
The Adipose Tissue-Derived Secretome (ADS) in Obesity Uniquely Induces L-Type Amino Acid Transporter 1 (LAT1) and mTOR Signaling in Estrogen-Receptor-Positive Breast Cancer Cells

 , and
, and
Abstract
:1. Introduction
2. Results
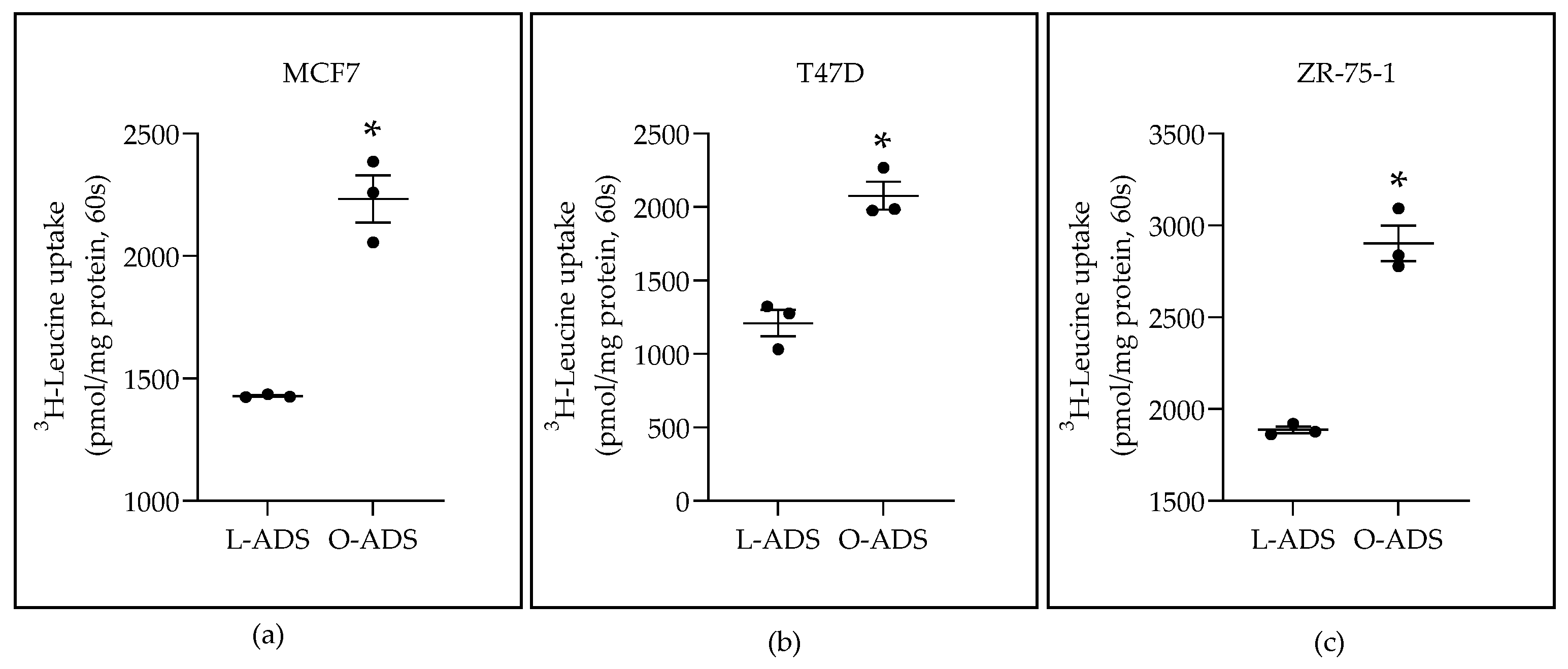
2.1. Obese-ADS Stimulates LAT1 Activity
2.2. Obese-ADS Increases LAT1 Affinity for Leucine
2.3. LAT1 Mediates Obese-ADS Induction of mTOR
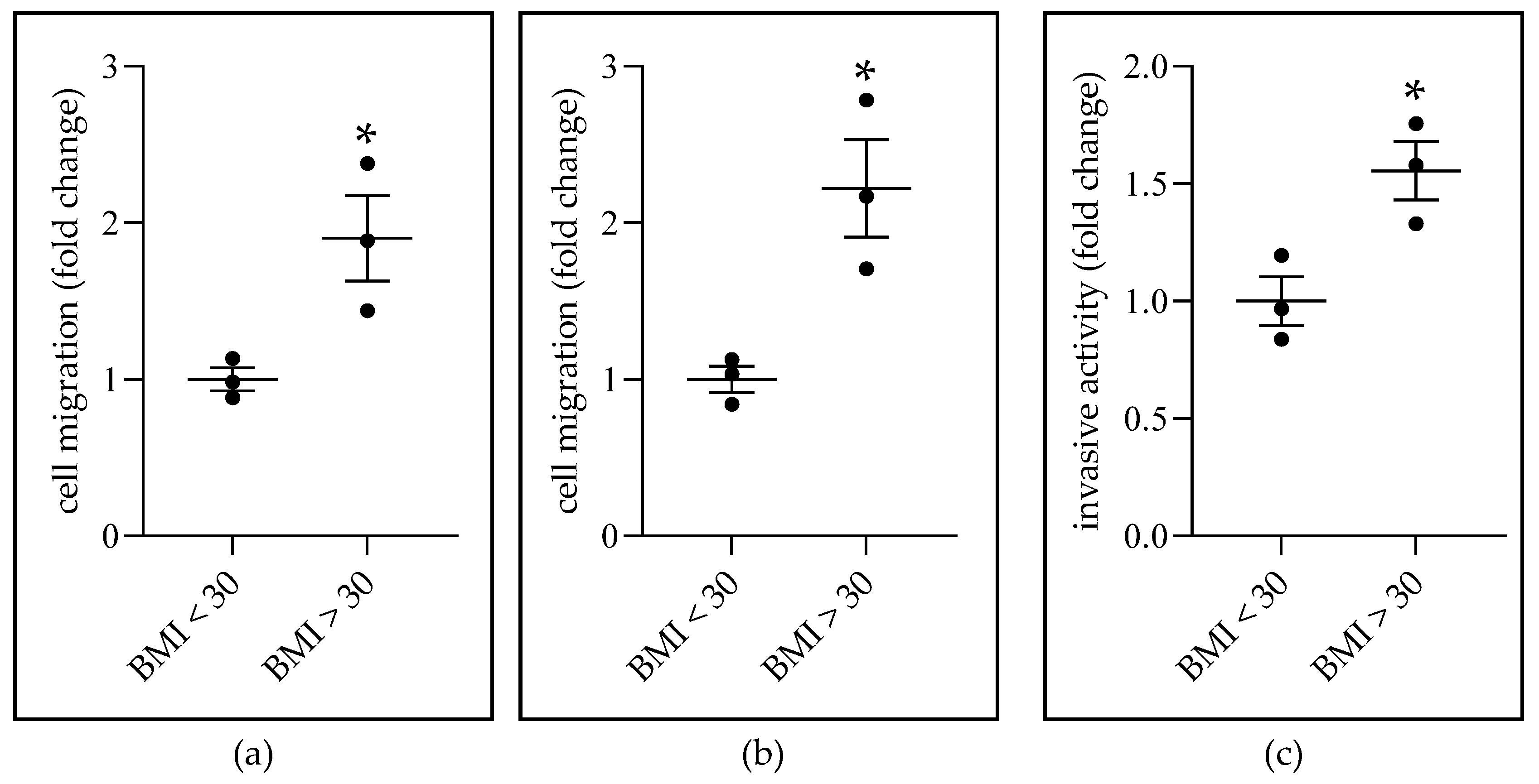
2.4. Obese-ADS Stimulates BC Cell Migration and Invasiveness
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Mouse Obesity Model
4.3. Human Samples
4.4. AT-Derived Secretome (ADS) System
4.5. Western Blotting Studies
4.6. Leucine Uptake Study
4.7. Kinetics Study for Leucine Uptake
4.8. Migration Assay
4.9. Invasion Assay
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Flegal, K.M.; Kruszon-Moran, D.; Carroll, M.D.; Fryar, C.D.; Ogden, C.L. Trends in obesity among adults in the United States, 2005 to 2014. JAMA 2016, 315, 2284–2291. [Google Scholar] [CrossRef] [Green Version]
- Hales, C.M.; Carroll, M.D.; Fryar, C.D.; Ogden, C.L. Prevalence of obesity among adults and youth: United States, 2015–2016. NCHS Data Brief. 2017, 288, 1–8. [Google Scholar]
- Neuhouser, M.L.; Aragaki, A.K.; Prentice, R.L.; Manson, J.E.; Chlebowski, R.; Carty, C.L.; Ochs-Balcom, H.M.; Thomson, C.A.; Caan, B.J.; Tinker, L.F.; et al. Overweight, obesity, and postmenopausal invasive breast cancer risk: A secondary analysis of the women’s health initiative randomized clinical trials. JAMA Oncol. 2015, 1, 611–621. [Google Scholar] [CrossRef]
- Suzuki, R.; Orsini, N.; Saji, S.; Key, T.J.; Wolk, A. Body weight and incidence of breast cancer defined by estrogen and progesterone receptor status—A meta-analysis. Int. J. Cancer 2009, 124, 698–712. [Google Scholar] [CrossRef]
- Yang, X.R.; Chang-Claude, J.; Goode, E.L.; Couch, F.J.; Nevanlinna, H.; Milne, R.L.; Gaudet, M.; Schmidt, M.K.; Broeks, A.; Cox, A.; et al. Associations of breast cancer risk factors with tumor subtypes: A pooled analysis from the breast cancer association consortium studies. J. Natl. Cancer Inst. 2011, 103, 250–263. [Google Scholar] [CrossRef]
- Calle, E.E.; Rodriguez, C.; Walker-Thurmond, K.; Thun, M.J. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N. Engl. J. Med. 2003, 348, 1625–1638. [Google Scholar] [CrossRef] [Green Version]
- Ewertz, M.; Jensen, M.B.; Gunnarsdóttir, K.; Højris, I.; Jakobsen, E.H.; Nielsen, D.; Stenbygaard, L.E.; Tange, U.B.; Cold, S. Effect of obesity on prognosis after early-stage breast cancer. J. Clin. Oncol. 2011, 29, 25–31. [Google Scholar] [CrossRef]
- Goodman, N.F.; Cobin, R.H.; Ginzburg, S.B.; Katz, I.A.; Woode, D.E. American association of clinical endocrinologists medical guidelines for clinical practice for the diagnosis and treatment of menopause: Executive summary of recommendations. Endocr. Pract. 2011, 17, 949–954. [Google Scholar] [CrossRef]
- Funcke, J.B.; Scherer, P.E. Beyond adiponectin and leptin: Adipose tissue-derived mediators of inter-organ communication. J. Lipid Res. 2019, 60, 1648–1684. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Morley, T.S.; Kim, M.; Clegg, D.J.; Scherer, P.E. Obesity and cancer--mechanisms underlying tumour progression and recurrence. Nat. Rev. Endocrinol. 2014, 10, 455–465. [Google Scholar] [CrossRef] [Green Version]
- Lengyel, E.; Makowski, L.; DiGiovanni, J.; Kolonin, M.G. Cancer as a Matter of fat: The crosstalk between adipose tissue and tumors. Trends Cancer 2018, 4, 374–384. [Google Scholar] [CrossRef]
- Iyengar, N.M.; Gucalp, A.; Dannenberg, A.J.; Hudis, C.A. Obesity and cancer mechanisms: Tumor microenvironment and inflammation. J. Clin. Oncol. 2016, 34, 4270–4276. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR signaling in growth, metabolism, and disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [Green Version]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef] [Green Version]
- Bracho-Valdés, I.; Moreno-Alvarez, P.; Valencia-Martínez, I.; Robles-Molina, E.; Chávez-Vargas, L.; Vázquez-Prado, J. mTORC1- and mTORC2-interacting proteins keep their multifunctional partners focused. IUBMB Life 2011, 63, 896–914. [Google Scholar] [CrossRef] [Green Version]
- Jhanwar-Uniyal, M.; Amin, A.G.; Cooper, J.B.; Das, K.; Schmidt, M.H.; Murali, R. Discrete signaling mechanisms of mTORC1 and mTORC2: Connected yet apart in cellular and molecular aspects. Adv. Biol. Regul. 2017, 64, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. mTOR signaling. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef]
- Wolfson, R.L.; Sabatini, D.M. The dawn of the age of amino acid sensors for the mTORC1 pathway. Cell Metab. 2017, 26, 301–309. [Google Scholar] [CrossRef] [Green Version]
- Sengupta, S.; Peterson, T.R.; Sabatini, D.M. Regulation of the mTOR complex 1 pathway by nutrients, growth factors, and stress. Mol. Cell 2010, 40, 310–322. [Google Scholar] [CrossRef] [Green Version]
- Jewell, J.L.; Russell, R.C.; Guan, K.L. Amino acid signalling upstream of mTOR. Nat. Rev. Mol. Cell Biol. 2013, 14, 133–139. [Google Scholar] [CrossRef] [Green Version]
- Melick, C.H.; Jewell, J.L. Regulation of mTORC1 by upstream stimuli. Genes 2020, 11, 989. [Google Scholar] [CrossRef] [PubMed]
- Jewell, J.L.; Kim, Y.C.; Russell, R.C.; Yu, F.X.; Park, H.W.; Plouffe, S.W.; Tagliabracci, V.S.; Guan, K.L. Differential regulation of mTORC1 by leucine and glutamine. Science 2015, 347, 194–198. [Google Scholar] [CrossRef] [Green Version]
- Lamming, D.W.; Sabatini, D.M. A central role for mTOR in lipid homeostasis. Cell Metab. 2013, 18, 465–469. [Google Scholar] [CrossRef] [Green Version]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [Green Version]
- Afshin, A.; Forouzanfar, M.H.; Reitsma, M.B.; Sur, P.; Estep, K.; Lee, A.; Marczak, L.; Mokdad, A.H.; Moradi-Lakeh, M.; Naghavi, M.; et al. Health effects of overweight and obesity in 195 countries over 25 years. N. Engl. J. Med. 2017, 377, 13–27. [Google Scholar] [CrossRef]
- Um, S.H.; Frigerio, F.; Watanabe, M.; Picard, F.; Joaquin, M.; Sticker, M.; Fumagalli, S.; Allegrini, P.R.; Kozma, S.C.; Auwerx, J.; et al. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature 2004, 431, 200–205. [Google Scholar] [CrossRef]
- Newgard, C.B.; An, J.; Bain, J.R.; Muehlbauer, M.J.; Stevens, R.D.; Lien, L.F.; Haqq, A.M.; Shah, S.H.; Arlotto, M.; Slentz, C.A.; et al. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab. 2009, 9, 311–326. [Google Scholar] [CrossRef] [Green Version]
- Wolfson, R.L.; Chantranupong, L.; Saxton, R.A.; Shen, K.; Scaria, S.M.; Cantor, J.R.; Sabatini, D.M. Sestrin2 is a leucine sensor for the mTORC1 pathway. Science 2016, 351, 43–48. [Google Scholar] [CrossRef] [Green Version]
- Tomblin, J.K.; Arthur, S.; Primerano, D.A.; Chaudhry, A.R.; Fan, J.; Denvir, J.; Salisbury, T.B. Aryl hydrocarbon receptor (AHR) regulation of l-type amino acid transporter 1 (LAT-1) expression in MCF-7 and MDA-MB-231 breast cancer cells. Biochem. Pharmacol. 2016, 106, 94–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shennan, D.B.; Thomson, J.; Gow, I.F.; Travers, M.T.; Barber, M.C. L-leucine transport in human breast cancer cells (MCF-7 and MDA-MB-231): Kinetics, regulation by estrogen and molecular identity of the transporter. Biochim. Biophys. Acta 2004, 1664, 206–216. [Google Scholar] [CrossRef] [Green Version]
- Saito, Y.; Li, L.; Coyaud, E.; Luna, A.; Sander, C.; Raught, B.; Asara, J.M.; Brown, M.; Muthuswamy, S.K. LLGL2 rescues nutrient stress by promoting leucine uptake in ER(+) breast cancer. Nature 2019, 569, 275–279. [Google Scholar] [CrossRef] [PubMed]
- Prasad, P.D.; Wang, H.; Huang, W.; Kekuda, R.; Rajan, D.P.; Leibach, F.H.; Ganapathy, V. Human LAT1, a subunit of system L amino acid transporter: Molecular cloning and transport function. Biochem. Biophys. Res. Commun. 1999, 255, 283–288. [Google Scholar] [CrossRef] [Green Version]
- Mastroberardino, L.; Spindler, B.; Pfeiffer, R.; Skelly, P.J.; Loffing, J.; Shoemaker, C.B.; Verrey, F. Amino-acid transport by heterodimers of 4F2hc/CD98 and members of a permease family. Nature 1998, 395, 288–291. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, R.; Rossier, G.; Spindler, B.; Meier, C.; Kühn, L.; Verrey, F. Amino acid transport of y+L-type by heterodimers of 4F2hc/CD98 and members of the glycoprotein-associated amino acid transporter family. EMBO J. 1999, 18, 49–57. [Google Scholar] [CrossRef] [Green Version]
- Yan, R.; Zhao, X.; Lei, J.; Zhou, Q. Structure of the human LAT1-4F2hc heteromeric amino acid transporter complex. Nature 2019, 568, 127–130. [Google Scholar] [CrossRef] [PubMed]
- Salisbury, T.B.; Arthur, S. The regulation and function of the l-type amino acid transporter 1 (LAT1) in cancer. Int. J. Mol. Sci. 2018, 19, 2373. [Google Scholar] [CrossRef] [Green Version]
- Ackermann, T.; Tardito, S. Cell culture medium formulation and its implications in cancer metabolism. Trends Cancer 2019, 5, 329–332. [Google Scholar] [CrossRef]
- Wang, Q.; Tiffen, J.; Bailey, C.G.; Lehman, M.L.; Ritchie, W.; Fazli, L.; Metierre, C.; Feng, Y.J.; Li, E.; Gleave, M.; et al. Targeting amino acid transport in metastatic castration-resistant prostate cancer: Effects on cell cycle, cell growth, and tumor development. J. Natl. Cancer Inst. 2013, 105, 1463–1473. [Google Scholar] [CrossRef]
- Oda, K.; Hosoda, N.; Endo, H.; Saito, K.; Tsujihara, K.; Yamamura, M.; Sakata, T.; Anzai, N.; Wempe, M.F.; Kanai, Y.; et al. L-type amino acid transporter 1 inhibitors inhibit tumor cell growth. Cancer Sci. 2010, 101, 173–179. [Google Scholar] [CrossRef]
- Madak-Erdogan, Z.; Band, S.; Zhao, Y.C.; Smith, B.P.; Kulkoyluoglu-Cotul, E.; Zuo, Q.; Santaliz Casiano, A.; Wrobel, K.; Rossi, G.; Smith, R.L.; et al. Free fatty acids rewire cancer metabolism in obesity-associated breast cancer via estrogen receptor and mTOR signaling. Cancer Res. 2019, 79, 2494–2510. [Google Scholar] [CrossRef] [Green Version]
- Fuentes-Mattei, E.; Velazquez-Torres, G.; Phan, L.; Zhang, F.; Chou, P.C.; Shin, J.H.; Choi, H.H.; Chen, J.S.; Zhao, R.; Chen, J.; et al. Effects of obesity on transcriptomic changes and cancer hallmarks in estrogen receptor-positive breast cancer. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef]
- Cheng, T.D.; Omilian, A.R.; Yao, S.; Sanchez, P.V.; Polk, L.Z.; Zhang, W.; Datta, S.; Bshara, W.; Ondracek, R.P.; Davis, W.; et al. Body fatness and mTOR pathway activation of breast cancer in the women’s circle of health study. NPJ Breast Cancer 2020, 6, 45. [Google Scholar] [CrossRef]
- Mihály, Z.; Kormos, M.; Lánczky, A.; Dank, M.; Budczies, J.; Szász, M.A.; Győrffy, B. A meta-analysis of gene expression-based biomarkers predicting outcome after tamoxifen treatment in breast cancer. Breast Cancer Res. Treat. 2013, 140, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Häfliger, P.; Charles, R.P. The l-type amino acid transporter LAT1-an emerging target in cancer. Int. J. Mol. Sci. 2019, 20, 2428. [Google Scholar] [CrossRef] [Green Version]
- Cormerais, Y.; Giuliano, S.; LeFloch, R.; Front, B.; Durivault, J.; Tambutté, E.; Massard, P.A.; de la Ballina, L.R.; Endou, H.; Wempe, M.F.; et al. Genetic disruption of the multifunctional CD98/LAT1 complex demonstrates the key role of essential amino acid transport in the control of mTORC1 and tumor growth. Cancer Res. 2016, 76, 4481–4492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okano, N.; Naruge, D.; Kawai, K.; Kobayashi, T.; Nagashima, F.; Endou, H.; Furuse, J. First-in-human phase I study of JPH203, an l-type amino acid transporter 1 inhibitor, in patients with advanced solid tumors. Investig. New Drugs 2020, 38, 1495–1506. [Google Scholar] [CrossRef] [PubMed]
- Häfliger, P.; Graff, J.; Rubin, M.; Stooss, A.; Dettmer, M.S.; Altmann, K.H.; Gertsch, J.; Charles, R.P. The LAT1 inhibitor JPH203 reduces growth of thyroid carcinoma in a fully immunocompetent mouse model. J. Exp. Clin. Cancer Res. 2018, 37, 234. [Google Scholar] [CrossRef]
- Hillers, L.E.; D’Amato, J.V.; Chamberlin, T.; Paderta, G.; Arendt, L.M. Obesity-activated adipose-derived stromal cells promote breast cancer growth and invasion. Neoplasia 2018, 20, 1161–1174. [Google Scholar] [CrossRef]
- Hornbeck, P.V.; Kornhauser, J.M.; Latham, V.; Murray, B.; Nandhikonda, V.; Nord, A.; Skrzypek, E.; Wheeler, T.; Zhang, B.; Gnad, F. 15 years of phosphositeplus®: Integrating post-translationally modified sites, disease variants and isoforms. Nucleic Acids Res. 2019, 47, D433–D441. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Kinetic Parameters | L-ADS | O-ADS |
|---|---|---|
| Vmax (pmol/mg protein.30 s) | 15.55 | 15.87 |
| Km (µM) | 163.6 ± 6 | 82 ± 4 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thompson, C.; Rahman, M.M.; Singh, S.; Arthur, S.; Sierra-Bakhshi, C.; Russell, R.; Denning, K.; Sundaram, U.; Salisbury, T. The Adipose Tissue-Derived Secretome (ADS) in Obesity Uniquely Induces L-Type Amino Acid Transporter 1 (LAT1) and mTOR Signaling in Estrogen-Receptor-Positive Breast Cancer Cells. Int. J. Mol. Sci. 2021, 22, 6706. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22136706
Thompson C, Rahman MM, Singh S, Arthur S, Sierra-Bakhshi C, Russell R, Denning K, Sundaram U, Salisbury T. The Adipose Tissue-Derived Secretome (ADS) in Obesity Uniquely Induces L-Type Amino Acid Transporter 1 (LAT1) and mTOR Signaling in Estrogen-Receptor-Positive Breast Cancer Cells. International Journal of Molecular Sciences. 2021; 22(13):6706. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22136706
Chicago/Turabian StyleThompson, Chelsea, M Motiur Rahman, Soudamani Singh, Subha Arthur, Cecilia Sierra-Bakhshi, Rebecca Russell, Krista Denning, Uma Sundaram, and Travis Salisbury. 2021. "The Adipose Tissue-Derived Secretome (ADS) in Obesity Uniquely Induces L-Type Amino Acid Transporter 1 (LAT1) and mTOR Signaling in Estrogen-Receptor-Positive Breast Cancer Cells" International Journal of Molecular Sciences 22, no. 13: 6706. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22136706