
Aldosterone Negatively Regulates Nrf2 Activity: An Additional Mechanism Contributing to Oxidative Stress and Vascular Dysfunction by Aldosterone

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Results
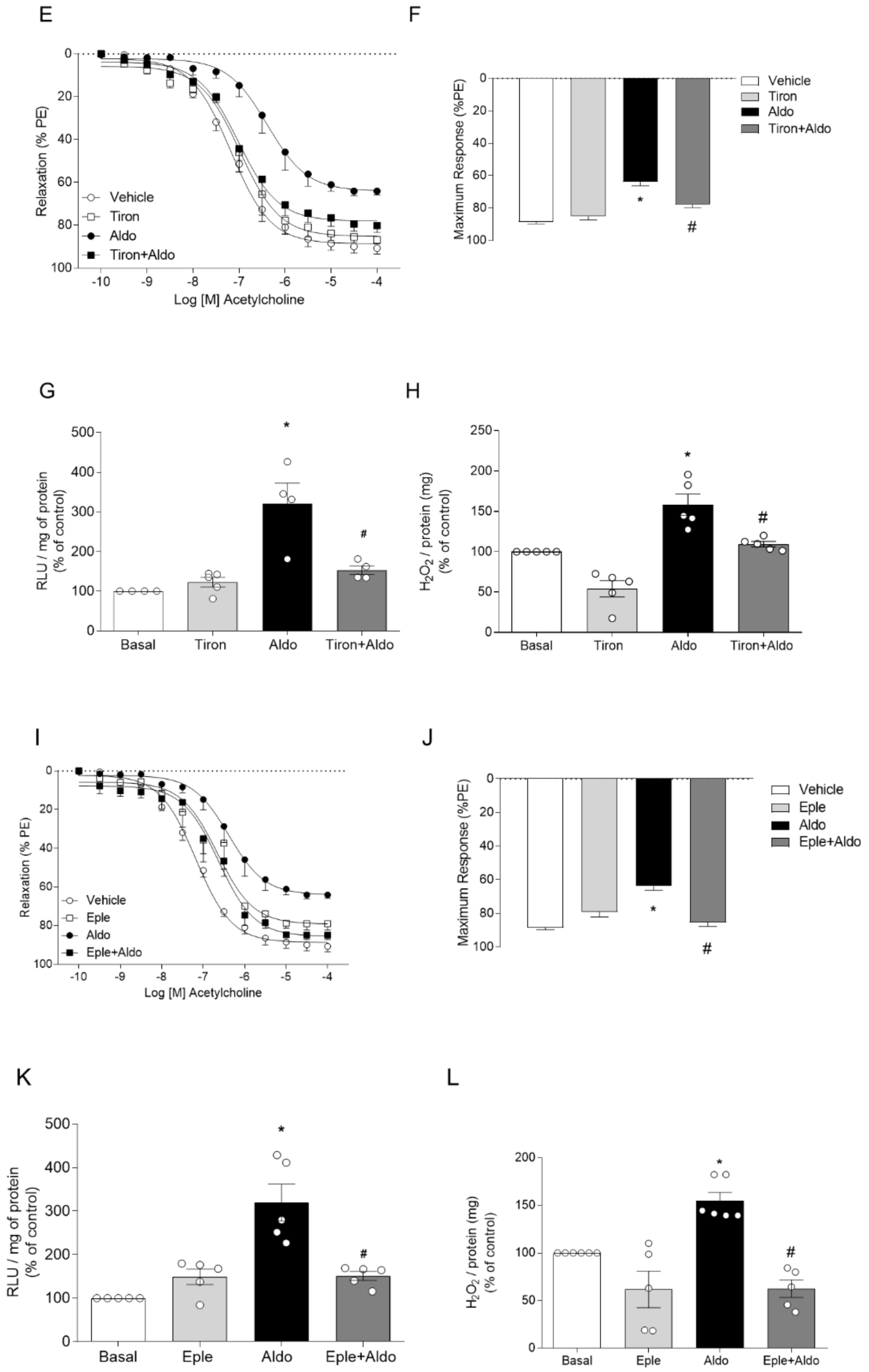
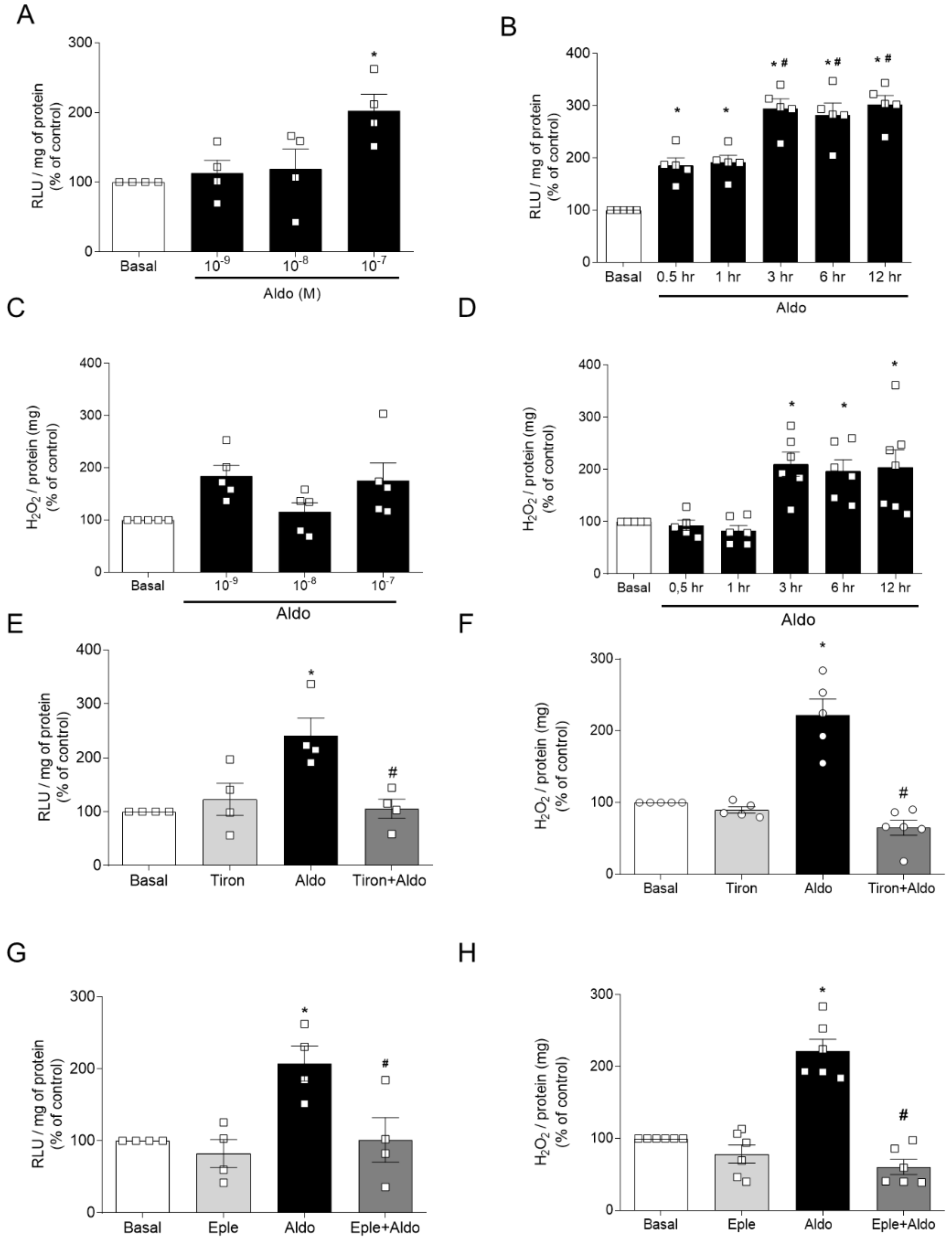
2.1. Aldosterone, via Mineralocorticoid Receptor (MR) Activation, Induces ROS Generation and Vascular Dysfunction
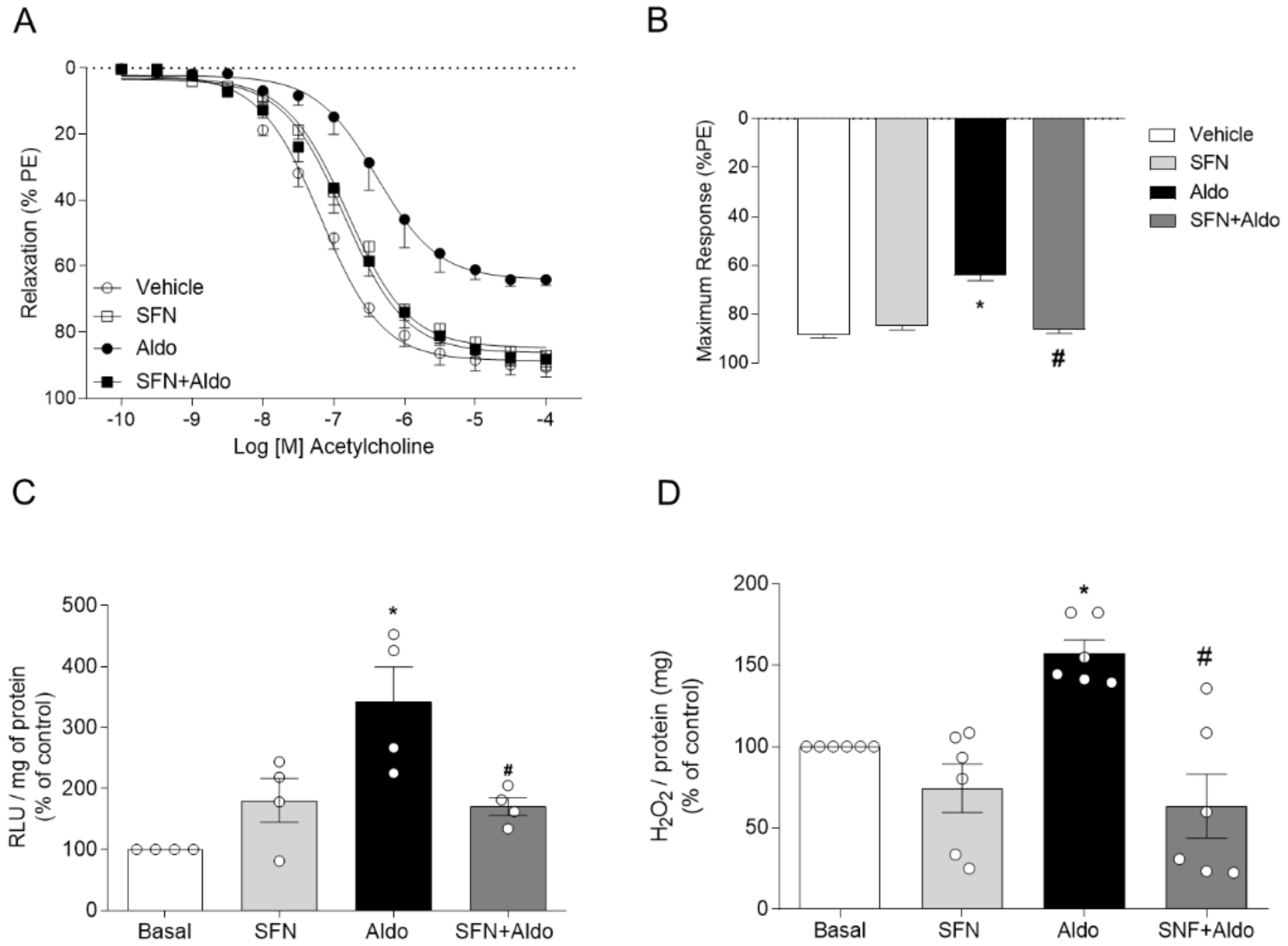
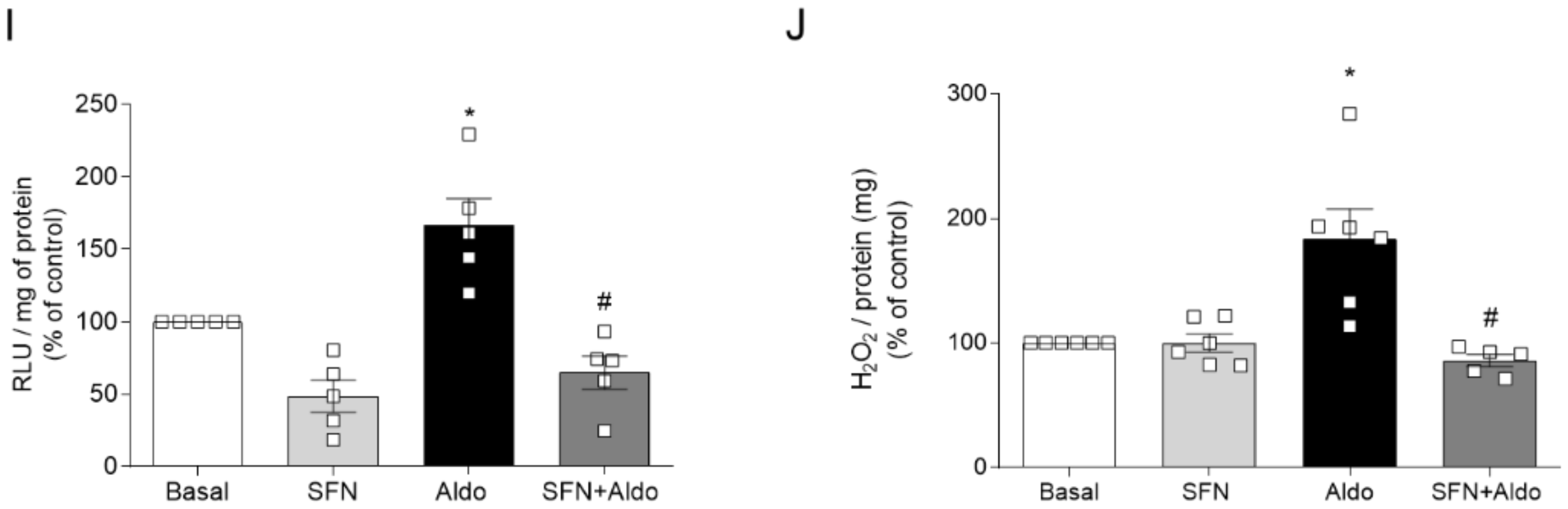
2.2. Pharmacological Activation of Nrf2 Prevents Aldosterone-Induced Vascular Dysfunction and Oxidative Stress
2.3. Aldosterone Induces ROS Generation in Human Endothelial Cells
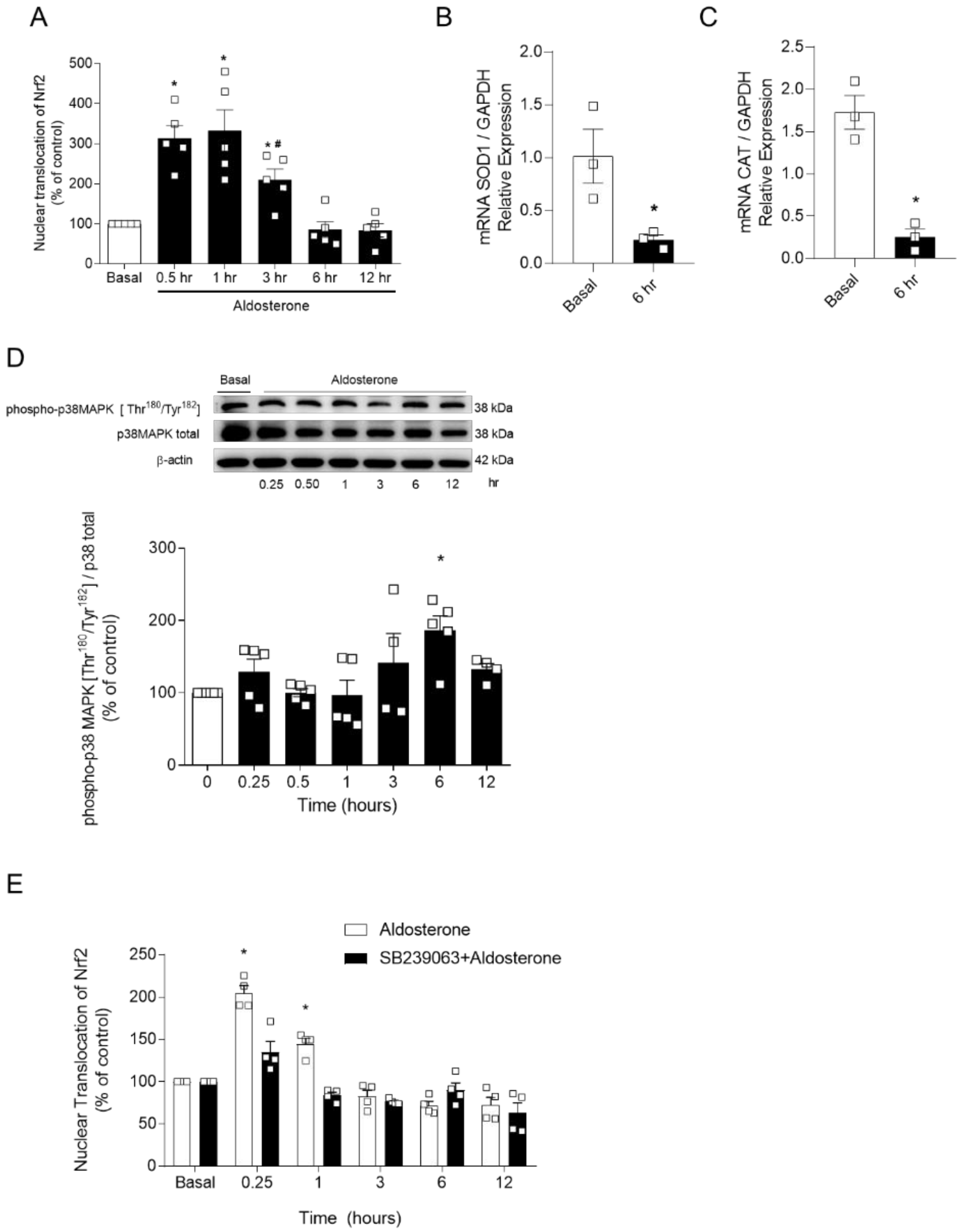
2.4. p38 MAPK Inhibition Does Not Prevent Aldo-Induced Decreased Nrf2 Signaling
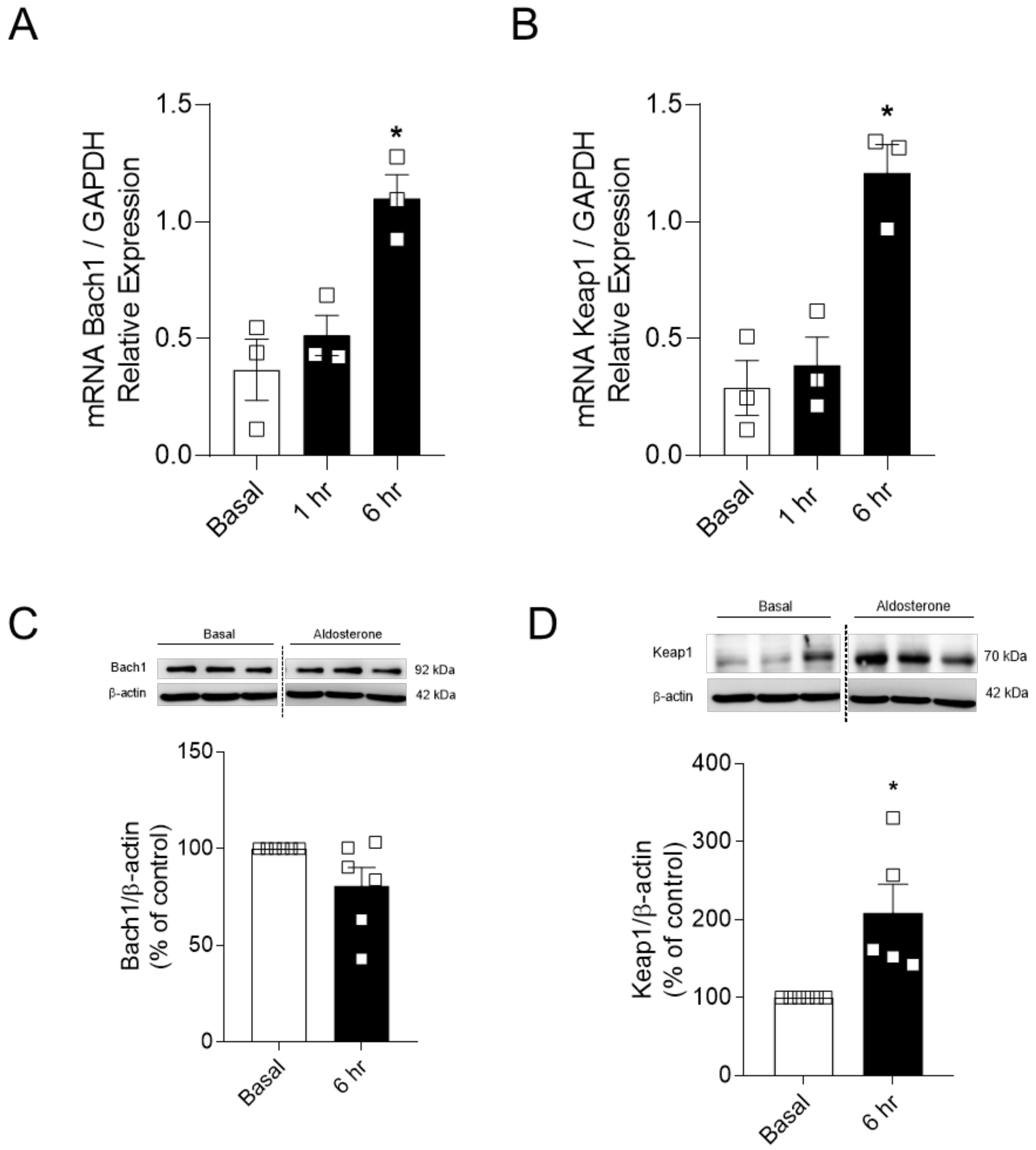
2.5. Aldosterone-Induced Reduced Nrf2 Activity Depends on the Expression of Nrf2-Negative Regulators
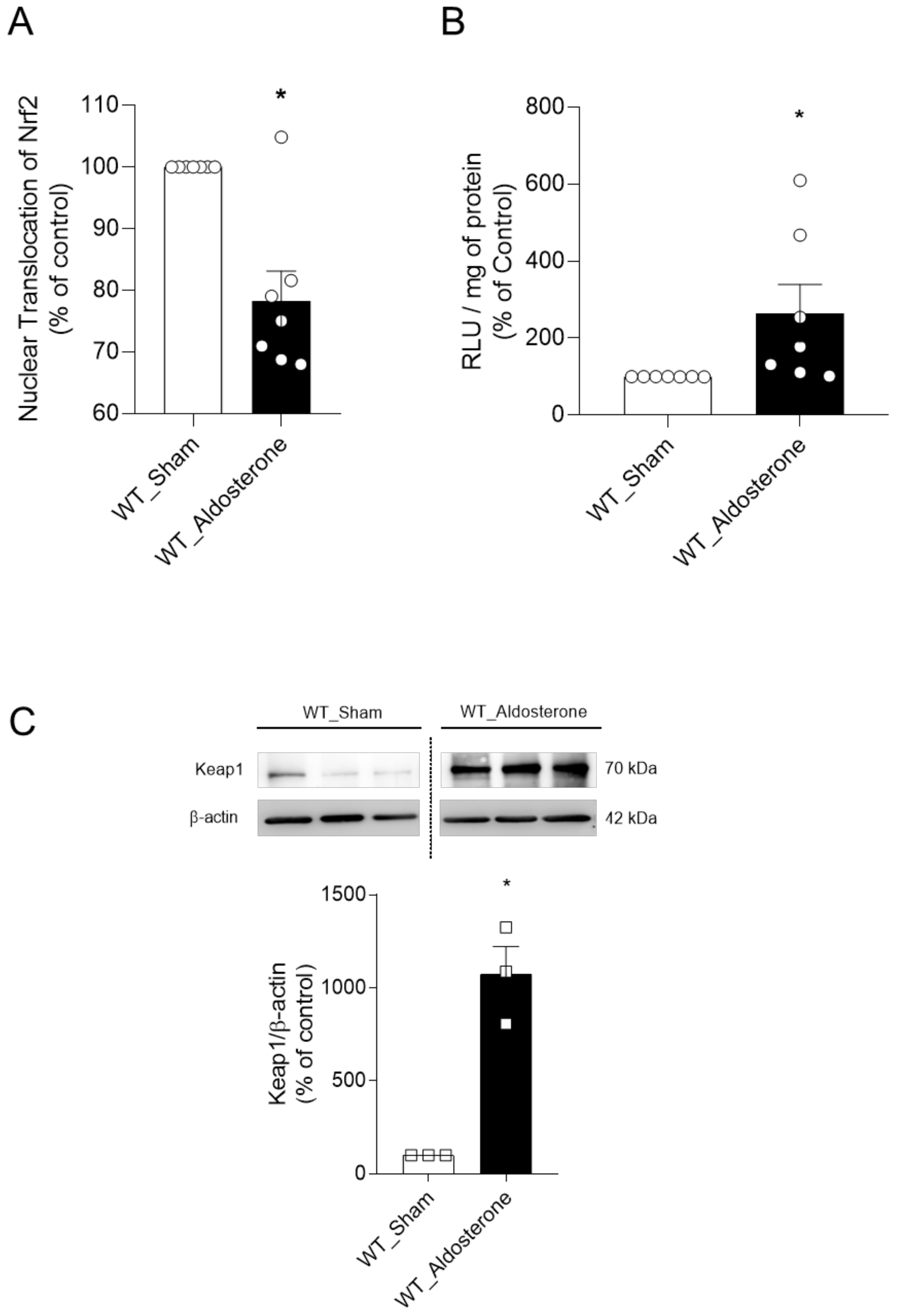
2.6. In Vivo, Aldosterone Treatment Decreases Vascular Nrf2 Activity, and Increases Oxidative Stress and the Protein Expression of Keap1
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Animals
4.3. Functional Vascular Studies
4.4. Cultured Endothelial Cells
4.5. Lucigenin-Enhanced Chemiluminescence
4.6. Amplex Red
4.7. Nrf2 Translocation Assay
4.8. Gene Expression Analysis by RT-qPCR
4.9. Western Blot
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACh | Acetylcholine |
| Aldo | Aldosterone |
| ARE | Antioxidant response element |
| BM | Bardoxolone methyl |
| BSA | Bovine serum albumin |
| Cul3 | Cullin 3 |
| Eple | Eplerenone |
| H2O2 | Hydrogen peroxide |
| Keap1 | Kelch-like ECH-associated protein 1 |
| MR | Mineralocorticoid receptor |
| NO | Nitric oxide |
| ONOO- | Peroxynitrite |
| Nrf2 | Nuclear factor erythroid 2-related factor 2 |
| O2·- | Superoxide anion |
| OS | Oxidative stress |
| PA | Primary aldosteronism |
| pEC50 | Negative logarithm of the EC50 |
| PE | Phenylephrine |
| RLU | Relative luminescence units |
| ROS | Reactive oxygen species |
| SEM | Standard error of the mean |
| siRNA | Small interfering RNA |
| SFN | L-Sulforaphane |
| TBS | Tris-buffered saline |
| Vehicle | Ethanol |
| VSMC | Vascular smooth muscle cell |
References
- Laragh, J.H. Hormones and the Pathogenesis of Congestive Heart Failure: Vasopressin, Aldosterone, And Angiotensin Ii. Circulation 1962, 25, 1015–1023. [Google Scholar] [CrossRef] [Green Version]
- Clark, D., 3rd; Ahmed, M.I.; Calhoun, D.A. Resistant Hypertension and Aldosterone: An Update. Can. J. Cardiol. 2012, 28, 318–325. [Google Scholar] [CrossRef]
- Mcfarlane, S.I.; Sowers, J.R. Aldosterone Function in Diabetes Mellitus: Effects on Cardiovascular and Renal Disease. J. Clin. Endocrinol. Metab. 2003, 88, 516–523. [Google Scholar] [CrossRef]
- Kawarazaki, W.; Fujita, T. The Role of Aldosterone in Obesity-Related Hypertension. Am. J. Hypertens. 2016, 29, 415–423. [Google Scholar] [CrossRef] [Green Version]
- Krug, A.W.; Ehrhart-Bornstein, M. Aldosterone and Metabolic Syndrome. Hypertension 2008, 51, 1252–1258. [Google Scholar] [CrossRef] [Green Version]
- Briet, M.; Schiffrin, E.L. Vascular Actions of Aldosterone. J. Vasc. Res. 2013, 50, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Kolkhof, P.; Bärfacker, L. 30 Years of the Mineralocorticoid Receptor: Mineralocorticoid Receptor Antagonists: 60 Years Of Research and Development. J. Endocrinol. 2017, 234, T125–T140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitt, B.; Zannad, F.; Remme, W.J.; Cody, R.; Castaigne, A.; Perez, A.; Palensky, J.; Wittes, J. The Effect of Spironolactone on Morbidity and Mortality in Patients with Severe Heart Failure. Randomized Aldactone Evaluation Study Investigators. N. Engl. J. Med. 1999, 341, 709–717. [Google Scholar] [CrossRef] [Green Version]
- Pitt, B.; Remme, W.; Zannad, F.; Neaton, J.; Martinez, F.; Roniker, B.; Bittman, R.; Hurley, S.; Kleiman, J.; Gatlin, M. Eplerenone, a Selective Aldosterone Blocker, in Patients with Left Ventricular Dysfunction after Myocardial Infarction. N. Engl. J. Med. 2003, 348, 1309–1321. [Google Scholar] [CrossRef] [PubMed]
- Schieber, M.; Chandel, N.S. Ros Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sies, H.; Jones, D.P. Reactive Oxygen Species (Ros) as Pleiotropic Physiological Signalling Agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Moldogazieva, N.T.; Mokhosoev, I.M.; Feldman, N.B.; Lutsenko, S.V. Ros And Rns Signalling: Adaptive Redox Switches through Oxidative/Nitrosative Protein Modifications. Free Radic. Res. 2018, 52, 507–543. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef] [PubMed]
- Callera, G.E.; Touyz, R.M.; Tostes, R.C.; Yogi, A.; He, Y.; Malkinson, S.; Schiffrin, E.L. Aldosterone Activates Vascular P38map Kinase and Nadph Oxidase via c-Src. Hypertension 2005, 45, 773–779. [Google Scholar] [CrossRef] [Green Version]
- Chrissobolis, S.; Drummond, G.R.; Faraci, F.M.; Sobey, C.G. Chronic Aldosterone Administration Causes Nox2-Mediated Increases in Reactive Oxygen Species Production and Endothelial Dysfunction in the Cerebral Circulation. J. Hypertens. 2014, 32, 1815–1821. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, H.; Kobara, M.; Abe, M.; Tanaka, N.; Gouda, E.; Toba, H.; Yamada, H.; Tatsumi, T.; Nakata, T.; Matsubara, H. Aldosterone Nongenomically Produces Nadph Oxidase–Dependent Reactive Oxygen Species And Induces Myocyte Apoptosis. Hypertens. Res. 2008, 31, 363–375. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.; Wang, X.; Li, Y.; Li, X.; Zhang, S.; Hao, L. Aldosterone Promotes Renal Interstitial Fibrosis via the Aif-1/Akt/Mtor Signaling Pathway. Mol. Med. Rep. 2019, 20, 4033–4044. [Google Scholar] [CrossRef] [Green Version]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 Represses Nuclear Activation of Antioxidant Responsive Elements by Nrf2 Through Binding to the Amino-Terminal Neh2 Domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef] [Green Version]
- Kaspar, J.W.; Niture, S.K.; Jaiswal, A.K. Nrf2:Inrf2 (Keap1) Signaling in Oxidative Stress. Free Radic. Biol. Med. 2009, 47, 1304–1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cullinan, S.B.; Gordan, J.D.; Jin, J.; Harper, J.W.; Diehl, J.A. The Keap1-Btb Protein is an Adaptor that Bridges Nrf2 to A Cul3-Based E3 Ligase: Oxidative Stress Sensing by A Cul3-Keap1 Ligase. Mol. Cell. Biol. 2004, 24, 8477–8486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bloom, D.A.; Jaiswal, A.K. Phosphorylation of Nrf2 at Ser40 by Protein Kinase C in Response to Antioxidants Leads to the Release of Nrf2 from Inrf2, but is not Required for Nrf2 Stabilization/Accumulation in the Nucleus and Transcriptional Activation of Antioxidant Response Element-Mediated Nad(P)H:Quinone Oxidoreductase-1 Gene Expression. J. Biol. Chem. 2003, 278, 44675–44682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niture, S.K.; Khatri, R.; Jaiswal, A.K. Regulation of Nrf2-An Update. Free Radic. Biol. Med. 2014, 66, 36–44. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Xu, Y.; Qiu, Z.; Jiang, L. Sulforaphane Attenuates Angiotensin Ii-Induced Vascular Smooth Muscle Cell Migration via Suppression of Nox4/Ros/Nrf2 Signaling. Int. J. Biol. Sci. 2019, 15, 148–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Zhou, S.; Guo, H.; Zhang, J.; Ma, T.; Zheng, Y.; Zhang, Z.; Cai, L. Protective Effects of Sulforaphane on Type 2 Diabetes-Induced Cardiomyopathy via Ampk-Mediated Activation of Lipid Metabolic Pathways and Nrf2 Function. Metab. Clin. Exp. 2020, 102, 154002. [Google Scholar] [CrossRef] [Green Version]
- Xin, Y.; Bai, Y.; Jiang, X.; Zhou, S.; Wang, Y.; Wintergerst, K.A.; Cui, T.; Ji, H.; Tan, Y.; Cai, L. Sulforaphane Prevents Angiotensin Ii-Induced Cardiomyopathy by Activation of Nrf2 via Stimulating the Akt/Gsk-3ß/Fyn Pathway. Redox Biol. 2018, 15, 405–417. [Google Scholar] [CrossRef]
- Bai, Y.; Cui, W.; Xin, Y.; Miao, X.; Barati, M.T.; Zhang, C.; Chen, Q.; Tan, Y.; Cui, T.; Zheng, Y.; et al. Prevention by Sulforaphane of Diabetic Cardiomyopathy is Associated with Up-Regulation of Nrf2 Expression and Transcription Activation. J. Mol. Cell. Cardiol. 2013, 57, 82–95. [Google Scholar] [CrossRef]
- Hisamichi, M.; Kamijo-Ikemori, A.; Sugaya, T.; Hoshino, S.; Kimura, K.; Shibagaki, Y. Role of Bardoxolone Methyl, a Nuclear Factor Erythroid 2-Related Factor 2 Activator, in Aldosterone- and Salt-Induced Renal Injury. Hypertens. Res. 2018, 41, 8–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bomback, A.S.; Klemmer, P.J. The Incidence and Implications of Aldosterone Breakthrough. Nat. Clin. Pract. Nephrol. 2007, 3, 486–492. [Google Scholar] [CrossRef]
- Goodfriend, T.L.; Kelley, D.E.; Goodpaster, B.H.; Winters, S.J. Visceral Obesity and Insulin Resistance are Associated with Plasma Aldosterone Levels in Women. Obes. Res. 1999, 7, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Ruhs, S.; Nolze, A.; Hübschmann, R.; Grossmann, C. 30 Years of the Mineralocorticoid Receptor: Nongenomic Effects via the Mineralocorticoid Receptor. J. Endocrinol. 2017, 234, T107–T124. [Google Scholar] [CrossRef] [Green Version]
- Sheng, L.; Yang, M.; Ding, W.; Zhang, M.; Niu, J.; Qiao, Z.; Gu, Y. Epidermal Growth Factor Receptor Signaling Mediates Aldosterone-Induced Profibrotic Responses in Kidney. Exp. Cell Res. 2016, 346, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Gros, R.; Ding, Q.; Liu, B.; Chorazyczewski, J.; Feldman, R.D. Aldosterone Mediates its Rapid Effects in Vascular Endothelial Cells through Gper Activation. Am. J. Physiol. Cell Physiol. 2013, 304, C532–C540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinh, Q.N.; Vinh, A.; Kim, H.A.; Saini, N.; Broughton, B.R.S.; Chrissobolis, S.; Diep, H.; Judkins, C.P.; Drummond, G.R.; Sobey, C.G. Aldosterone-Induced Hypertension ss Sex-Dependent, Mediated by T Cells and Sensitive to Gper Activation. Cardiovasc. Res. 2020, 117, 960–970. [Google Scholar] [CrossRef]
- Mittler, R. Ros Are Good. Trends Plant Sci. 2017, 22, 11–19. [Google Scholar] [CrossRef] [Green Version]
- Costa, T.J.; Barros, P.R.; Arce, C.; Santos, J.D.; Da Silva-Neto, J.; Egea, G.; Dantas, A.P.; Tostes, R.C.; Jiménez-Altayó, F. The Homeostatic Role of Hydrogen Peroxide, Superoxide Anion and Nitric Oxide in The Vasculature. Free Radic. Biol. Med. 2021, 162, 615–635. [Google Scholar] [CrossRef]
- Miyata, K.; Rahman, M.; Shokoji, T.; Nagai, Y.; Zhang, G.X.; Sun, G.P.; Kimura, S.; Yukimura, T.; Kiyomoto, H.; Kohno, M.; et al. Aldosterone Stimulates Reactive Oxygen Species Production through Activation of Nadph Oxidase in Rat Mesangial Cells. J. Am. Soc. Nephrol. Jasn 2005, 16, 2906–2912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rude, M.K.; Duhaney, T.A.; Kuster, G.M.; Judge, S.; Heo, J.; Colucci, W.S.; Siwik, D.A.; Sam, F. Aldosterone Stimulates Matrix Metalloproteinases and Reactive Oxygen Species in Adult Rat Ventricular Cardiomyocytes. Hypertension 2005, 46, 555–561. [Google Scholar] [CrossRef] [Green Version]
- Sanz-Rosa, D.; Oubiña, M.P.; Cediel, E.; De Las Heras, N.; Aragoncillo, P.; Balfagón, G.; Cachofeiro, V.; Lahera, V. Eplerenone Reduces Oxidative Stress and Enhances Enos in Shr: Vascular Functional and Structural Consequences. Antioxid. Redox Signal. 2005, 7, 1294–1301. [Google Scholar] [CrossRef]
- Ferreira, N.S.; Bruder-Nascimento, T.; Pereira, C.A.; Zanotto, C.Z.; Prado, D.S.; Silva, J.F.; Rassi, D.M.; Foss-Freitas, M.C.; Alves-Filho, J.C.; Carlos, D.; et al. Nlrp3 Inflammasome and Mineralocorticoid Receptors are Associated with Vascular Dysfunction in Type 2 Diabetes Mellitus. Cells 2019, 8, 1595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, N.S.; Cau, S.B.; Silva, M.A.; Manzato, C.P.; Mestriner, F.L.; Matsumoto, T.; Carneiro, F.S.; Tostes, R.C. Diabetes Impairs the Vascular Effects of Aldosterone Mediated by G Protein-Coupled Estrogen Receptor Activation. Front. Pharmacol. 2015, 6, 34. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Zhang, B.; Zhou, K.; Chen, M.; Wang, M.; Jia, Y.; Song, Y.; Li, Y.; Wen, A. Dietary Ellagic Acid Improves Oxidant-Induced Endothelial Dysfunction and Atherosclerosis: Role Of Nrf2 Activation. Int. J. Cardiol. 2014, 175, 508–514. [Google Scholar] [CrossRef]
- Xue, M.; Qian, Q.; Adaikalakoteswari, A.; Rabbani, N.; Babaei-Jadidi, R.; Thornalley, P.J. Activation of Nf-E2-Related Factor-2 Reverses Biochemical Dysfunction of Endothelial Cells Induced by Hyperglycemia Linked to Vascular Disease. Diabetes 2008, 57, 2809–2817. [Google Scholar] [CrossRef] [Green Version]
- Lopes, R.A.; Neves, K.B.; Tostes, R.C.; Montezano, A.C.; Touyz, R.M. Downregulation of Nuclear Factor Erythroid 2-Related Factor and Associated Antioxidant Genes Contributes to Redox-Sensitive Vascular Dysfunction in Hypertension. Hypertension 2015, 66, 1240–1250. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Cha, Y.N.; Surh, Y.J. A Protective Role of Nuclear Factor-Erythroid 2-Related Factor-2 (Nrf2) in Inflammatory Disorders. Mutat. Res. 2010, 690, 12–23. [Google Scholar] [CrossRef]
- Ramsey, C.P.; Glass, C.A.; Montgomery, M.B.; Lindl, K.A.; Ritson, G.P.; Chia, L.A.; Hamilton, R.L.; Chu, C.T.; Jordan-Sciutto, K.L. Expression of Nrf2 in Neurodegenerative Diseases. J. Neuropathol. Exp. Neurol. 2007, 66, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Da Costa, R.M.; Rodrigues, D.; Pereira, C.A.; Silva, J.F.; Alves, J.V.; Lobato, N.S.; Tostes, R.C. Nrf2 as a Potential Mediator of Cardiovascular Risk In Metabolic Diseases. Front. Pharmacol. 2019, 10, 382. [Google Scholar] [CrossRef] [Green Version]
- Satta, S.; Mahmoud, A.M.; Wilkinson, F.L.; Yvonne Alexander, M.; White, S.J. The Role of Nrf2 In Cardiovascular Function and Disease. Oxid. Med. Cell. Longev. 2017, 2017, 9237263. [Google Scholar] [CrossRef]
- Queisser, N.; Oteiza, P.I.; Link, S.; Hey, V.; Stopper, H.; Schupp, N. Aldosterone Activates Transcription Factor Nrf2 in Kidney Cells both In Vitro and In Vivo. Antioxid. Redox Signal. 2014, 21, 2126–2142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keum, Y.S.; Yu, S.; Chang, P.P.; Yuan, X.; Kim, J.H.; Xu, C.; Han, J.; Agarwal, A.; Kong, A.N. Mechanism of Action of Sulforaphane: Inhibition of P38 Mitogen-Activated Protein Kinase Isoforms Contributing to the Induction of Antioxidant Response Element-Mediated Heme Oxygenase-1 in Human Hepatoma Hepg2 Cells. Cancer Res. 2006, 66, 8804–8813. [Google Scholar] [CrossRef] [Green Version]
- Neves, K.B.; Montezano, A.C.; Alves-Lopes, R.; Bruder-Nascimento, T.; Costa, R.M.; Costa, R.S.; Touyz, R.M.; Tostes, R.C. Upregulation of Nrf2 and Decreased Redox Signaling Contribute to Renoprotective Effects of Chemerin Receptor Blockade in Diabetic Mice. Int. J. Mol. Sci. 2018, 19, 2454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, L.; Zhan, P.; Wang, Q.; Wang, C.; Liu, Y.; Yu, Z.; Zhang, S. Curcumin Upregulates the Nrf2 System by Repressing Inflammatory Signaling-Mediated Keap1 Expression in Insulin-Resistant Conditions. Biochem. Biophys. Res. Commun. 2019, 514, 691–698. [Google Scholar] [CrossRef] [PubMed]
- Palsamy, P.; Subramanian, S. Resveratrol Protects Diabetic Kidney by Attenuating Hyperglycemia-Mediated Oxidative Stress and Renal Inflammatory Cytokines via Nrf2-Keap1 Signaling. Biochim. Et Biophys. Acta 2011, 1812, 719–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Vehicle | Tiron | Eple | Aldosterone | Tiron + Aldosterone | Eple + Aldosterone |
|---|---|---|---|---|---|
| 7.18 ± 0.04 | 6.99 ± 0.08 | 6.66 ± 0.11 | 6.38 ± 0.10 * | 7.00 ± 0.07 # | 6.63 ± 0.08 |
| Vehicle | SFN | Aldosterone | SFN + Aldosterone |
|---|---|---|---|
| 7.18 ± 0.04 | 6.80 ± 0.06 | 6.38 ± 0.10 * | 6.85 ± 0.05 # |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodrigues, D.; Costa, T.J.; Silva, J.F.; Neto, J.T.d.O.; Alves, J.V.; Fedoce, A.G.; Costa, R.M.; Tostes, R.C. Aldosterone Negatively Regulates Nrf2 Activity: An Additional Mechanism Contributing to Oxidative Stress and Vascular Dysfunction by Aldosterone. Int. J. Mol. Sci. 2021, 22, 6154. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22116154
Rodrigues D, Costa TJ, Silva JF, Neto JTdO, Alves JV, Fedoce AG, Costa RM, Tostes RC. Aldosterone Negatively Regulates Nrf2 Activity: An Additional Mechanism Contributing to Oxidative Stress and Vascular Dysfunction by Aldosterone. International Journal of Molecular Sciences. 2021; 22(11):6154. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22116154
Chicago/Turabian StyleRodrigues, Daniel, Tiago J. Costa, Josiane F. Silva, José Teles de Oliveira Neto, Juliano V. Alves, Aline G. Fedoce, Rafael Menezes Costa, and Rita C. Tostes. 2021. "Aldosterone Negatively Regulates Nrf2 Activity: An Additional Mechanism Contributing to Oxidative Stress and Vascular Dysfunction by Aldosterone" International Journal of Molecular Sciences 22, no. 11: 6154. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22116154