
S-Equol Protects Chondrocytes against Sodium Nitroprusside-Caused Matrix Loss and Apoptosis through Activating PI3K/Akt Pathway

 , ,
, ,
Abstract
:1. Introduction
2. Results
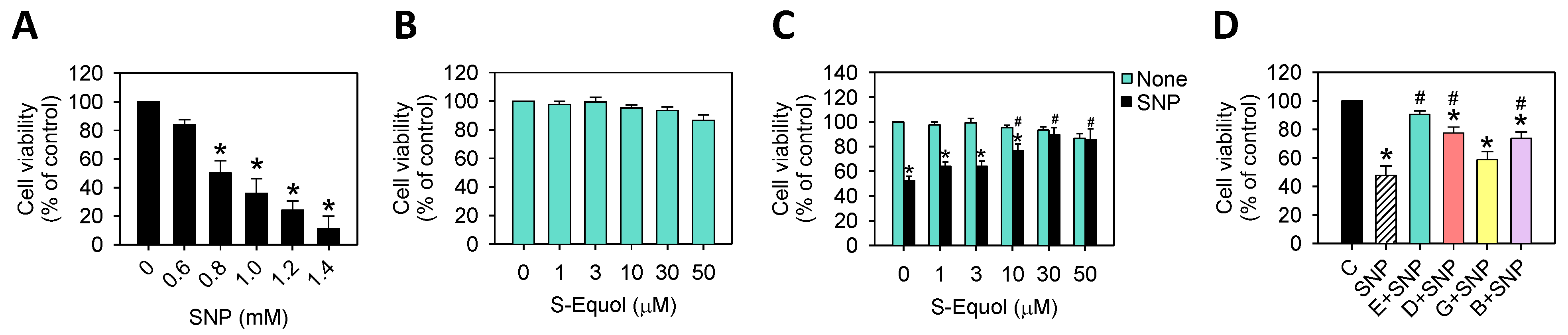
2.1. S-Equol Reduces SNP-Induced Cell Death
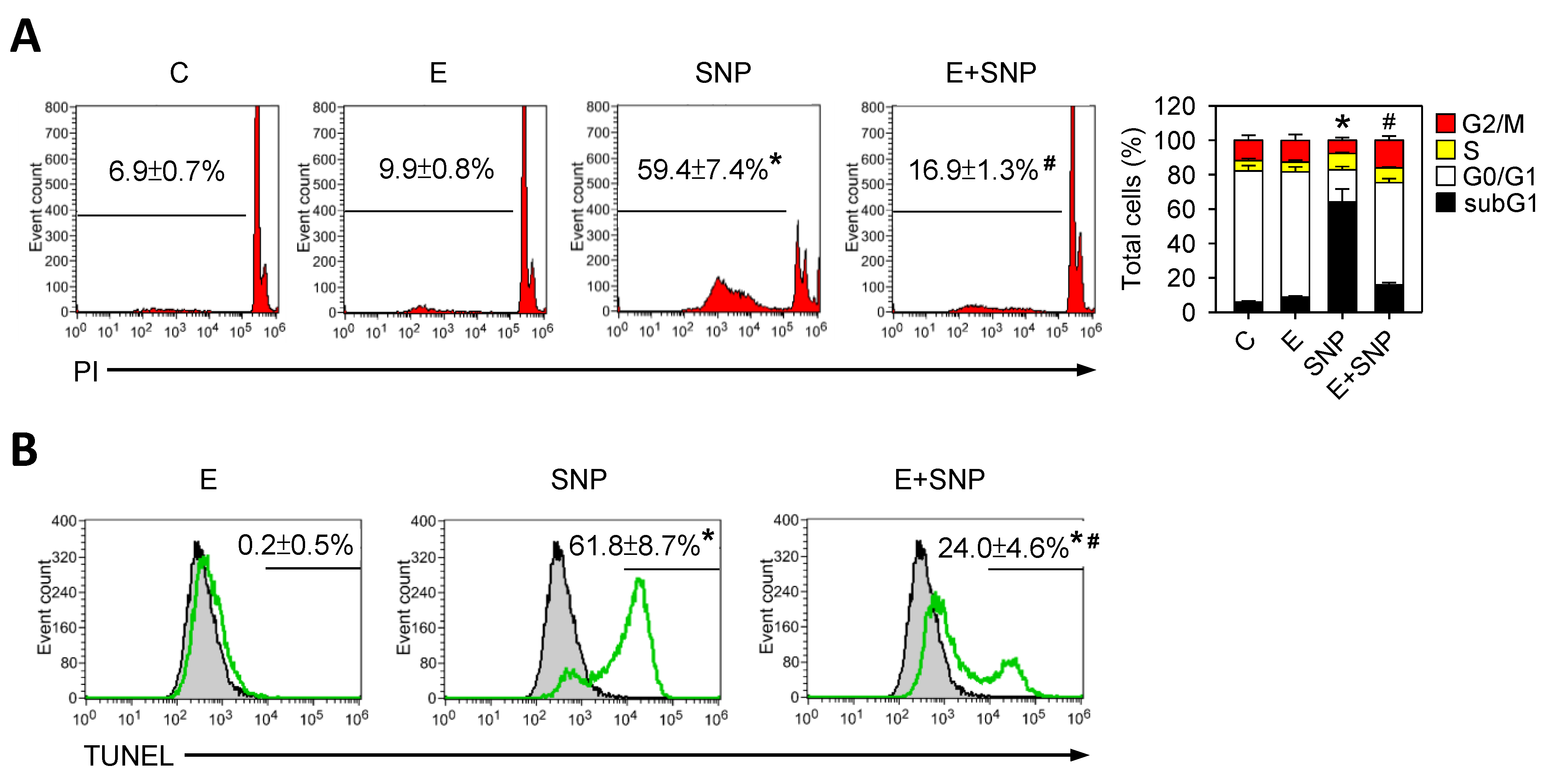
2.2. S-Equol Decreases SNP-Induced Apoptosis
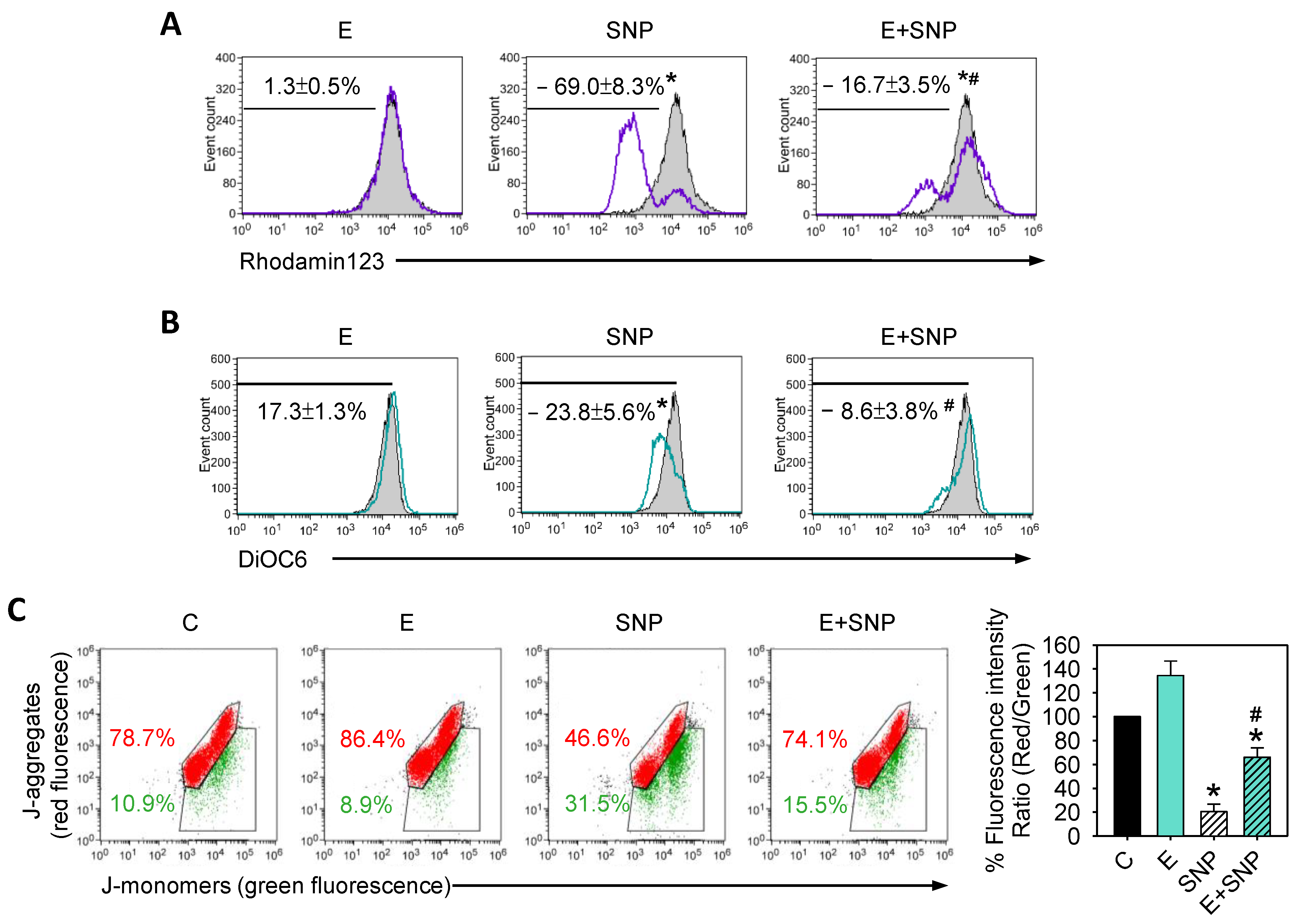
2.3. S-Equol Diminishes the SNP-Caused Decrease of Mitochondrial Membrane Potential (ΔΨM)
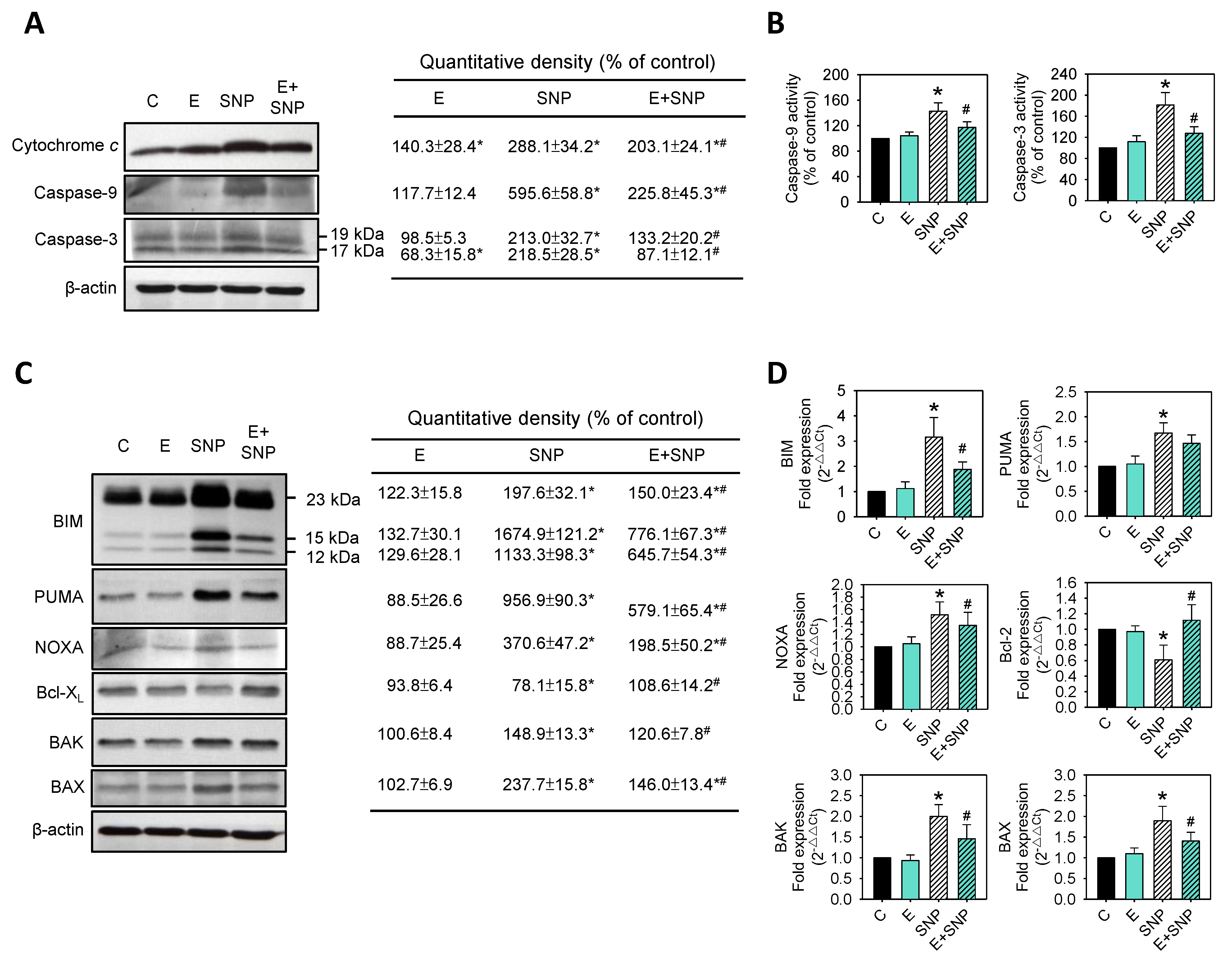
2.4. Effect of SNP/S-Equol on Apoptosis-Related Proteins Expressions
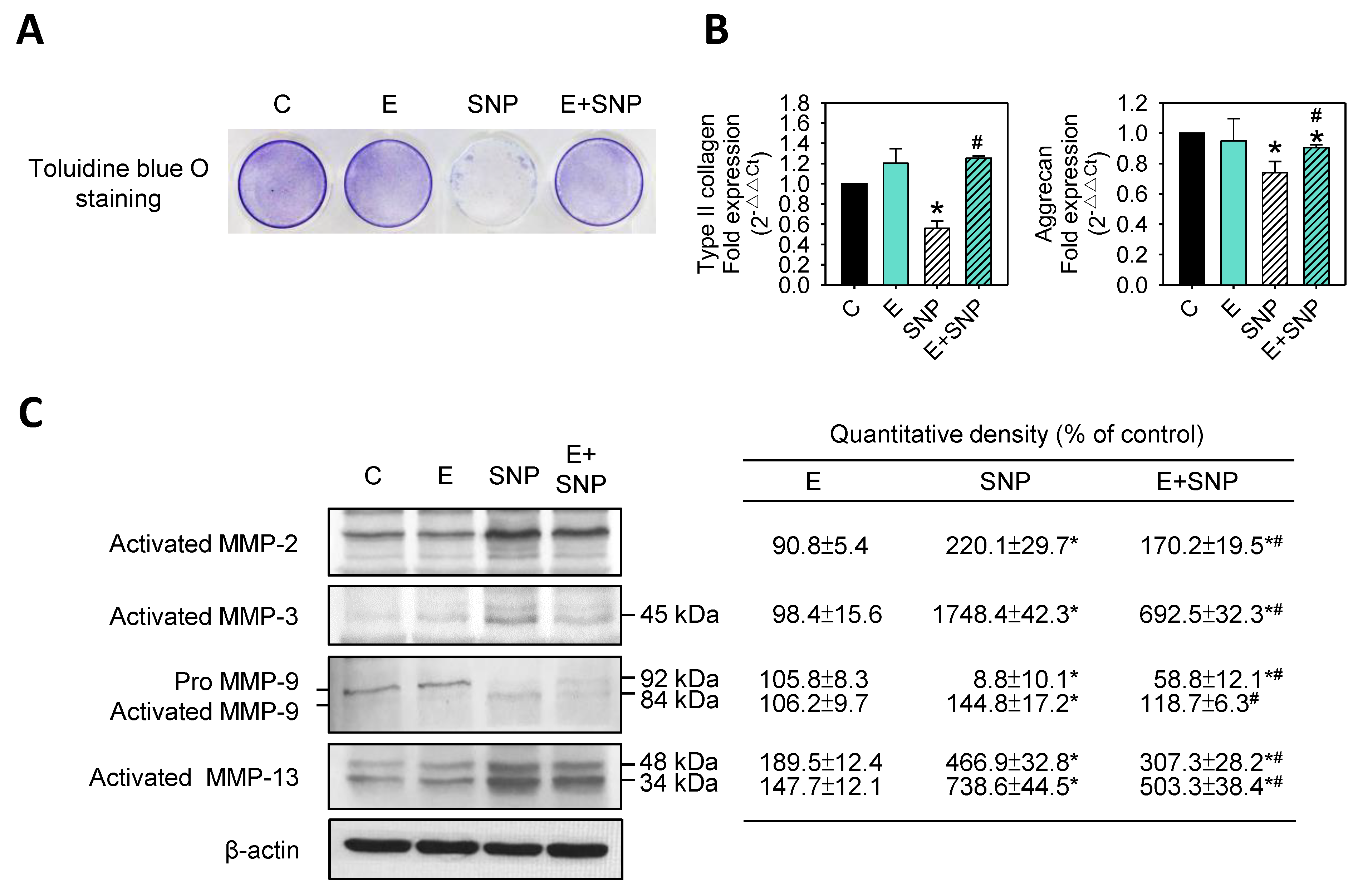
2.5. S-Equol Inhibits SNP-Induced Matrix Degradation
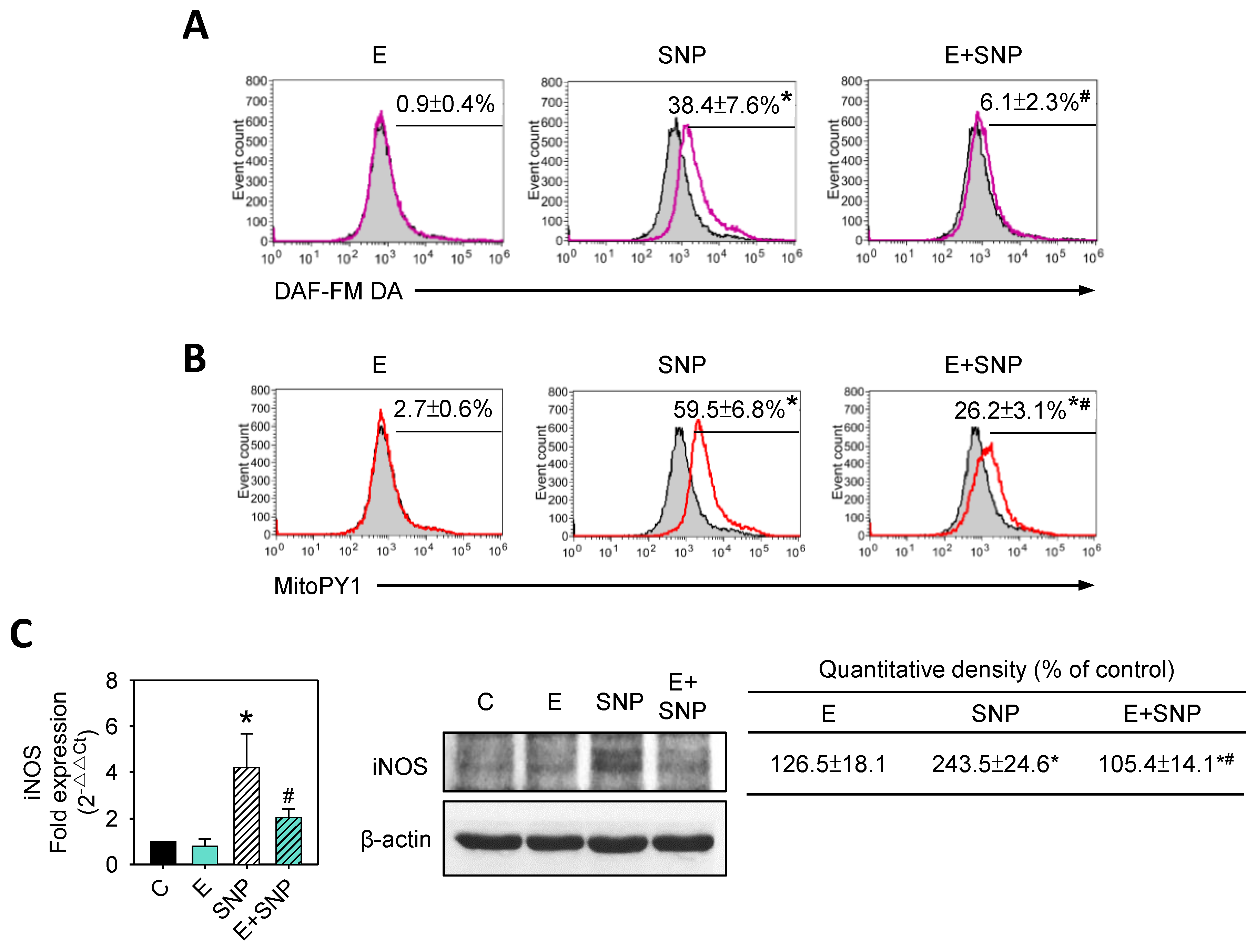
2.6. S-Equol Reduces SNP-Induced NO and H2O2 Production
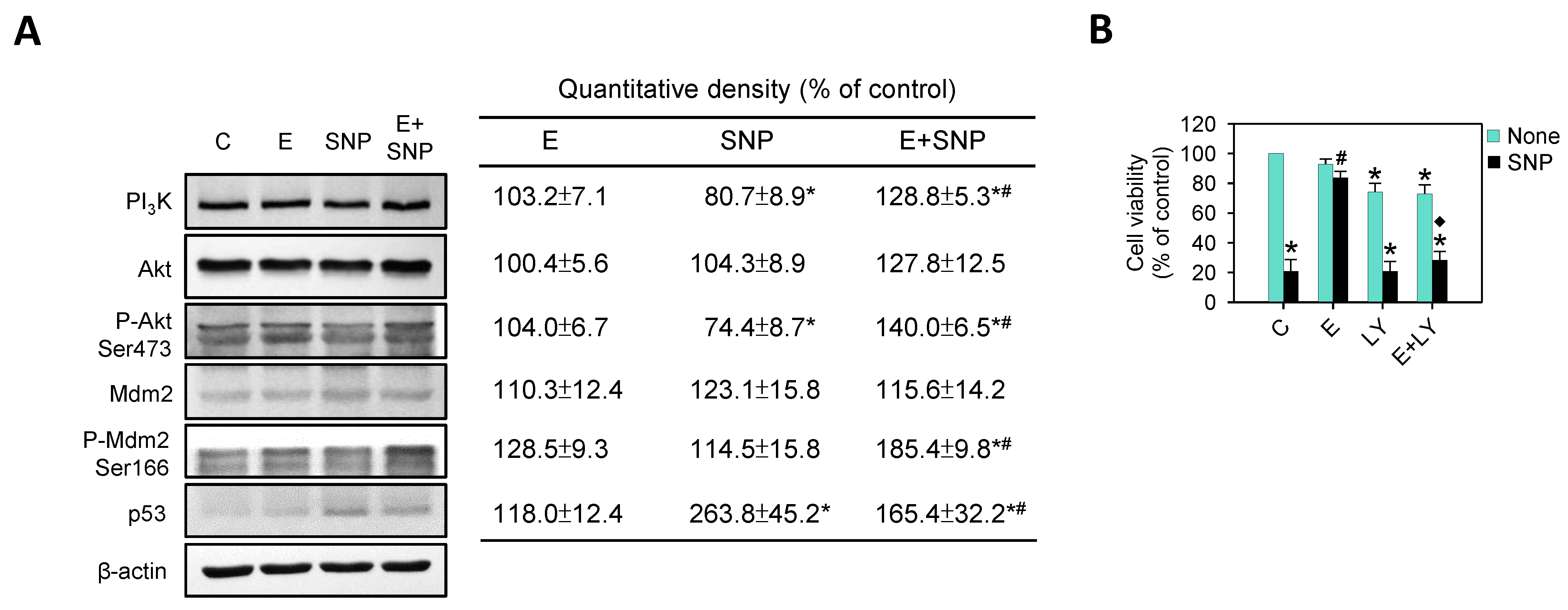
2.7. The Effect of SNP/S-Equol on PI3K-Akt Pathway
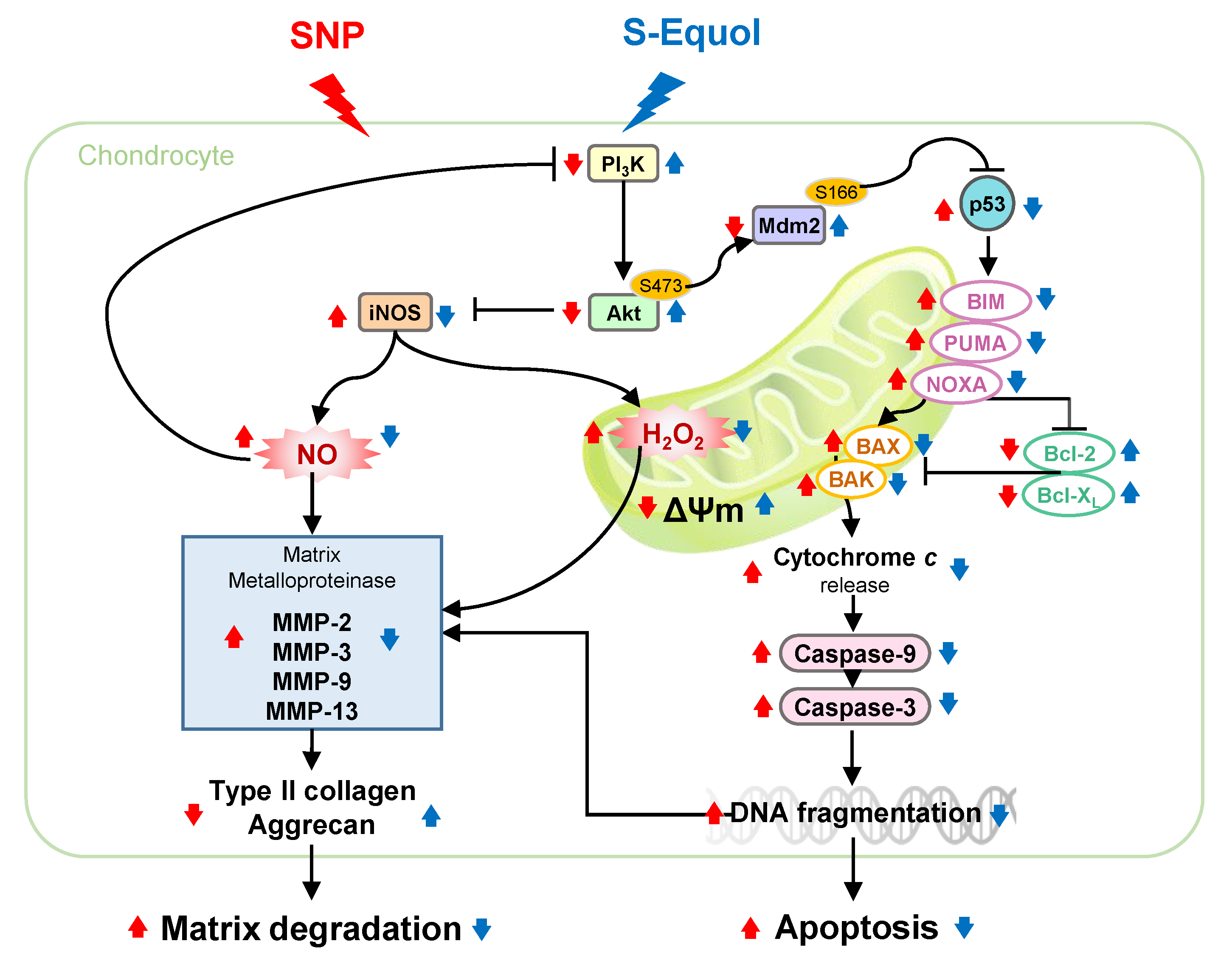
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Isolation and Expansion of Rat Primary Chondrocytes
4.3. Cell Culture
4.4. Cell Viability Assay
4.5. Effects PI3K Inhibitors on Cell Viability
4.6. Toluidine Blue O Staining
4.7. Measurement of NO and H2O2 Levels
4.8. Western Blot Analysis
4.9. Quantitative Real-Time PCR Analysis (qPCR)
4.10. TUNEL Assay
4.11. Cell Cycle Progression Analysis
4.12. Detection of Mitochondrial Membrane Potential (ΔΨM)
4.13. Measurement of Caspase-9 and Caspase-3 Activities
4.14. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Spencer, L.J.; Christopher, J.L.M. GBD 2017 Disease and Injury Incidence and Prevalence Collaborators, Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1789–1858. [Google Scholar]
- Prieto-Alhambra, D.; Judge, A.; Javaid, M.K.; Cooper, C.; Diez-Perez, A.; Arden, N.K. Incidence and risk factors for clinically diagnosed knee, hip and hand osteoarthritis: Influences of age, gender and osteoarthritis affecting other joints. Ann. Rheum. Dis. 2014, 73, 1659–1664. [Google Scholar] [CrossRef] [Green Version]
- Brooks, P.M. The burden of musculoskeletal disease—A global perspective. Clin. Rheumatol. 2006, 25, 778–781. [Google Scholar] [CrossRef]
- Stevens-Lapsley, J.E.; Kohrt, W.M. Osteoarthritis in women: Effects of estrogen, obesity and physical activity. Womens Health 2010, 6, 601–615. [Google Scholar] [CrossRef]
- Kalson, N.S.; Gikas, P.D.; Briggs, T.W. Current strategies for knee cartilage repair. Int. J. Clin. Pract. 2010, 64, 1444–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, H.S.; Kim, H.A. Chondrocyte Apoptosis in the Pathogenesis of Osteoarthritis. Int. J. Mol. Sci. 2015, 16, 26035–26054. [Google Scholar] [CrossRef] [PubMed]
- Park, D.R.; Kim, J.; Kim, G.M.; Lee, H.; Kim, M.; Hwang, D.; Lee, H.; Kim, H.S.; Kim, W.; Park, M.C.; et al. Osteoclast-associated receptor blockade prevents articular cartilage destruction via chondrocyte apoptosis regulation. Nat. Commun. 2020, 11, 4343. [Google Scholar] [CrossRef]
- Chiu, P.R.; Hu, Y.C.; Huang, T.C.; Hsieh, B.S.; Yeh, J.P.; Cheng, H.L.; Huang, L.W.; Chang, K.L. Vitamin C Protects Chondrocytes against Monosodium Iodoacetate-Induced Osteoarthritis by Multiple Pathways. Int. J. Mol. Sci. 2016, 18, 38. [Google Scholar] [CrossRef] [Green Version]
- Huang, T.C.; Chang, W.T.; Hu, Y.C.; Hsieh, B.S.; Cheng, H.L.; Yen, J.H.; Chiu, P.R.; Chang, K.L. Zinc Protects Articular Chondrocytes through Changes in Nrf2-Mediated Antioxidants, Cytokines and Matrix Metalloproteinases. Nutrients 2018, 10, 471. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.W.; Huang, T.C.; Hu, Y.C.; Hsieh, B.S.; Chiu, P.R.; Cheng, H.L.; Chang, K.L. Zinc protects chondrocytes from monosodium iodoacetate-induced damage by enhancing ATP and mitophagy. Biochem. Biophys Res. Commun. 2020, 521, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Bourgonje, A.R.; Abdulle, A.E.; Al-Rawas, A.M.; Al-Maqbali, M.; Al-Saleh, M.; Enriquez, M.B.; Al-Siyabi, S.; Al-Hashmi, K.; Al-Lawati, I.; Bulthuis, M.L.C.; et al. Systemic Oxidative Stress Is Increased in Postmenopausal Women and Independently Associates with Homocysteine Levels. Int. J. Mol. Sci. 2020, 21, 314. [Google Scholar] [CrossRef] [Green Version]
- Lepetsos, P.; Papavassiliou, A.G. ROS/oxidative stress signaling in osteoarthritis. Biochim. Biophys Acta 2016, 1862, 576–591. [Google Scholar] [CrossRef]
- Zahan, O.M.; Serban, O.; Gherman, C.; Fodor, D. The evaluation of oxidative stress in osteoarthritis. Med. Pharm. Rep. 2020, 93, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Bolduc, J.A.; Collins, J.A.; Loeser, R.F. Reactive oxygen species, aging and articular cartilage homeostasis. Free Radic Biol. Med. 2019, 132, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Setchell, K.D.; Clerici, C. Equol: Pharmacokinetics and biological actions. J. Nutr. 2010, 140, 1363S–1368S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kostelac, D.; Rechkemmer, G.; Briviba, K. Phytoestrogens modulate binding response of estrogen receptors alpha and beta to the estrogen response element. J. Agric. Food Chem. 2003, 51, 7632–7635. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, S.; Suzuki, T. Isolation and characterization of a novel equol-producing bacterium from human feces. Biosci. Biotechnol. Biochem. 2008, 72, 2660–2666. [Google Scholar] [CrossRef] [Green Version]
- Rafii, F. The role of colonic bacteria in the metabolism of the natural isoflavone daidzin to equol. Metabolites 2015, 5, 56–73. [Google Scholar] [CrossRef] [Green Version]
- Charlier, E.; Relic, B.; Deroyer, C.; Malaise, O.; Neuville, S.; Collee, J.; Malaise, M.G.; De Seny, D. Insights on Molecular Mechanisms of Chondrocytes Death in Osteoarthritis. Int. J. Mol. Sci. 2016, 17, 2146. [Google Scholar] [CrossRef] [Green Version]
- Quan, Y.Y.; Qin, G.Q.; Huang, H.; Liu, Y.H.; Wang, X.P.; Chen, T.S. Dominant roles of Fenton reaction in sodium nitroprusside-induced chondrocyte apoptosis. Free Radic Biol. Med. 2016, 94, 135–144. [Google Scholar] [CrossRef]
- Lin, J.; Wu, G.; Chen, J.; Fu, C.; Hong, X.; Li, L.; Liu, X.; Wu, M. Electroacupuncture inhibits sodium nitroprussidemediated chondrocyte apoptosis through the mitochondrial pathway. Mol. Med. Rep. 2018, 18, 4922–4930. [Google Scholar]
- Maneiro, E.; Lopez-Armada, M.J.; de Andres, M.C.; Carames, B.; Martin, M.A.; Bonilla, A.; Del Hoyo, P.; Galdo, F.; Arenas, J.; Blanco, F.J. Effect of nitric oxide on mitochondrial respiratory activity of human articular chondrocytes. Ann. Rheum Dis. 2005, 64, 388–395. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Wu, F.; Li, X.; Liu, Y.; Fan, L.; Li, H. Mitochondrial dependent pathway is involved in the protective effects of carboxymethylated chitosan on nitric oxide-induced apoptosis in chondrocytes. BMC Complement. Med. Ther. 2020, 20, 23. [Google Scholar] [CrossRef]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial membrane potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef]
- Roufayel, R. Regulation of stressed-induced cell death by the Bcl-2 family of apoptotic proteins. Mol. Membr. Biol. 2016, 33, 89–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leibowitz, B.; Yu, J. Mitochondrial signaling in cell death via the Bcl-2 family. Cancer Biol. Ther. 2010, 9, 417–422. [Google Scholar] [CrossRef] [Green Version]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Y.; Sinkeviciute, D.; He, Y.; Karsdal, M.; Henrotin, Y.; Mobasheri, A.; Onnerfjord, P.; Bay-Jensen, A. The minor collagens in articular cartilage. Protein Cell 2017, 8, 560–572. [Google Scholar] [CrossRef] [Green Version]
- Mehana, E.E.; Khafaga, A.F.; El-Blehi, S.S. The role of matrix metalloproteinases in osteoarthritis pathogenesis: An updated review. Life Sci. 2019, 234, 116786. [Google Scholar] [CrossRef]
- Liang, Q.; Wang, X.P.; Chen, T.S. Resveratrol protects rabbit articular chondrocyte against sodium nitroprusside-induced apoptosis via scavenging ROS. Apoptosis 2014, 19, 1354–1363. [Google Scholar] [CrossRef]
- Wink, D.A.; Hines, H.B.; Cheng, R.Y.; Switzer, C.H.; Flores-Santana, W.; Vitek, M.P.; Ridnour, L.A.; Colton, C.A. Nitric oxide and redox mechanisms in the immune response. J. Leukoc Biol. 2011, 89, 873–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, T.; Tan, B.X.; Lane, D. How the Other Half Lives: What p53 Does When It Is Not Being a Transcription Factor. Int. J. Mol. Sci. 2019, 21, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meek, D.W.; Knippschild, U. Posttranslational modification of MDM2. Mol. Cancer Res. 2003, 1, 1017–1026. [Google Scholar]
- Shi, D.; Gu, W. Dual Roles of MDM2 in the Regulation of p53: Ubiquitination Dependent and Ubiquitination Independent Mechanisms of MDM2 Repression of p53 Activity. Genes Cancer 2012, 3, 240–248. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.M.; Kim, S.J. Simvastatin prevents articular chondrocyte dedifferentiation induced by nitric oxide by inhibiting the expression of matrix metalloproteinases 1 and 13. Exp. Biol. Med. 2018, 243, 1165–1172. [Google Scholar] [CrossRef] [Green Version]
- Forstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef] [PubMed]
- Fan, D.X.; Yang, X.H.; Li, Y.N.; Guo, L. 17beta-Estradiol on the Expression of G-Protein Coupled Estrogen Receptor (GPER/GPR30) Mitophagy, and the PI3K/Akt Signaling Pathway in ATDC5 Chondrocytes In Vitro. Med. Sci. Monit. 2018, 24, 1936–1947. [Google Scholar] [CrossRef]
- Huang, J.G.; Xia, C.; Zheng, X.P.; Yi, T.T.; Wang, X.Y.; Song, G.; Zhang, B. 17beta-Estradiol promotes cell proliferation in rat osteoarthritis model chondrocytes via PI3K/Akt pathway. Cell Mol. Biol. Lett. 2011, 16, 564–575. [Google Scholar] [CrossRef]
- Zhang, T.; Liang, X.; Shi, L.; Wang, L.; Chen, J.; Kang, C.; Zhu, J.; Mi, M. Estrogen receptor and PI3K/Akt signaling pathway involvement in S-(-)equol-induced activation of Nrf2/ARE in endothelial cells. PLoS ONE 2013, 8, e79075. [Google Scholar] [CrossRef]
- Manfredi, J.J. The Mdm2-p53 relationship evolves: Mdm2 swings both ways as an oncogene and a tumor suppressor. Genes Dev. 2010, 24, 1580–1589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottlieb, T.M.; Leal, J.F.; Seger, R.; Taya, Y.; Oren, M. Cross-talk between Akt, p53 and Mdm2: Possible implications for the regulation of apoptosis. Oncogene 2002, 21, 1299–1303. [Google Scholar] [CrossRef]
- Gosset, M.; Berenbaum, F.; Thirion, S.; Jacques, C. Primary culture and phenotyping of murine chondrocytes. Nat. Protoc. 2008, 3, 1253–1260. [Google Scholar] [CrossRef]
- Hsieh, B.S.; Huang, L.W.; Su, S.J.; Cheng, H.L.; Hu, Y.C.; Hung, T.C.; Chang, K.L. Combined arginine and ascorbic acid treatment induces apoptosis in the hepatoma cell line HA22T/VGH and changes in redox status involving the pentose phosphate pathway and reactive oxygen and nitrogen species. J. Nutr. Biochem. 2011, 22, 234–241. [Google Scholar] [CrossRef]
- Huang, T.C.; Chiu, P.R.; Chang, W.T.; Hsieh, B.S.; Huang, Y.C.; Cheng, H.L.; Huang, L.W.; Hu, Y.C.; Chang, K.L. Epirubicin induces apoptosis in osteoblasts through death-receptor and mitochondrial pathways. Apoptosis 2018, 23, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.L.; Su, S.J.; Huang, L.W.; Hsieh, B.S.; Hu, Y.C.; Hung, T.C.; Chang, K.L. Arecoline induces HA22T/VGH hepatoma cells to undergo anoikis—Involvement of STAT3 and RhoA activation. Mol. Cancer 2010, 9, 126. [Google Scholar] [CrossRef] [Green Version]
- Chang, K.L.; Hung, T.C.; Hsieh, B.S.; Chen, Y.H.; Chen, T.F.; Cheng, H.L. Zinc at pharmacologic concentrations affects cytokine expression and induces apoptosis of human peripheral blood mononuclear cells. Nutrition 2006, 22, 465–474. [Google Scholar] [CrossRef]







 : enhanced by SNP; Red
: enhanced by SNP; Red  : decreased by SNP; Blue : enhanced by S-Equol; Blue : decreased by S-Equol.
: enhanced by SNP; Red : decreased by SNP; Blue : enhanced by S-Equol; Blue : decreased by S-Equol.
: decreased by SNP; Blue : enhanced by S-Equol; Blue : decreased by S-Equol.
: enhanced by SNP; Red : decreased by SNP; Blue : enhanced by S-Equol; Blue : decreased by S-Equol.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | NCBI Reference Sequence | Primer Sequence (5’->3’) |
|---|---|---|
| β-actin | EF156276.1 | F: ACTATCGGCAATGAGCGGTTCC R: AGCACTGTGTTGGCATAGAGGTC |
| Type II collagen | NM_012929.1 | F: CGAACCCAAAGGACCCAAAT R: TCCGGACTGTGAGGTTAGG |
| Aggrecan | NM_022190.1 | F: CGAGTGAACAGCATCTACC R: GAGTCATTGGAGCGAAGG |
| iNOS (NOS2) | NM_012611.3 | F: GCTACACTTCCAACGCAACA R: ATGGTGAACACGTTCTTGGC |
| BIM | NM_171988.2 | F: GCAAACGATTACCGAGAGGC R: TCCAGACCAGACGGAAGATG |
| PUMA | NM_173837.2 | F: AAGAGCAACATCGACACCGA R: TCCAGGATCCCTGGGTAAGG |
| NOXA | NM_001008385.1 | F: GAGTGCACCGGACATAACTG R: GCTTGGGCTTCTTCTCATCG |
| Bcl-2 | NM_016993.1 | F: CTGGGGATGACTTCTCTCGT R: GGACATCTCTGCAAAGTCGC |
| BAK | NM_053812.1 | F: CACCCATGCTCCTGTTATGC R: CCCACCCTTGCTACATTTGG |
| BAX | NM_017059.2 | F: TTGGCGATGAACTGGACAAC R: GTAGAAAAGGGCAACCACCC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, L.-W.; Huang, T.-C.; Hu, Y.-C.; Hsieh, B.-S.; Cheng, H.-L.; Chiu, P.-R.; Chang, K.-L. S-Equol Protects Chondrocytes against Sodium Nitroprusside-Caused Matrix Loss and Apoptosis through Activating PI3K/Akt Pathway. Int. J. Mol. Sci. 2021, 22, 7054. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22137054
Huang L-W, Huang T-C, Hu Y-C, Hsieh B-S, Cheng H-L, Chiu P-R, Chang K-L. S-Equol Protects Chondrocytes against Sodium Nitroprusside-Caused Matrix Loss and Apoptosis through Activating PI3K/Akt Pathway. International Journal of Molecular Sciences. 2021; 22(13):7054. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22137054
Chicago/Turabian StyleHuang, Li-Wen, Tzu-Ching Huang, Yu-Chen Hu, Bau-Shan Hsieh, Hsiao-Ling Cheng, Pu-Rong Chiu, and Kee-Lung Chang. 2021. "S-Equol Protects Chondrocytes against Sodium Nitroprusside-Caused Matrix Loss and Apoptosis through Activating PI3K/Akt Pathway" International Journal of Molecular Sciences 22, no. 13: 7054. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22137054