
Pro-Inflammatory Microenvironment Modulates the Transfer of Mutated TP53 Mediated by Tumor Exosomes


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
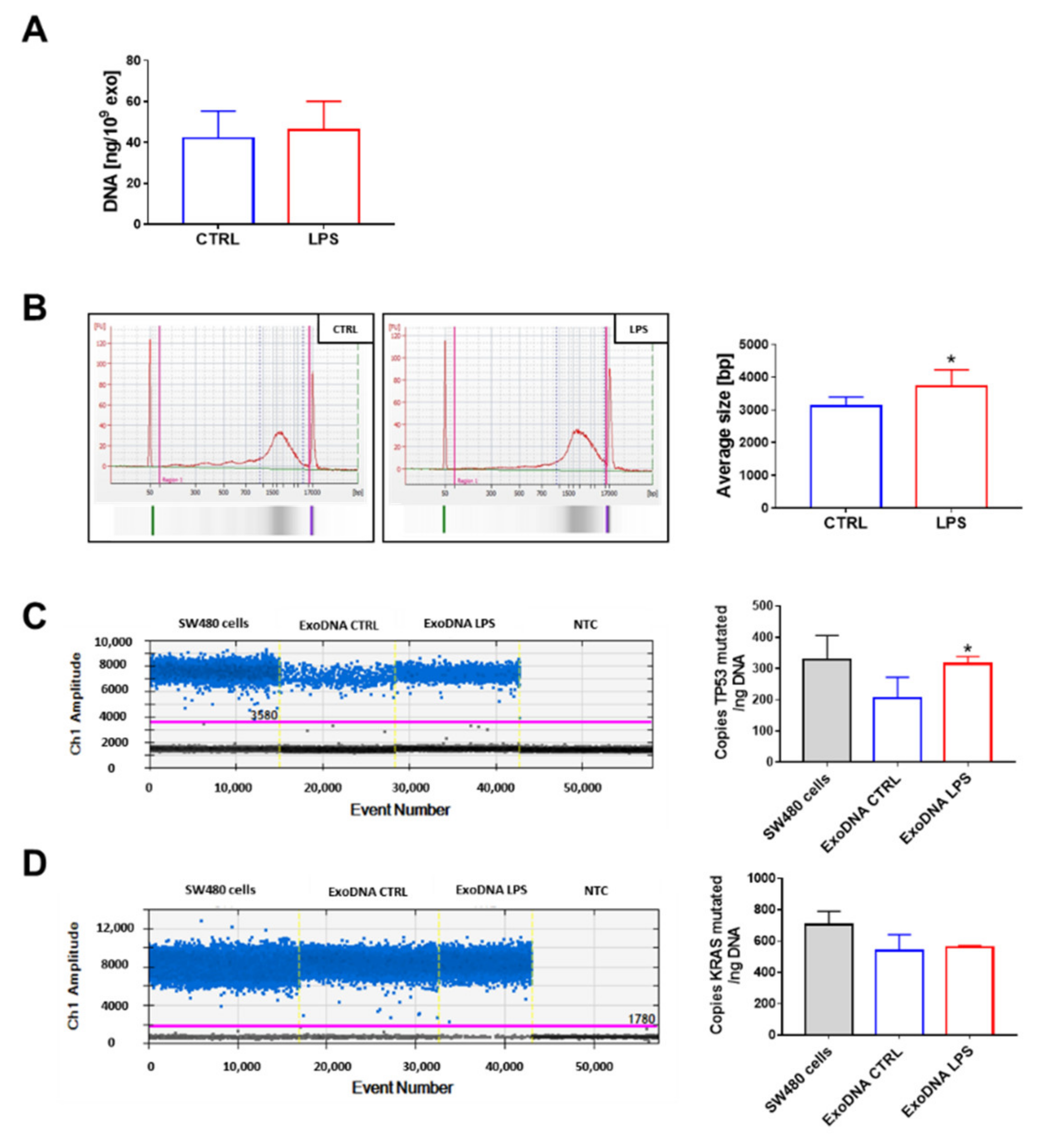
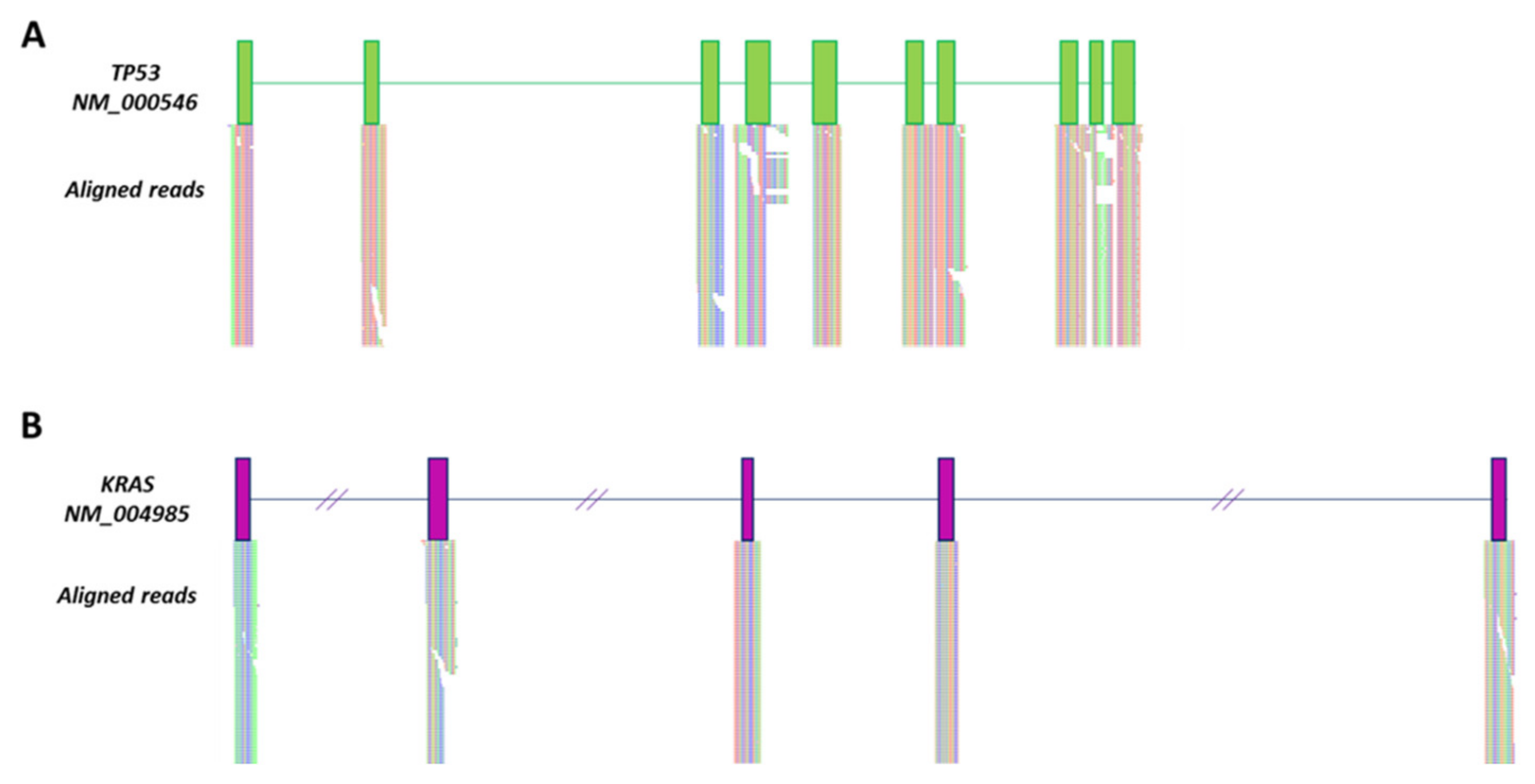
2.1. Detection of TP53 c.818G > A and KRAS c.35G > T Mutations in Exosomal DNA of SW480 Cells
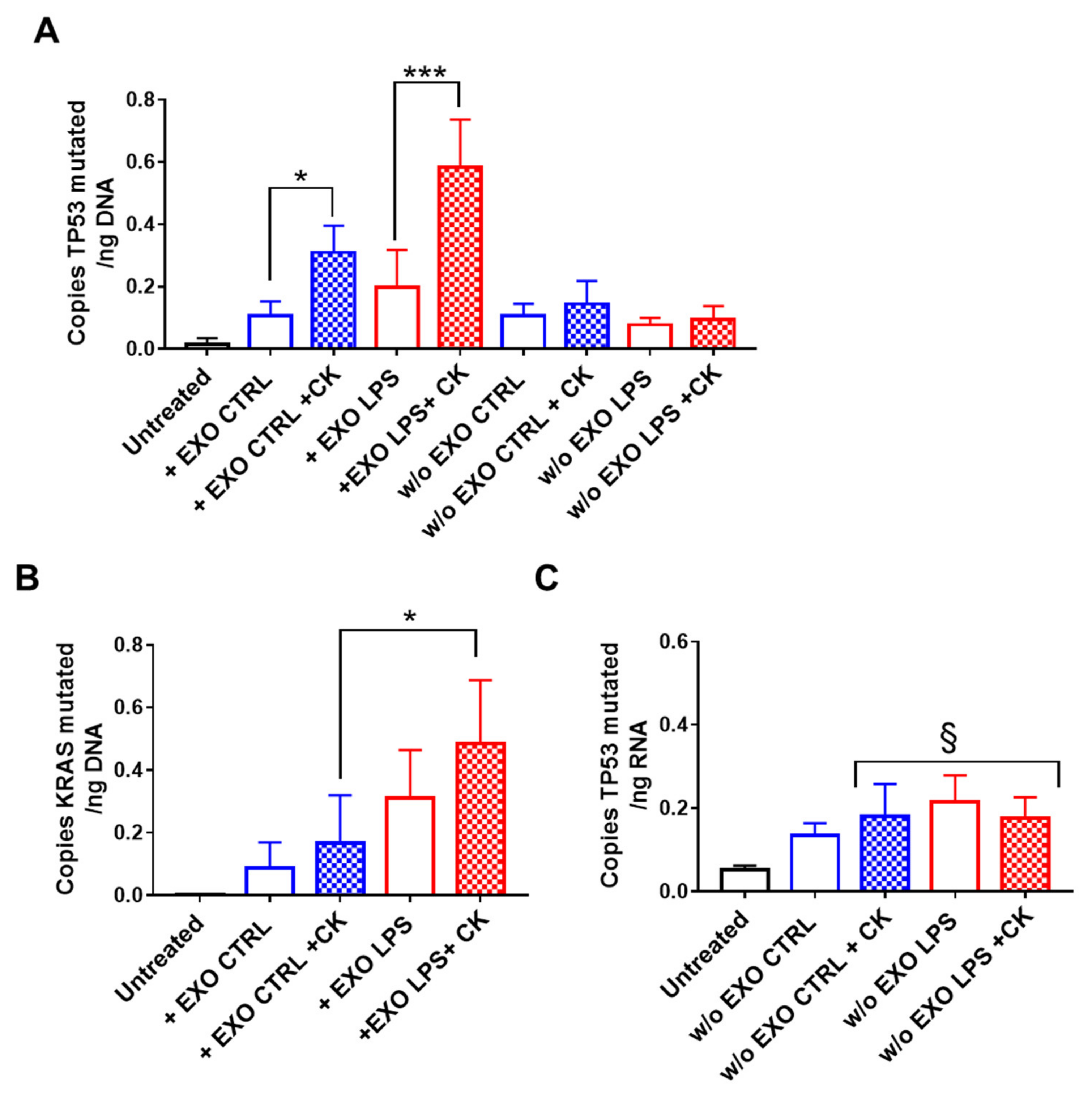
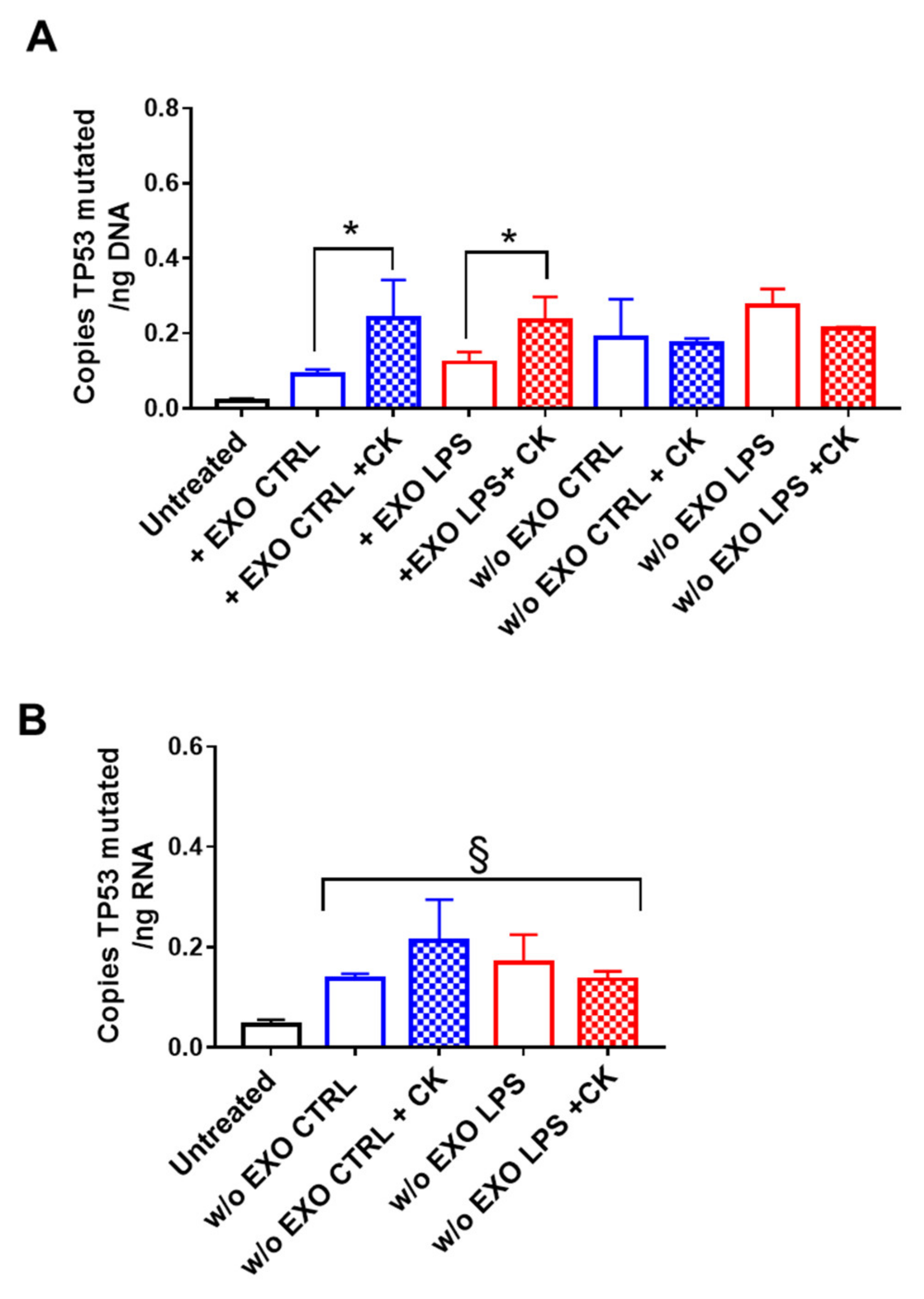
2.2. Pro-Inflammatory Cytokines Potentiate Oncogenic DNA Transfer by Exosomes in Normal Colon and Liver Epithelial Cells
3. Discussion
4. Materials and Methods
4.1. Culture and Treatment of Cell Lines
4.2. Exosomal DNA Extraction and Analysis
4.3. Library Preparation and Next-Generation Sequencing
4.4. Data Analysis and Variant Prioritization
4.5. Droplet Digital PCR (ddPCR)
4.6. Exosomes Transfer Assay
4.7. Statistical Methods
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jan, A.T.; Rahman, S.; Khan, S.; Tasduq, S.A.; Choi, I. Biology, Pathophysiological Role, and Clinical Implications of Exosomes: A Critical Appraisal. Cells 2019, 8, 99. [Google Scholar] [CrossRef] [Green Version]
- Wei, H.; Chen, Q.; Lin, L.; Sha, C.; Li, T.; Liu, Y.; Yin, X.; Xu, Y.; Chen, L.; Gao, W.; et al. Regulation of exosome production and cargo sorting. Int. J. Biol. Sci. 2021, 17, 163–177. [Google Scholar] [CrossRef]
- Kahlert, C.; Melo, S.; Protopopov, A.; Tang, J.; Seth, S.; Koch, M.; Zhang, J.; Weitz, J.; Chin, L.; Futreal, A.; et al. Identification of Double-stranded Genomic DNA Spanning All Chromosomes with Mutated KRAS and p53 DNA in the Serum Exosomes of Patients with Pancreatic Cancer. J. Biol. Chem. 2014, 289, 3869–3875. [Google Scholar] [CrossRef] [Green Version]
- Fernando, M.R.; Jiang, C.; Krzyzanowski, G.D.; Ryan, W.L. New evidence that a large proportion of human blood plasma cell-free DNA is localized in exosomes. PLoS ONE 2017, 12, e0183915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, A.; Okada, R.; Nagao, K.; Kawamata, Y.; Hanyu, A.; Yoshimoto, S.; Takasugi, M.; Watanabe, S.; Kanemaki, M.T.; Obuse, C.; et al. Exosomes maintain cellular homeostasis by excreting harmful DNA from cells. Nat. Commun. 2017, 8, 15287. [Google Scholar] [CrossRef] [Green Version]
- Yokoi, A.; Villar-Prados, A.; Oliphint, P.A.; Zhang, J.; Song, X.; De Hoff, P.; Morey, R.; Liu, J.; Roszik, J.; Clise-Dwyer, K.; et al. Mechanisms of nuclear content loading to exosomes. Sci. Adv. 2019, 5, eaax8849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elzanowska, J.; Semira, C.; Costa-Silva, B. DNA in extracellular vesicles: Biological and clinical aspects. Mol. Oncol. 2020, 15, 1701–1714. [Google Scholar] [CrossRef]
- Thakur, B.K.; Zhang, H.; Becker, A.; Matei, I.; Huang, Y.; Costa-Silva, B.; Zheng, Y.; Hoshino, A.; Brazier, H.; Xiang, J.; et al. Double-stranded DNA in exosomes: A novel biomarker in cancer detection. Cell Res. 2014, 24, 766–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A.; Johnson, A. Exosome DNA: Critical regulator of tumor immunity and a diagnostic biomarker. J. Cell. Physiol. 2020, 235, 1921–1932. [Google Scholar] [CrossRef]
- Kraus, T.F.J. Emergence of exosomal DNA in molecular neuropathology. Lab. Med. 2018, 42, 9–22. [Google Scholar] [CrossRef] [Green Version]
- Balaj, L.; Lessard, R.; Dai, L.; Cho, Y.-J.; Pomeroy, S.L.; Breakefield, X.O.; Skog, J. Tumour microvesicles contain retrotransposon elements and amplified oncogene sequences. Nat. Commun. 2011, 2, 180. [Google Scholar] [CrossRef]
- Lázaro-Ibáñez, E.; Sanz-Garcia, A.; Visakorpi, T.; Escobedo-Lucea, C.; Siljander, P.; Ayuso-Sacido, Á.; Yliperttula, M. Different gDNA content in the subpopulations of prostate cancer extracellular vesicles: Apoptotic bodies, microvesicles, and exosomes. Prostate 2014, 74, 1379–1390. [Google Scholar] [CrossRef]
- Qu, X.; Li, Q.; Yang, J.; Zhao, H.; Wang, F.; Zhang, F.; Zhang, S.; Zhang, H.; Wang, R.; Wang, Q.; et al. Double-Stranded DNA in Exosomes of Malignant Pleural Effusions as a Novel DNA Source for EGFR Mutation Detection in Lung Adenocarcinoma. Front. Oncol. 2019, 9, 931. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, Y.; Guan, X.; Zhao, J.; Shen, L.; Liu, J. Exosomal double-stranded DNA as a biomarker for the diagnosis and preoperative assessment of pheochromocytoma and paraganglioma. Mol. Cancer 2018, 17, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, S.; Cornils, K.; Speiseder, T.; Badbaran, A.; Reimer, R.; Indenbirken, D.; Grundhoff, A.; Brunswig-Spickenheier, B.; Alawi, M.; Lange, C. Indication of Horizontal DNA Gene Transfer by Extracellular Vesicles. PLoS ONE 2016, 11, e0163665. [Google Scholar] [CrossRef] [Green Version]
- Sansone, P.; Savini, C.; Kurelac, I.; Chang, Q.; Amato, L.B.; Strillacci, A.; Stepanova, A.; Iommarini, L.; Mastroleo, C.; Daly, L.; et al. Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer. Proc. Natl. Acad. Sci. USA 2017, 114, E9066–E9075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, T.H.; Chennakrishnaiah, S.; Audemard, E.; Montermini, L.; Meehan, B.; Rak, J. Oncogenic ras-driven cancer cell vesiculation leads to emission of double-stranded DNA capable of interacting with target cells. Biochem. Biophys. Res. Commun. 2014, 451, 295–301. [Google Scholar] [CrossRef] [Green Version]
- Cai, J.; Wu, G.; Jose, P.A.; Zeng, C. Functional transferred DNA within extracellular vesicles. Exp. Cell Res. 2016, 349, 179–183. [Google Scholar] [CrossRef]
- Cai, J.; Han, Y.; Ren, H.; Chen, C.; He, D.; Zhou, L.; Eisner, G.M.; Asico, L.D.; Jose, P.A.; Zeng, C. Extracellular vesicle-mediated transfer of donor genomic DNA to recipient cells is a novel mechanism for genetic influence between cells. J. Mol. Cell Biol. 2013, 5, 227. [Google Scholar] [CrossRef] [Green Version]
- Wan, M.; Ning, B.; Spiegel, S.; Lyon, C.J.; Hu, T.Y. Tumor-derived exosomes (TDEs): How to avoid the sting in the tail. Med. Res. Rev. 2020, 40, 385–412. [Google Scholar] [CrossRef]
- Domenis, R.; Cifù, A.; Fabris, M.; Niazi, K.R.; Soon-Shiong, P.; Curcio, F. Tumor Exosomes Mediate the Horizontal Transfer of DNA Gene Mutation. FASEB J. 2020, 34. [Google Scholar] [CrossRef]
- Kothandan, V.K.; Kothandan, S.; Kim, D.H.; Byun, Y.; Lee, Y.-K.; Park, I.-K.; Hwang, S.R. Crosstalk between Stress Granules, Exosomes, Tumour Antigens, and Immune Cells: Significance for Cancer Immunity. Vaccines 2020, 8, 172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domenis, R.; Cifù, A.; Marinò, D.; Fabris, M.; Niazi, K.R.; Soon-Shiong, P.; Curcio, F. Toll-like Receptor-4 Activation Boosts the Immunosuppressive Properties of Tumor Cells-derived Exosomes. Sci. Rep. 2019, 9, 8457. [Google Scholar] [CrossRef] [PubMed]
- Osaki, M.; Okada, F. Exosomes and Their Role in Cancer Progression. Yonago Acta Med. 2019, 62, 182–190. [Google Scholar] [CrossRef] [Green Version]
- Cai, J.; Guan, W.; Tan, X.; Chen, C.; Li, L.; Wang, N.; Zou, X.; Zhou, F.; Wang, J.; Pei, F.; et al. SRY gene transferred by extracellular vesicles accelerates atherosclerosis by promotion of leucocyte adherence to endothelial cells. Clin. Sci. 2015, 129, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.H.; Chennakrishnaiah, S.; Meehan, B.; Montermini, L.; Garnier, D.; D’Asti, E.; Hou, W.; Magnus, N.; Gayden, T.; Jabado, N.; et al. Barriers to horizontal cell transformation by extracellular vesicles containing oncogenic H-ras. Oncotarget 2016, 7, 51991–52002. [Google Scholar] [CrossRef] [Green Version]
- Cai, J.; Wu, G.; Tan, X.; Han, Y.; Chen, C.; Li, C.; Wang, N.; Zou, X.; Chen, X.; Zhou, F.; et al. Transferred BCR/ABL DNA from K562 Extracellular Vesicles Causes Chronic Myeloid Leukemia in Immunodeficient Mice. PLoS ONE 2014, 9, e105200. [Google Scholar] [CrossRef] [Green Version]
- Bergsmedh, A.; Szeles, A.; Henriksson, M.; Bratt, A.; Folkman, M.J.; Spetz, A.-L.; Holmgren, L. Horizontal transfer of oncogenes by uptake of apoptotic bodies. Proc. Natl. Acad. Sci. USA 2001, 98, 6407–6411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luhtala, N.; Hunter, T. Failure to detect functional transfer of active K-Ras protein from extracellular vesicles into recipient cells in culture. PLoS ONE 2018, 13, e0203290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, X.; Fang, L.; Bai, J.; Wang, Z. Characteristics and Changes of DNA in Extracellular Vesicles. DNA Cell Biol. 2020, 39, 1486–1493. [Google Scholar] [CrossRef] [PubMed]
- Barıs, I.C.; Hacıoglu, S.; Turk, N.S.; Cetın, G.O.; Zencır, S.; Bagcı, G.; Caner, V. Expression and DNA methylation profiles of EZH2-target genes in plasma exosomes and matched primary tumor tissues of the patients with diffuse large B-cell lymphoma. Clin. Transl. Oncol. 2021, 23, 1152–1166. [Google Scholar] [CrossRef]
- Stobiecka, M.; Ratajczak, K.; Jakiela, S. Toward early cancer detection: Focus on biosensing systems and biosensors for an anti-apoptotic protein survivin and survivin mRNA. Biosens. Bioelectron. 2019, 137, 58–71. [Google Scholar] [CrossRef]
- Stefanius, K.; Servage, K.A.; Santos, M.D.S.; Gray, H.F.; Toombs, J.E.; Chimalapati, S.; Kim, M.S.; Malladi, V.S.; Brekken, R.A.; Orth, K. Human pancreatic cancer cell exosomes, but not human normal cell exosomes, act as an initiator in cell transformation. Elife 2019, 8, e40226. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Che, S.P.Y.; Kurywchak, P.; Tavormina, J.L.; Gansmo, L.B.; De Sampaio, P.C.; Tachezy, M.; Bockhorn, M.; Gebauer, F.; Haltom, A.R.; et al. Detection of mutant KRAS and TP53 DNA in circulating exosomes from healthy individuals and patients with pancreatic cancer. Cancer Biol. Ther. 2017, 18, 158–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Codrich, M.; Comelli, M.; Malfatti, M.C.; Mio, C.; Ayyildiz, D.; Zhang, C.; Kelley, M.R.; Terrosu, G.; Pucillo, C.E.M.; Tell, G. Inhibition of APE1-endonuclease activity affects cell metabolism in colon cancer cells via a p53-dependent pathway. DNA Repair 2019, 82, 102675. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Domenis, R.; Cifù, A.; Mio, C.; Fabris, M.; Curcio, F. Pro-Inflammatory Microenvironment Modulates the Transfer of Mutated TP53 Mediated by Tumor Exosomes. Int. J. Mol. Sci. 2021, 22, 6258. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22126258
Domenis R, Cifù A, Mio C, Fabris M, Curcio F. Pro-Inflammatory Microenvironment Modulates the Transfer of Mutated TP53 Mediated by Tumor Exosomes. International Journal of Molecular Sciences. 2021; 22(12):6258. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22126258
Chicago/Turabian StyleDomenis, Rossana, Adriana Cifù, Catia Mio, Martina Fabris, and Francesco Curcio. 2021. "Pro-Inflammatory Microenvironment Modulates the Transfer of Mutated TP53 Mediated by Tumor Exosomes" International Journal of Molecular Sciences 22, no. 12: 6258. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22126258