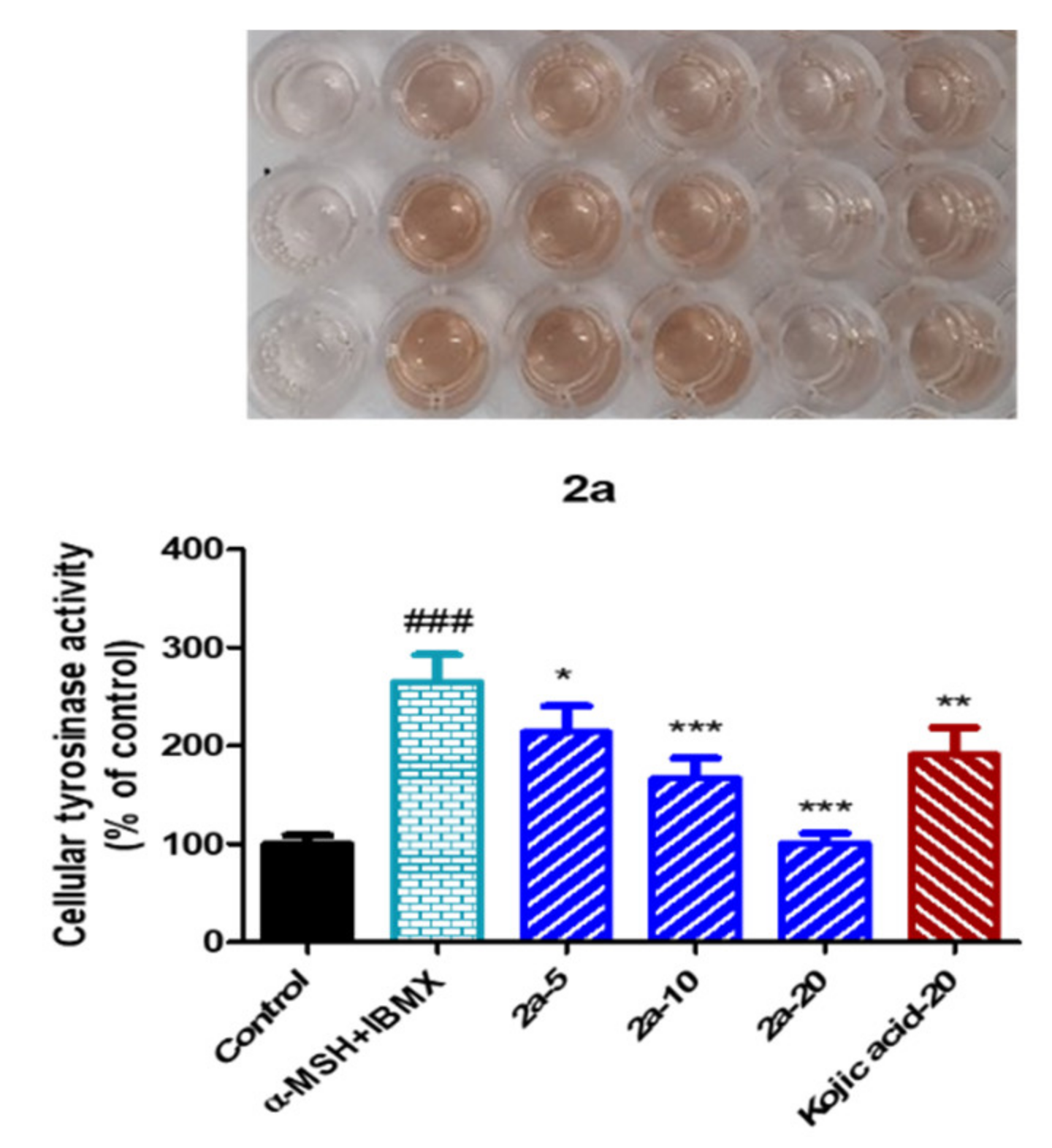
2.11. Tyrosinase Inhibitory Activity of the Reduced Urolithin Analog 2a in α-MSH- Plus IBMX-Stimulated B16F10 Melanoma Cells
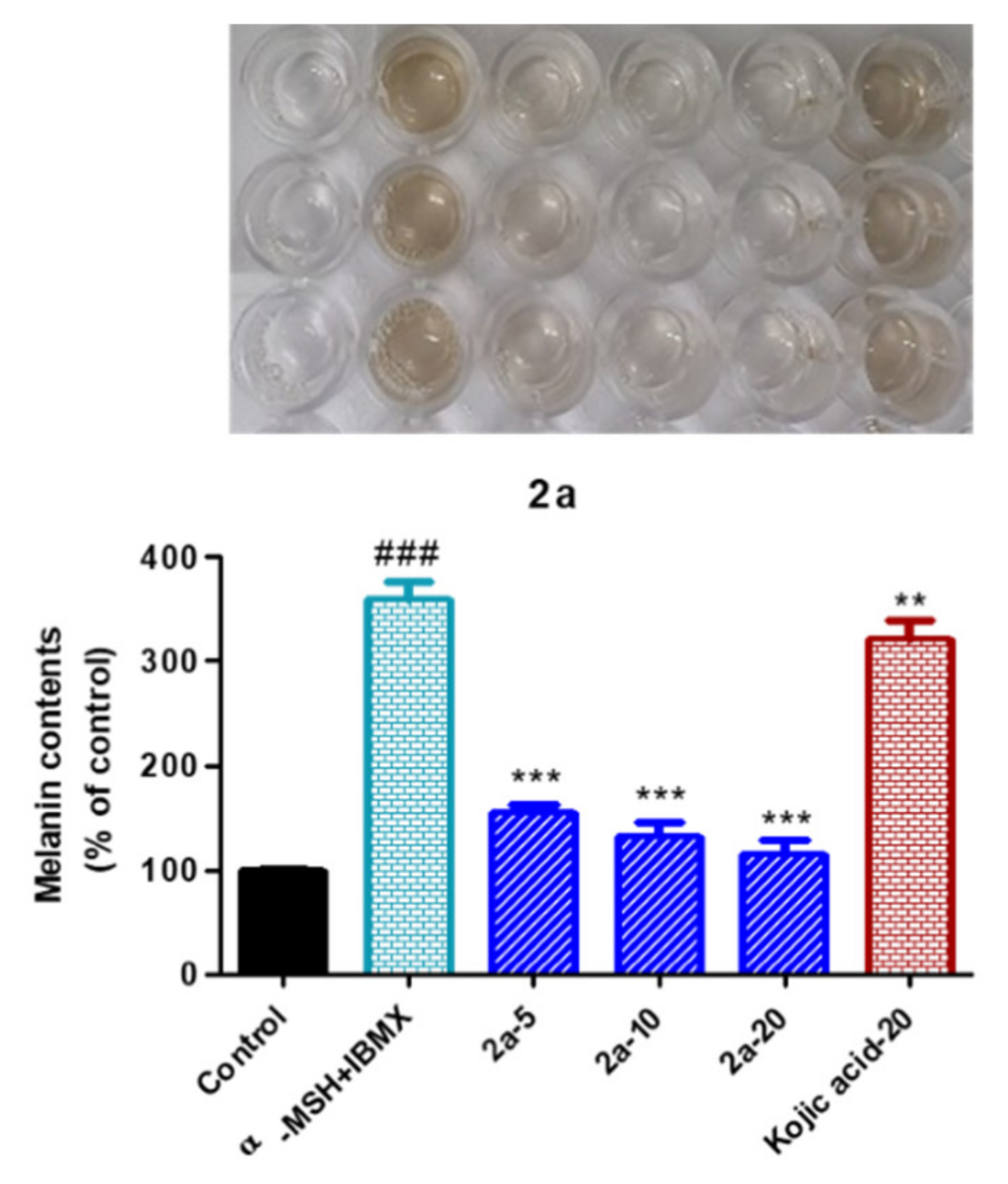
2.12. Inhibition of Melanin Production by the Reduced Urolithin Analog 2a in α-MSH Plus IBMX Stimulated B16F10 Melanoma Cells
3. Conclusions
4. Materials and Methods
4.1. General Methods
4.1.1. General Procedure Used for Synthesizing Urolithin Derivatives 1a, 1c, and 1g–1h
4.1.2. General Procedure Used for Synthesizing Urolithin Derivatives 1b, 1d, 1i, and 1j
4.1.3. General Synthetic Procedure for Urolithin Derivatives 1e and 1f
4.1.4. General Synthetic Procedure for o-phenylbenzyl Alcohol Derivatives 2a–2i
4.2. Biological Studies
4.2.1. Mushroom Tyrosinase Assay
4.2.2. Kinetic Analysis Studies: Lineweaver–Burk Plots
4.2.3. Docking and Molecular Modeling Studies of 1c, 1h, 2a, and Kojic acid in Mushroom Tyrosinase
4.2.4. Docking and Molecular Studies of 1c, 1h, 2a, and Kojic Acid Using Human Tyrosinase Homology Model
4.2.5. B16F10 Melanoma Cell Culture
4.2.6. Cell Cytotoxicity Studies: EZ-Cytox Assay
4.2.7. Cellular Tyrosinase Inhibition in B16F10 Melanoma Cells
4.2.8. Melanin Content Assays in B16F10 Melanoma Cells
4.2.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Akira, H.; Mamiko, K.; Tsuypshi, O. Tyrosinase inhibitors. U.S. Patent 2014112878A1, 24 April 2014. [Google Scholar]
- Kidson, S.; de Haan, J. Effect of thymidine analogs on tyrosinase activity and mRNA accumulation in mouse melanoma cells. Exp. Cell Res. 1990, 188, 36–41. [Google Scholar] [CrossRef]
- Imokawa, G. Analysis of initial melanogenesis including tyrosinase transfer and melanosome differentiation through interrupted melanization by glutathione. J. Investig. Dermatol. 1989, 93, 100–107. [Google Scholar] [CrossRef] [Green Version]
- Petrescu, S.M.; Branza-Nichita, N.; Negroiu, G.; Petrescu, A.J.; Dwek, R.A. Tyrosinase and glycoprotein folding: Roles of chaperones that recognize glycans. Biochemistry 2000, 39, 5229–5237. [Google Scholar] [CrossRef]
- Ando, H.; Kondoh, H.; Ichihashi, M.; Hearing, V.J. Approaches to identify inhibitors of melanin biosynthesis via the quality control of tyrosinase. J. Investig. Dermatol. 2007, 127, 751–761. [Google Scholar] [CrossRef] [Green Version]
- Saeki, H.; Oikawa, A. Synthesis and degradation of tyrosinase in cultured melanoma cells. J. Cell. Physiol. 1980, 104, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.S.; Vani, M.G.; Wang, S.Y.; Liao, J.W.; Hsu, L.S.; Yang, H.L.; Hseu, Y.C. In vitro and in vivo studies disclosed the depigmenting effects of gallic acid: A novel skin lightening agent for hyperpigmentary skin diseases. Biofactors 2013, 39, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Arndt, K.A.; Fitzpatrick, T.B. Topical use of hydroquinone as a depigmenting agent. JAMA 1965, 194, 965–967. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, T.; Arndt, K.; el Mofty, A.; Pathak, M. Hydroquinone and psoralens in the therapy of hypermelanosis and vitiligo. Arch. Dermatol. 1966, 93, 589–600. [Google Scholar] [CrossRef]
- Heilgemeir, G.; Balda, B. Irreversible toxic depigmentation. Observations following use of hydroquinonemonobenzylether-containing skin bleaching preparations. MMW Munch. Med. Wochenschr. 1981, 123, 47. [Google Scholar]
- Kligman, A.M.; Willis, I. A new formula for depigmenting human skin. Arch. Dermatol. 1975, 111, 40–48. [Google Scholar] [CrossRef]
- Gonçalez, M.; Correa, M.A.; Chorilli, M. Skin delivery of kojic acid-loaded nanotechnology-based drug delivery systems for the treatment of skin aging. BioMed Res. Int. 2013, 2013. [Google Scholar] [CrossRef]
- Breathnach, A.C.; Nazzaro-Porro, M.; Passi, S.; Zina, G. Azelaic acid therapy in disorders of pigmentation. Clin. Dermatol. 1989, 7, 106–119. [Google Scholar] [CrossRef]
- Garcia-Lopez, M. Double-blind comparison of azelaic acid and hydroquinone in the treatment of melisma. Acta Derm. Venereol. 1989, 143, 58–61. [Google Scholar]
- Khemis, A.; Kaiafa, A.; Queille-Roussel, C.; Duteil, L.; Ortonne, J. Evaluation of efficacy and safety of rucinol serum in patients with melasma: A randomized controlled trial. Br. J. Dermatol. 2007, 156, 997–1004. [Google Scholar] [CrossRef] [PubMed]
- Köpke, D.; Mueller, R.H.; Pyo, S.M. Phenylethyl resorcinol smartLipids for skin brightening–Increased loading & chemical stability. Eur. J. Pharm. Sci. 2019, 137, 104992. [Google Scholar] [PubMed]
- Mann, T.; Scherner, C.; Röhm, K.-H.; Kolbe, L. Structure-activity relationships of thiazolyl resorcinols, potent and selective inhibitors of human tyrosinase. Int. J. Mol. Sci. 2018, 19, 690. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.-J.; Uyama, H. Tyrosinase inhibitors from natural and synthetic sources: Structure, inhibition mechanism and perspective for the future. Cell. Mol. Life Sci. CMLS 2005, 62, 1707–1723. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.-S. An updated review of tyrosinase inhibitors. Int. J. Mol. Sci. 2009, 10, 2440–2475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabanes, J.; Chazarra, S.; Garcia-Carmona, F. Kojic acid, a cosmetic skin whitening agent, is a slow-binding inhibitor of catecholase activity of tyrosinase. J. Pharm. Pharmacol. 1994, 46, 982–985. [Google Scholar] [CrossRef]
- Shin, N.-H.; Ryu, S.Y.; Choi, E.J.; Kang, S.-H.; Chang, I.-M.; Min, K.R.; Kim, Y. Oxyresveratrol as the potent inhibitor on dopa oxidase activity of mushroom tyrosinase. Biochem. Biophys. Res. Commun. 1998, 243, 801–803. [Google Scholar] [CrossRef] [PubMed]
- Khatib, S.; Nerya, O.; Musa, R.; Shmuel, M.; Tamir, S.; Vaya, J. Chalcones as potent tyrosinase inhibitors: The importance of a 2, 4-substituted resorcinol moiety. Bioorg. Med. Chem. 2005, 13, 433–441. [Google Scholar] [CrossRef]
- Jung, H.J.; Lee, M.J.; Park, Y.J.; Noh, S.G.; Lee, A.K.; Moon, K.M.; Lee, E.K.; Bang, E.J.; Park, Y.J.; Kim, S.J. A novel synthetic compound, (Z)-5-(3-hydroxy-4-methoxybenzylidene)-2-iminothiazolidin-4-one (MHY773) inhibits mushroom tyrosinase. Biosci. Biotechnol. Biochem. 2018, 82, 759–767. [Google Scholar] [CrossRef]
- Kim, S.J.; Yang, J.; Lee, S.; Park, C.; Kang, D.; Akter, J.; Ullah, S.; Kim, Y.-J.; Chun, P.; Moon, H.R. The tyrosinase inhibitory effects of isoxazolone derivatives with a (Z)-β-phenyl-α, β-unsaturated carbonyl scaffold. Bioorg. Med. Chem. 2018, 26, 3882–3889. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.K.D.H.; Ullah, S.; Yun, H.Y.; Chun, P.; Moon, H.R. Design, synthesis, and antimelanogenic effects of (2-substituted phenyl-1, 3-dithiolan-4-yl) methanol derivatives. Drug Des. Dev. Ther. 2017, 11, 827. [Google Scholar] [CrossRef] [Green Version]
- Yun, H.Y.; Kim, S.S.D.H.; Ullah, S.; Kim, S.J.; Kim, Y.-J.; Yoo, J.-W.; Jung, Y.; Chun, P.; Moon, H.R. Design, synthesis, and anti-melanogenic effects of (E)-2-benzoyl-3-(substituted phenyl) acrylonitriles. Drug Des. Dev. Ther. 2015, 9, 4259. [Google Scholar]
- Pillaiyar, T.; Manickam, M.; Namasivayam, V. Skin whitening agents: Medicinal chemistry perspective of tyrosinase inhibitors. J. Enzym. Inhib. Med. Chem. 2017, 32, 403–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pillaiyar, T.; Manickam, M.; Jung, S.H. Recent development of signaling pathways inhibitors of melanogenesis. Cell Signal 2017, 40, 99–115. [Google Scholar] [CrossRef] [PubMed]
- Pillaiyar, T.; Namasivayam, V.; Manickam, M.; Jung, S.H. Inhibitors of Melanogenesis: An Updated Review. J. Med. Chem. 2018, 61, 7395–7418. [Google Scholar] [CrossRef]
- Pillaiyar, T.; Manickam, M.; Jung, S.H. Downregulation of melanogenesis: Drug discovery and therapeutic options. Drug Discov. Today 2017, 22, 282–298. [Google Scholar] [CrossRef]
- Halaouli, S.; Asther, M.; Sigoillot, J.C.; Hamdi, M.; Lomascolo, A. Fungal tyrosinases: New prospects in molecular characteristics, bioengineering and biotechnological applications. J. Appl. Microbiol. 2006, 100, 219–232. [Google Scholar] [CrossRef]
- Boatman, R.J. Differences in the nephrotoxicity of hydroquinone among Fischer 344 and Sprague-Dawley rats and B6C3F1 mice. J Toxicol Environ Health. 1996, 9, 159–172. [Google Scholar] [CrossRef]
- Barneda-Zahonero, B.; Parra, M. Histone deacetylases and cancer. Mol. Oncol. 2012, 6, 579–589. [Google Scholar] [CrossRef] [Green Version]
- Afshin, A.; Forouzanfar, M.H.; Reitsma, M.B.; Sur, P.; Estep, K.; Lee, A.; Marczak, L.; Mokdad, A.H.; Moradi-Lakeh, M.; Naghavi, M.; et al. Health Effects of Overweight and Obesity in 195 Countries over 25 Years. N. Engl. J. Med. 2017, 377, 13–27. [Google Scholar] [PubMed]
- Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. Executive Summary of The Third Report of The National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, And Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III). JAMA 2001, 285, 2486–2497. [Google Scholar] [CrossRef] [PubMed]
- Bandgar, B.P.; Adsul, L.K.; Chavan, H.V.; Shringare, S.N.; Korbad, B.L.; Jalde, S.S.; Lonikar, S.V.; Nile, S.H.; Shirfule, A.L. Synthesis, biological evaluation, and molecular docking of N-{3-[3-(9-methyl-9H-carbazol-3-yl)-acryloyl]-phenyl}-benzamide/amide derivatives as xanthine oxidase and tyrosinase inhibitors. Bioorg. Med. Chem. 2012, 20, 5649–5657. [Google Scholar] [CrossRef]
- Cerda, B.; Tomas-Barberan, F.A.; Espin, J.C. Metabolism of antioxidant and chemopreventive ellagitannins from strawberries, raspberries, walnuts, and oak-aged wine in humans: Identification of biomarkers and individual variability. J. Agric. Food Chem. 2005, 53, 227–235. [Google Scholar] [CrossRef]
- Espin, J.C.; Larrosa, M.; García-Conesa, M.T.; Tomás-Barberán, F. Biological Significance of Urolithins, the Gut Microbial Ellagic Acid-Derived Metabolites: The Evidence So Far. Evid. Based Complement. Altern. Med. 2013, 2013, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, G.-H.; Chen, W.-Q.; Shen, Z.-Y. Urolithin A shows anti-atherosclerotic activity via activation of class B scavenger receptor and activation of Nef2 signaling pathway. Pharmacol. Rep. 2018, 70, 519–524. [Google Scholar] [CrossRef]
- Velagapudi, R.; Lepiarz, I.; El-Bakoush, A.; Katola, F.O.; Bhatia, H.; Fiebich, B.L.; Olajide, O.A. Induction of Autophagy and Activation of SIRT-1 Deacetylation Mechanisms Mediate Neuroprotection by the Pomegranate Metabolite Urolithin A in BV2 Microglia and Differentiated 3D Human Neural Progenitor Cells. Mol. Nutr. Food Res. 2019, 63, 1801237. [Google Scholar] [CrossRef] [Green Version]
- Saha, P.; Yeoh, B.S.; Singh, R.; Chandrasekar, B.; Vemula, P.K.; Haribabu, B.; Vijay-Kumar, M.; Jala, V.R. Gut microbiota conversion of dietary ellagic acid into bioactive phytoceutical Urolithin a inhibits heme peroxidases. PLoS ONE 2016, 11, e0156811. [Google Scholar] [CrossRef] [Green Version]
- Dell’Agli, M.; Galli, G.V.; Bulgari, M.; Basilico, N.; Romeo, S.; Bhattacharya, D.; Taramelli, D.; Bosisio, E. Ellagitannins of the fruit rind of pomegranate (Punica granatum) antagonize in vitro the host inflammatory response mechanisms involved in the onset of malaria. Malar. J. 2010, 9, 208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, G.; Park, J.-S.; Lee, E.-J.; Ahn, J.-H.; Kim, H.-S. Anti-inflammatory and antioxidant mechanisms of urolithin B in activated microglia. Phytomedicine 2019, 55, 50–57. [Google Scholar] [CrossRef]
- Bialonska, D.; Kasimsetty, S.G.; Khan, S.I.; Ferreira, D. Urolithins, Intestinal Microbial Metabolites of Pomegranate Ellagitannins, Exhibit Potent Antioxidant Activity in a Cell-Based Assay. J. Agric. Food Chem. 2009, 57, 10181–10186. [Google Scholar] [CrossRef] [PubMed]
- Larrosa, M.; González-Sarrías, A.; García-Conesa, M.T.; Tomás-Barberán, F.A.; Espín, J.C. Urolithins, ellagic acid-derived metabolites produced by human colonic microflora, exhibit estrogenic and antiestrogenic activities. J. Agric. Food Chem. 2006, 54, 1611–1620. [Google Scholar] [CrossRef]
- Seeram, N.P.; Heber, D. Therapeutic uses of urolithins. EP2068864A2, 8 November 2007. [Google Scholar]
- Yin, P.; Zhang, J.; Yan, L.; Yang, L.; Sun, L.; Shi, L.; Ma, C.; Liu, Y. Urolithin C, a gut metabolite of ellagic acid, induces apoptosis in PC12 cells through a mitochondria-mediated pathway. RSC Adv. 2017, 7, 17254–17263. [Google Scholar] [CrossRef] [Green Version]
- Gulcan, H.O.; Unlu, S.; Esiringu, I.; Ercetin, T.; Sahin, Y.; Oz, D.; Sahin, M.F. Design, synthesis and biological evaluation of novel 6H-benzo[c]chromen-6-one, and 7,8,9,10-tetrahydro-benzo[c]chromen-6-one derivatives as potential cholinesterase inhibitors. Bioorg. Med. Chem. 2014, 22, 5141–5154. [Google Scholar] [CrossRef]
- Norouzbahari, M.; Burgaz, E.V.; Ercetin, T.; Fallah, A.; Foroumadi, A.; Firoozpour, L.; Sahin, M.F.; Gazi, M.; Gulcan, H.O. Design, Synthesis and Characterization of Novel Urolithin Derivatives as Cholinesterase Inhibitor Agents. Lett. Drug Des. Discov. 2018, 15, 1131–1140. [Google Scholar] [CrossRef]
- Yuan, T.; Ma, H.; Liu, W.; Niesen, D.B.; Shah, N.; Crews, R.; Rose, K.N.; Vattem, D.A.; Seeram, N.P. Pomegranate’s Neuroprotective Effects against Alzheimer’s Disease Are Mediated by Urolithins, Its Ellagitannin-Gut Microbial Derived Metabolites. ACS Chem. Neurosci. 2016, 7, 26–33. [Google Scholar] [CrossRef] [PubMed]
- De Molina, A.R.; Vargas, T.; Molina, S.; Sánchez, J.; Martínez-Romero, J.; González-Vallinas, M.; Martín-Hernández, R.; Sánchez-Martínez, R.; de Cedrón, M.G.; Dávalos, A.; et al. The Ellagic Acid Derivative 4,4′-Di-O-Methylellagic Acid Efficiently Inhibits Colon Cancer Cell Growth through a Mechanism Involving WNT16. J. Pharmacol. Exp. Ther. 2015, 353, 433–444. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.; Jung, H.; Lee, H.; Yi, H.C.; Kwak, H.-K.; Hwang, K.T. Chemopreventive activity of ellagitannins and their derivatives from black raspberry seeds on HT-29 colon cancer cells. Food Funct. 2015, 6, 1675–1683. [Google Scholar] [CrossRef]
- Adams, L.S.; Zhang, Y.; Seeram, N.P.; Heber, D.; Chen, S. Pomegranate Ellagitannin–Derived Compounds Exhibit Antiproliferative and Antiaromatase Activity in Breast Cancer Cells in Vitro. Cancer Prev. Res. 2010, 3, 108–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seeram, N.P.; Aronson, W.J.; Zhang, Y.; Henning, S.M.; Moro, A.; Lee, R.-P.; Sartippour, M.; Harris, D.M.; Rettig, M.; Suchard, M.A.; et al. Pomegranate Ellagitannin-Derived Metabolites Inhibit Prostate Cancer Growth and Localize to the Mouse Prostate Gland. J. Agric. Food Chem. 2007, 55, 7732–7737. [Google Scholar] [CrossRef] [PubMed]
- Furlanetto, V.; Zagotto, G.; Pasquale, R.; Moro, S.; Gatto, B. Ellagic Acid and Polyhydroxylated Urolithins Are Potent Catalytic Inhibitors of Human Topoisomerase II: An In Vitro Study. J. Agric. Food Chem. 2012, 60, 9162–9170. [Google Scholar] [CrossRef] [PubMed]
- Roulier, B.; Pérès, B.; Haudecoeur, R. Advances in the Design of Genuine Human Tyrosinase Inhibitors for Targeting Melanogenesis and Related Pigmentations. J. Med. Chem. 2020, 63, 13428–13443. [Google Scholar] [CrossRef] [PubMed]
- Lai, X.; Wichers, H.J.; Soler-Lopez, M.; Dijkstra, B.W. Structure and Function of Human Tyrosinase and Tyrosinase-Related Proteins. Chem. A Eur. J. 2018, 24, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Mann, T.; Gerwat, W.; Batzer, J.; Eggers, K.; Scherner, C.; Wenck, H.; Stäb, F.; Hearing, V.J.; Röhm, K.-H.; Kolbe, L. Inhibition of human tyrosinase requires molecular motifs distinctively different from mushroom tyrosinase. J. Investig. Dermatol. 2018, 138, 1601–1608. [Google Scholar] [CrossRef] [Green Version]
- Kang, K.H.; Lee, B.; Son, S.; Yun, H.Y.; Moon, K.M.; Jeong, H.O.; Kim, D.H.; Lee, E.K.; Choi, Y.J.; Do, H.K. (Z)-2-(Benzo [d] thiazol-2-ylamino)-5-(substituted benzylidene) thiazol-4 (5H)-one Derivatives as Novel Tyrosinase Inhibitors. Biol. Pharm. Bull. 2015, 38, 1227–1233. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.R.; Lee, H.J.; Choi, Y.J.; Park, Y.J.; Woo, Y.; Kim, S.J.; Park, M.H.; Lee, H.W.; Chun, P.; Chung, H.Y. Benzylidene-linked thiohydantoin derivatives as inhibitors of tyrosinase and melanogenesis: Importance of the β-phenyl-α, β-unsaturated carbonyl functionality. Med. Chem. Comm. 2014, 5, 1410–1417. [Google Scholar] [CrossRef]
- Kim, S.H.; Ha, Y.M.; Moon, K.M.; Choi, Y.J.; Park, Y.J.; Jeong, H.O.; Chung, K.W.; Lee, H.J.; Chun, P.; Moon, H.R. Anti-melanogenic effect of (Z)-5-(2, 4-dihydroxybenzylidene) thiazolidine-2, 4-dione, a novel tyrosinase inhibitor. Arch. Pharmacal Res. 2013, 36, 1189–1197. [Google Scholar] [CrossRef]
- Chung, K.W.; Park, Y.J.; Choi, Y.J.; Park, M.H.; Ha, Y.M.; Uehara, Y.; Yoon, J.H.; Chun, P.; Moon, H.R.; Chung, H.Y. Evaluation of in vitro and in vivo anti-melanogenic activity of a newly synthesized strong tyrosinase inhibitor (E)-3-(2, 4 dihydroxybenzylidene) pyrrolidine-2, 5-dione (3-DBP). Biochim. Biophys. Acta Gen. Subj. 2012, 1820, 962–969. [Google Scholar] [CrossRef]
- Han, Y.K.; Park, Y.J.; Ha, Y.M.; Park, D.; Lee, J.Y.; Lee, N.; Yoon, J.H.; Moon, H.R.; Chung, H.Y. Characterization of a novel tyrosinase inhibitor,(2RS, 4R)-2-(2, 4-dihydroxyphenyl) thiazolidine-4-carboxylic acid (MHY384). Biochim. Biophys. Acta Gen. Subj. 2012, 1820, 542–549. [Google Scholar] [CrossRef]
- Ullah, S.; Park, Y.; Park, C.; Lee, S.; Kang, D.; Yang, J.; Akter, J.; Chun, P.; Moon, H.R. Antioxidant, anti-tyrosinase and anti-melanogenic effects of (E)-2, 3-diphenylacrylic acid derivatives. Bioorg. Med. Chem. 2019, 27, 2192–2200. [Google Scholar] [CrossRef]
- Ullah, S.; Akter, J.; Kim, S.J.; Yang, J.; Park, Y.; Chun, P.; Moon, H.R. The tyrosinase-inhibitory effects of 2-phenyl-1, 4-naphthoquinone analogs: Importance of the (E)-β-phenyl-α, β-unsaturated carbonyl scaffold of an endomethylene type. Med. Chem. Res. 2019, 28, 95–103. [Google Scholar] [CrossRef]
- Ullah, S.; Kang, D.; Lee, S.; Ikram, M.; Park, C.; Park, Y.; Yoon, S.; Chun, P.; Moon, H.R. Synthesis of cinnamic amide derivatives and their anti-melanogenic effect in α-MSH-stimulated B16F10 melanoma cells. Eur. J. Med. Chem. 2019, 161, 78–92. [Google Scholar] [CrossRef] [PubMed]
- Ullah, S.; Park, Y.; Ikram, M.; Lee, S.; Park, C.; Kang, D.; Yang, J.; Akter, J.; Yoon, S.; Chun, P. Design, synthesis and anti-melanogenic effect of cinnamamide derivatives. Bioorg. Med. Chem. 2018, 26, 5672–5681. [Google Scholar] [CrossRef]
- Pandey, J.; Jha, A.K.; Hajela, K. Synthesis and biological activities of some new dibenzopyranones and dibenzopyrans: Search for potential oestrogen receptor agonists and antagonists. Bioorg. Med. Chem. 2004, 12, 2239–2249. [Google Scholar] [CrossRef]
- Hurtley, W.R.H. CCXLIV—Replacement of halogen in orthobromo-benzoic acid. J. Chem. Soc. (Resumed) 1929, 1870–1873. [Google Scholar] [CrossRef]
- Wang, S.-T.; Chang, W.-C.; Hsu, C.; Su, N.-W. Antimelanogenic Effect of Urolithin A and Urolithin B, the Colonic Metabolites of Ellagic Acid, in B16 Melanoma Cells. J. Agric. Food Chem. 2017, 65, 6870–6876. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2016, 45, D158–D169. [Google Scholar]
- Lai, X.; Wichers, H.J.; Soler-Lopez, M.; Dijkstra, B.W. Structure of Human Tyrosinase Related Protein 1 Reveals a Binuclear Zinc Active Site Important for Melanogenesis. Angew. Chem. Int. Ed. 2017, 56, 9812–9815. [Google Scholar] [CrossRef] [PubMed]
- Hyun, S.K.; Lee, W.-H.; Jeong, D.M.; Kim, Y.; Choi, J.S. Inhibitory effects of kurarinol, kuraridinol, and trifolirhizin from Sophora flavescens on tyrosinase and melanin synthesis. Biol. Pharm. Bull. 2008, 31, 154–158. [Google Scholar] [CrossRef] [Green Version]
- No, J.K.; Soung, D.Y.; Kim, Y.J.; Shim, K.H.; Jun, Y.S.; Rhee, S.H.; Yokozawa, T.; Chung, H.Y. Inhibition of tyrosinase by green tea components. Life Sci. 1999, 65, PL241–PL246. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef] [Green Version]
- Moustakas, D.T.; Lang, P.T.; Pegg, S.; Pettersen, E.; Kuntz, I.D.; Brooijmans, N.; Rizzo, R.C. Development and validation of a modular, extensible docking program: DOCK 5. J. Comput. Aided Mol. Des. 2006, 20, 601–619. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.H.; Hyun, J.S.; Choi, J.; Choi, K.E.; Jee, J.G.; Park, S.J. Structural ensemble-based docking simulation and biophysical studies discovered new inhibitors of Hsp90 N-terminal domain. Sci Rep. 2018, 8, 368. [Google Scholar] [CrossRef] [Green Version]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein–ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farid, R.; Day, T.; Friesner, R.A.; Pearlstein, R.A. New insights about HERG blockade obtained from protein modeling, potential energy mapping, and docking studies. Bioorg. Med. Chem. 2006, 14, 3160–3173. [Google Scholar] [CrossRef]
- Jung, H.J.; Noh, S.G.; Park, Y.; Kang, D.; Chun, P.; Chung, H.Y.; Moon, H.R. In vitro and in silico insights into tyrosinase inhibitors with (E)-benzylidene-1-indanone derivatives. Comput. Struct. Biotechnol. J. 2019, 17, 1255–1264. [Google Scholar] [CrossRef]
- Bae, S.J.; Ha, Y.M.; Kim, J.-A.; Park, J.Y.; Ha, T.K.; Park, D.; Chun, P.; Park, N.H.; Moon, H.R.; Chung, H.Y. A novel synthesized tyrosinase inhibitor:(E)-2-((2, 4-dihydroxyphenyl) diazenyl) phenyl 4-methylbenzenesulfonate as an azo-resveratrol analog. Biosci. Biotechnol. Biochem. 2013, 77, 65–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.-G.; Chang, W.-L.; Lee, C.-J.; Lee, L.-T.; Shih, C.-M.; Wang, C.-C. Melanogenesis inhibition by gallotannins from Chinese galls in B16 mouse melanoma cells. Biol. Pharm. Bull. 2009, 32, 1447–1452. [Google Scholar] [CrossRef] [Green Version]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Compounds | R1 | R2 | R3 | Tyrosinase Inhibition (%) a | Log P |
| 1a (urolithin B) | H | OH | H | NI b | 2.77 |
| 1b | H | OMe | H | NI | 3.03 |
| 1c | H | OH | OH | 68.93 ± 0.67 | 2.38 |
| 1d | H | OMe | OMe | NI | 2.91 |
| 1e (urolithin A) | OH | OH | H | NI | 2.38 |
| 1f | OH | OH | OH | 12.68 ± 6.09 | 1.99 |
| 1g | OMe | OH | H | NI | 2.64 |
| 1h | OMe | OH | OH | 94.86 ± 0.16 | 2.25 |
| 1i | OMe | OMe | H | NI | 2.91 |
| 1j | OMe | OMe | OMe | NI | 2.78 |
| Kojic acid | 52.16 ± 2.05 | -2.45 | |||
 | |||||
|---|---|---|---|---|---|
| Compounds | R1 | R2 | R3 | Tyrosinase Inhibition (%) a | Log P c |
| 2a | H | OH | H | 75.92 ± 2.19 | 2.36 |
| 2b | H | OMe | H | NI b | 2.62 |
| 2c | H | OH | OH | 19.78 ± 15.84 | 1.97 |
| 2d | H | OMe | OMe | NI | 2.49 |
| 2e | OH | OH | H | NI | 1.97 |
| 2f | OMe | OH | H | 47.87 ± 12.81 | 2.23 |
| 2g | OMe | OMe | H | NI | 2.49 |
| 2h | OMe | OH | OH | 26.30 ± 0.75 | 1.84 |
| 2i | OMe | OMe | OMe | NI | 2.37 |
| Kojic acid | 52.16 ± 2.05 | −2.45 | |||
| Compound | Concentration (μM) | Inhibition (%) | Average of Inhibition (%) | IC50 (μM) | ||
|---|---|---|---|---|---|---|
| 1c | 1.56 | 2.61 | 4.01 | 2.57 | 3.06 ± 0.47 | 18.09 ± 0.25 |
| 3.13 | 9.61 | 10.84 | 9.94 | 10.13 ± 0.37 | ||
| 6.25 | 27.31 | 25.08 | 33.10 | 28.50 ± 2.39 | ||
| 12.50 | 43.74 | 45.18 | 48.37 | 45.76 ± 1.37 | ||
| 25.00 | 60.89 | 61.85 | 60.89 | 61.21 ± 0.32 | ||
| 1h | 0.78 | 15.68 | 15.61 | 18.94 | 16.74 ± 1.10 | 4.14 ± 0.10 |
| 1.56 | 28.27 | 24.63 | 31.11 | 28.00 ± 1.88 | ||
| 3.13 | 48.71 | 48.71 | 48.92 | 48.78 ± 0.07 | ||
| 6.25 | 63.98 | 63.95 | 66.79 | 64.91 ± 0.94 | ||
| 2a | 1.56 | 0.27 | 0.72 | 1.06 | 0.68 ± 0.23 | 15.69 ± 0.40 |
| 3.13 | 28.06 | 10.29 | 7.03 | 15.13 ± 6.53 | ||
| 6.25 | 32.25 | 31.66 | 31.22 | 31.71 ± 0.30 | ||
| 12.50 | 55.81 | 50.39 | 54.31 | 53.50 ± 1.62 | ||
| 25.00 | 69.81 | 68.71 | 69.37 | 69.30 ± 0.32 | ||
| Kojic acid | 12.50 | 14.68 | 20.50 | 23.41 | 19.53 ± 2.57 | 48.62 ± 3.38 |
| 25.00 | 37.46 | 42.27 | 42.44 | 40.72 ± 1.63 | ||
| 50.00 | 43.81 | 48.83 | 53.08 | 48.57 ± 2.68 | ||
| 1c | 1h | 2a | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Vmax (mM·min−1) | KM (mM) | Ki (M) | Vmax (mM·min−1) | KM (mM) | Ki (M) | Vmax (mM·min−1) | KM (mM) | Ki (M) | |
| 2.5 | - | - | - | 3.1 × 10−2 | 5.64 | 4.1 × 10−7 | - | - | - |
| 5 μM | - | - | - | 3.1 × 10−2 | 13.92 | 3.0 × 10−7 | - | - | - |
| 10 μM | 2.7 × 10−2 | 12.54 | 1.9 × 10−6 | 3.1 × 10−2 | 26.97 | 3.0 × 10−7 | 2.9 × 10−2 | 7.83 | 3.4 × 10−6 |
| 20 μM | 2.7 × 10−2 | 22.24 | 1.9 × 10−6 | - | - | - | 2.9 × 10−2 | 15.07 | 3.1 × 10−6 |
| 40 μM | 2.7 × 10−2 | 41.56 | 2.0 × 10−6 | - | - | - | 2.9 × 10−2 | 32.42 | 2.6 × 10−6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, S.; Choi, H.; Park, Y.; Jung, H.J.; Ullah, S.; Choi, I.; Kang, D.; Park, C.; Ryu, I.Y.; Jeong, Y.; et al. Urolithin and Reduced Urolithin Derivatives as Potent Inhibitors of Tyrosinase and Melanogenesis: Importance of the 4-Substituted Resorcinol Moiety. Int. J. Mol. Sci. 2021, 22, 5616. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22115616
Lee S, Choi H, Park Y, Jung HJ, Ullah S, Choi I, Kang D, Park C, Ryu IY, Jeong Y, et al. Urolithin and Reduced Urolithin Derivatives as Potent Inhibitors of Tyrosinase and Melanogenesis: Importance of the 4-Substituted Resorcinol Moiety. International Journal of Molecular Sciences. 2021; 22(11):5616. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22115616
Chicago/Turabian StyleLee, Sanggwon, Heejeong Choi, Yujin Park, Hee Jin Jung, Sultan Ullah, Inkyu Choi, Dongwan Kang, Chaeun Park, Il Young Ryu, Yeongmu Jeong, and et al. 2021. "Urolithin and Reduced Urolithin Derivatives as Potent Inhibitors of Tyrosinase and Melanogenesis: Importance of the 4-Substituted Resorcinol Moiety" International Journal of Molecular Sciences 22, no. 11: 5616. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22115616