
A Comparative Analysis of the In Vitro Anticancer Activity of Iridium(III) {η5-C5Me4R} Complexes with Variable R Groups

 , , ,
, , , 
Abstract
:
1. Introduction
2. Results and Discussion
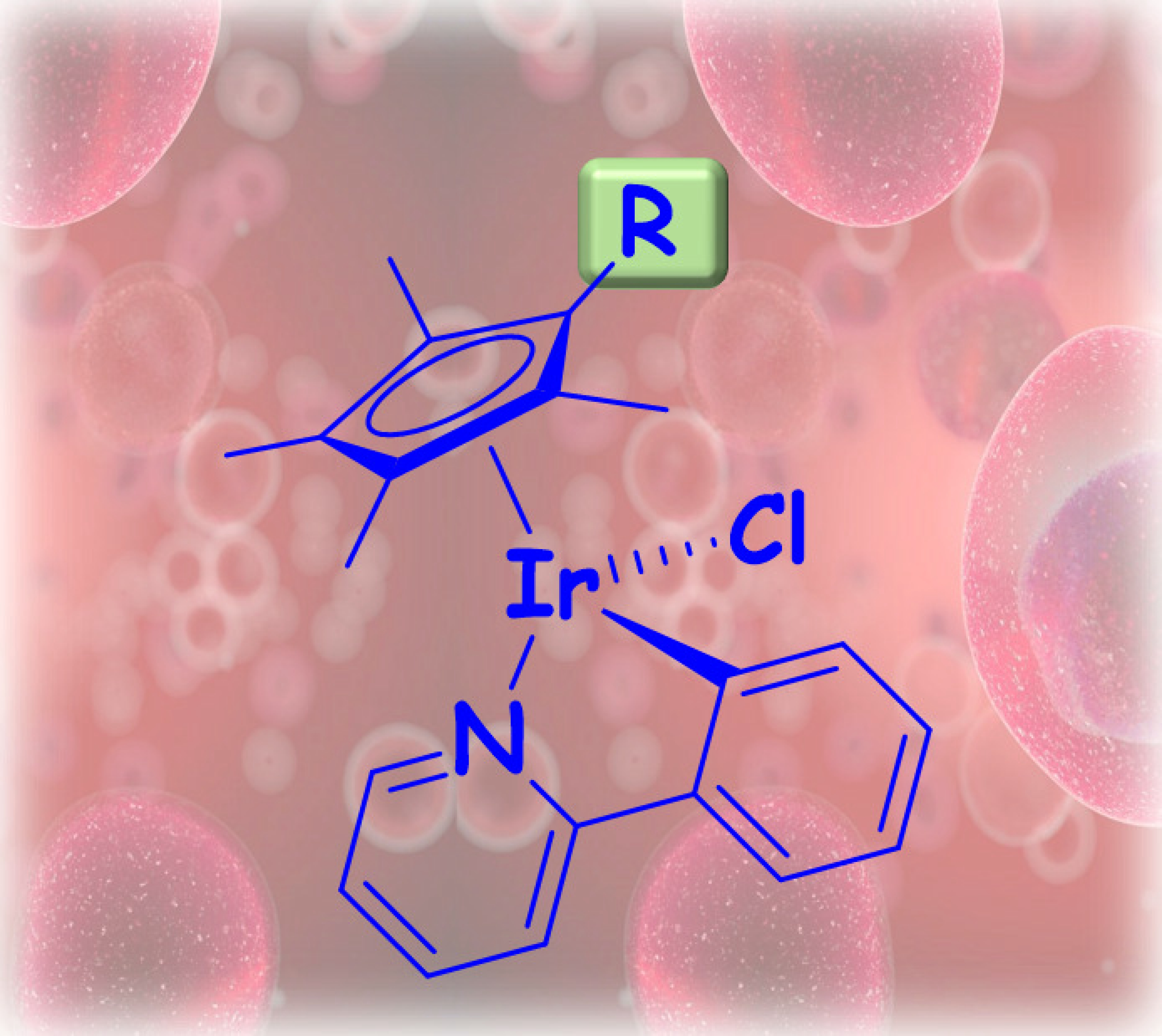
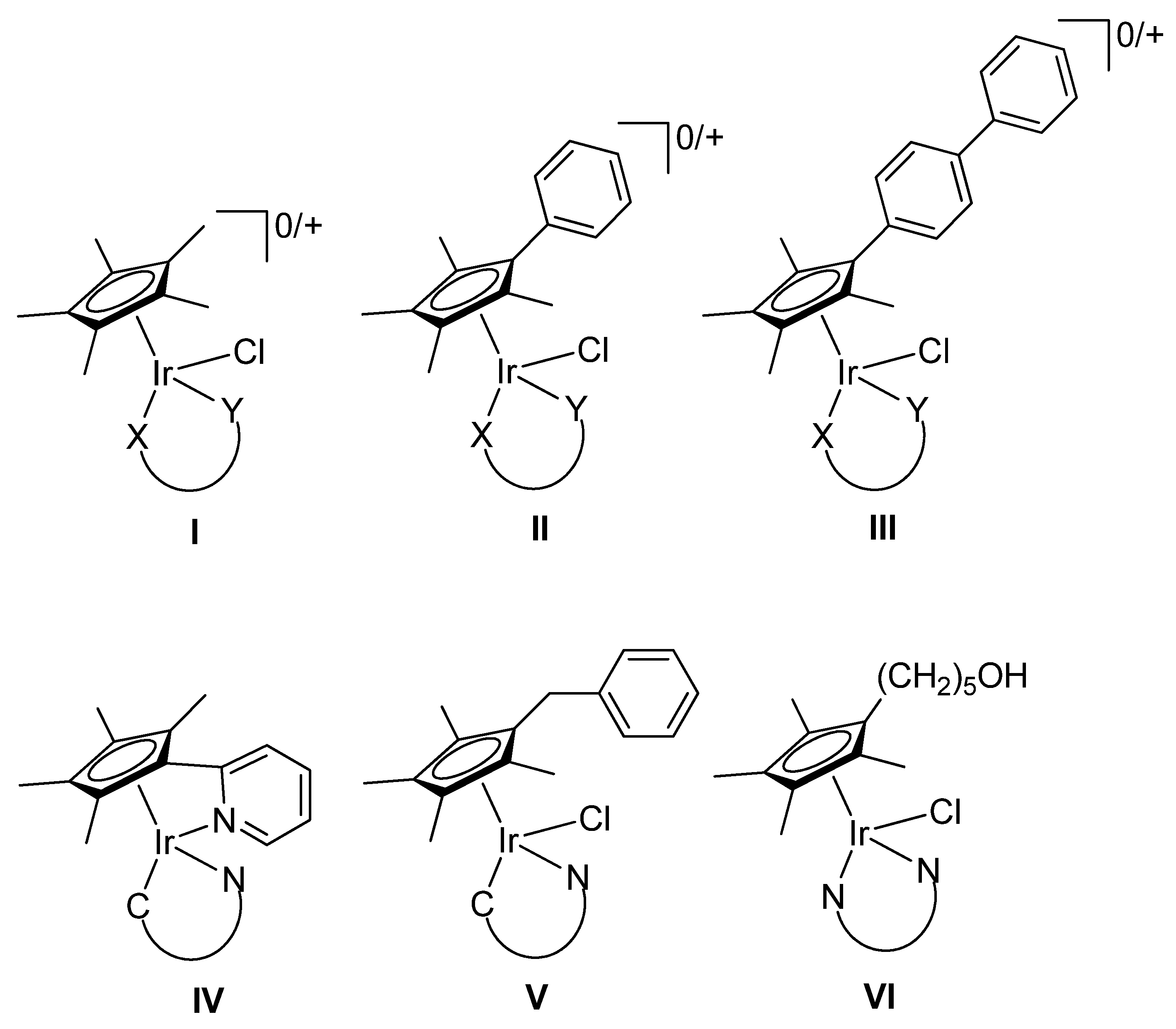
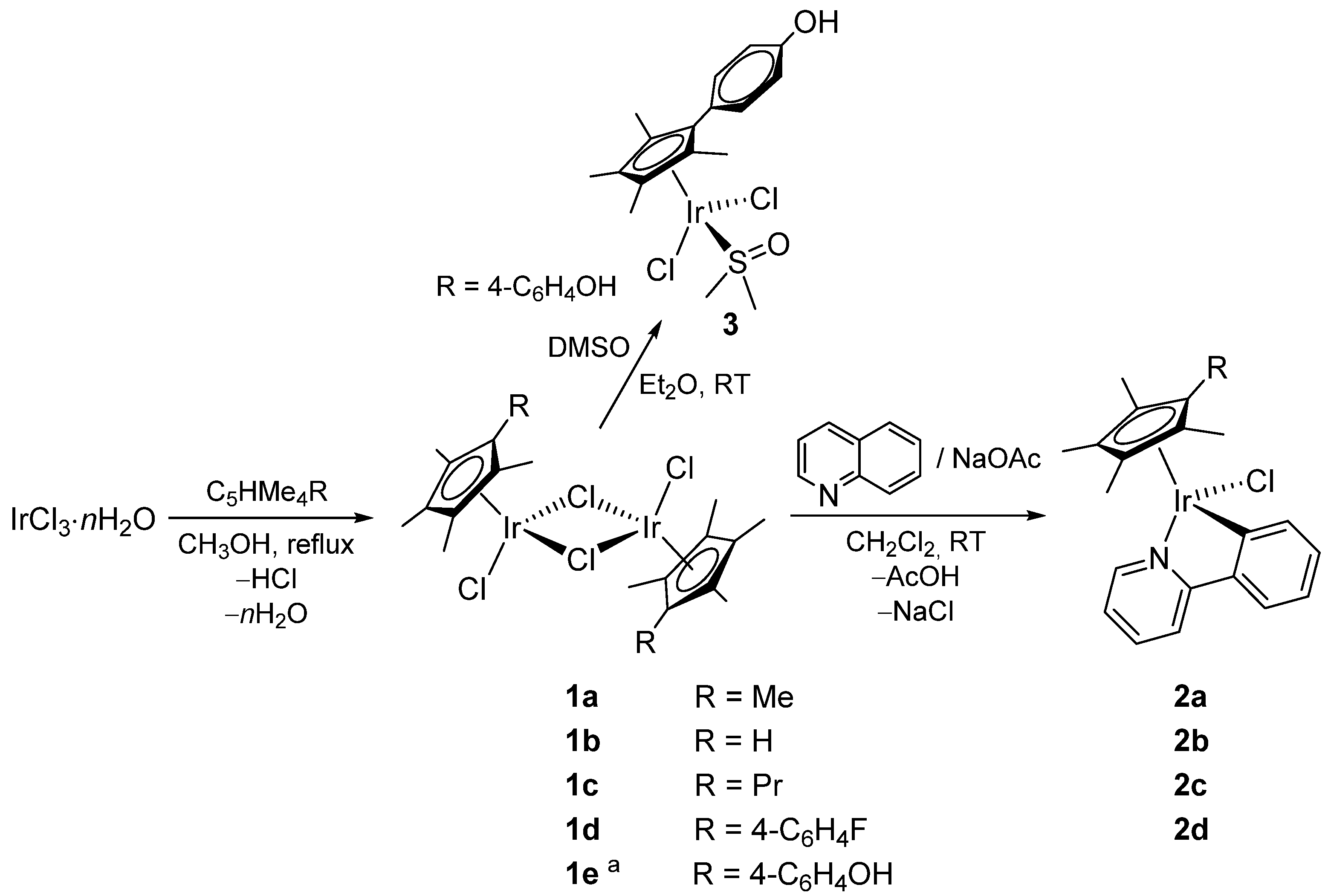
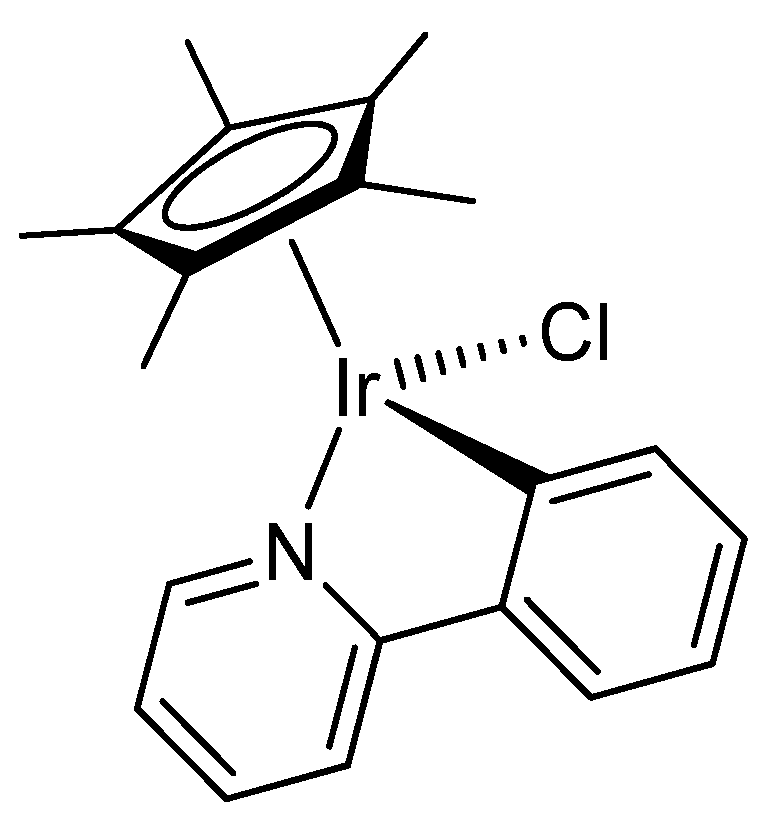
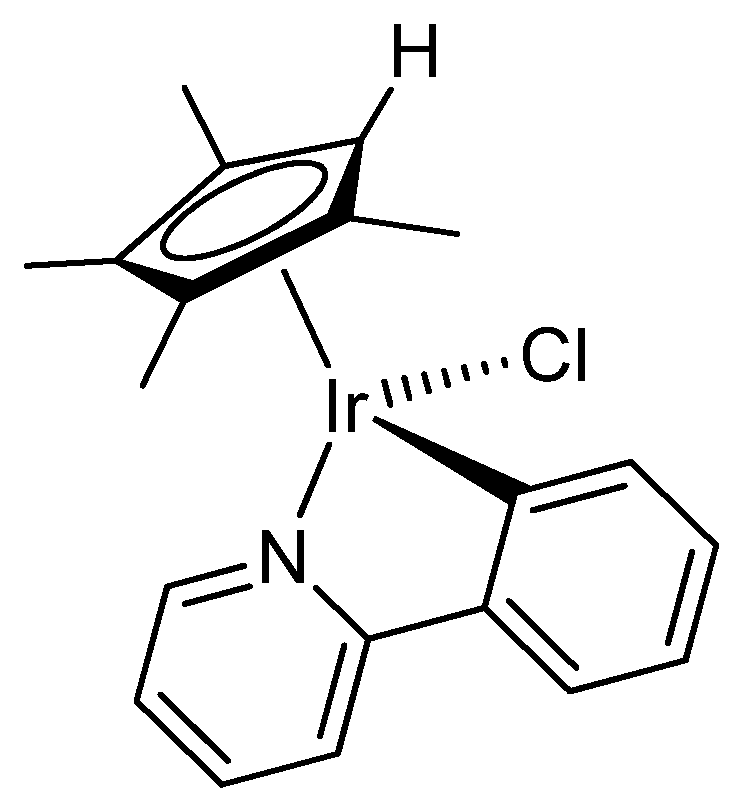
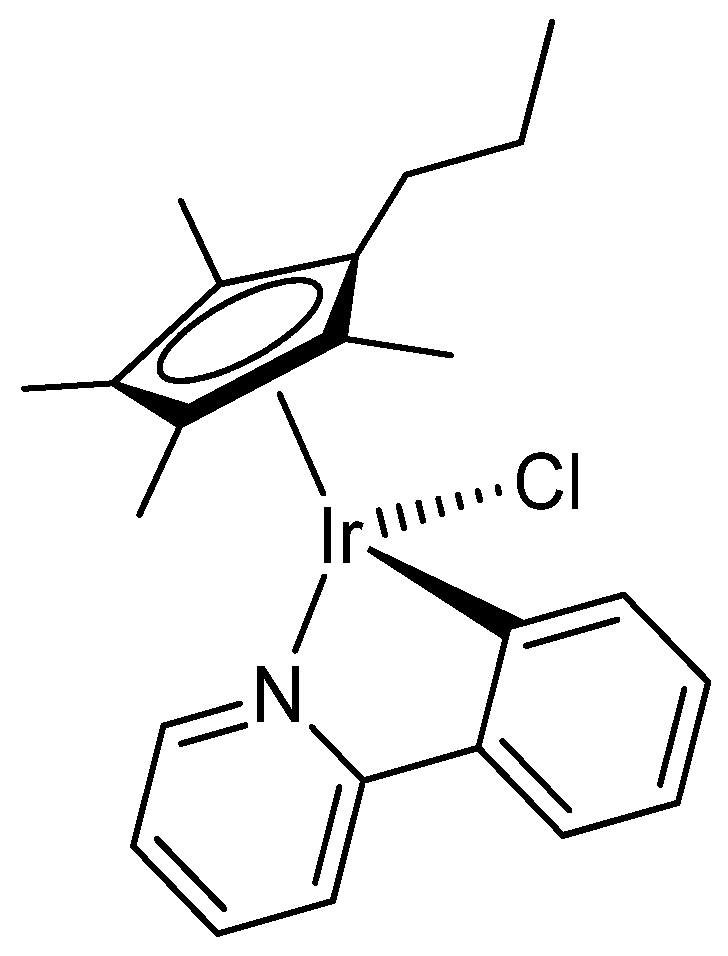
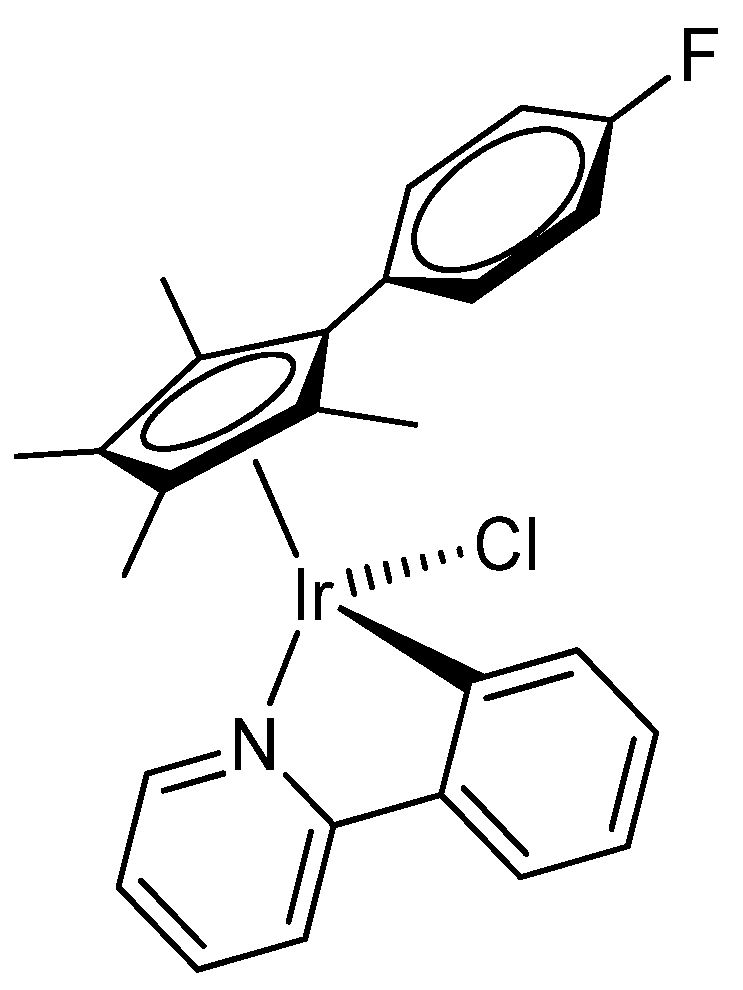
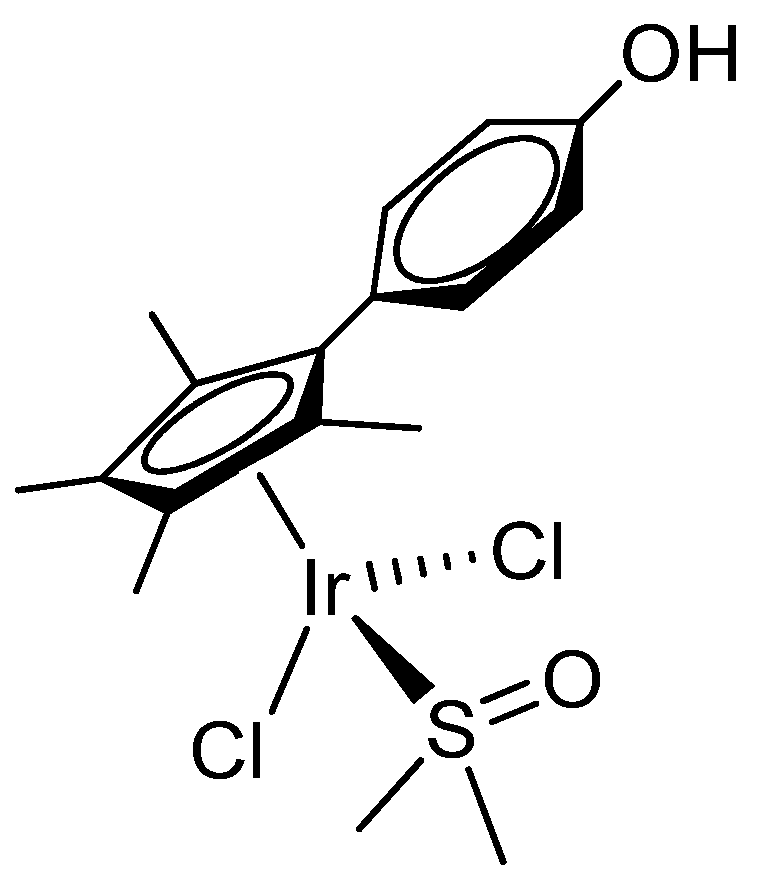
2.1. Synthesis and Characterization of Compounds
2.2. Cytotoxicity
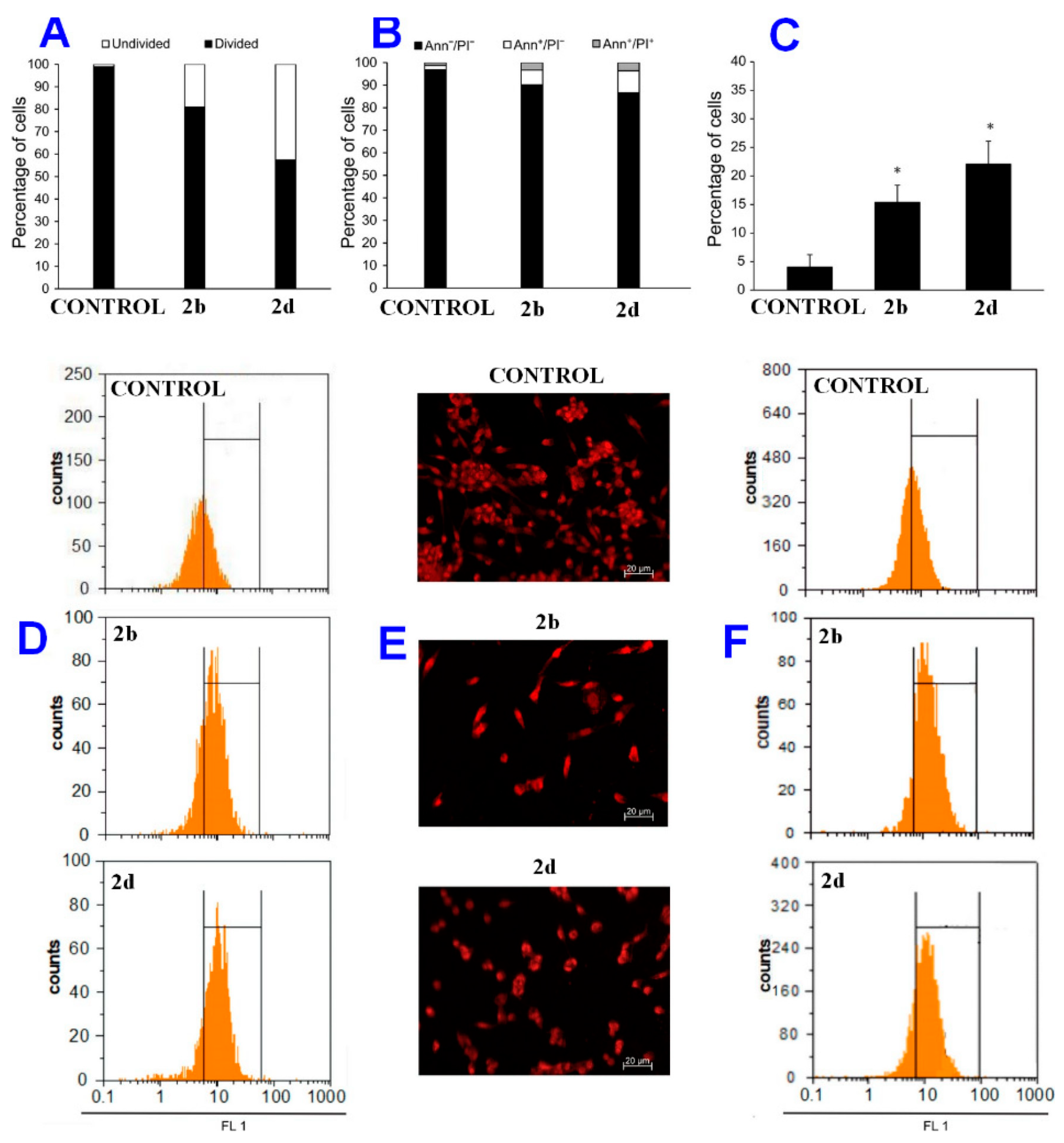
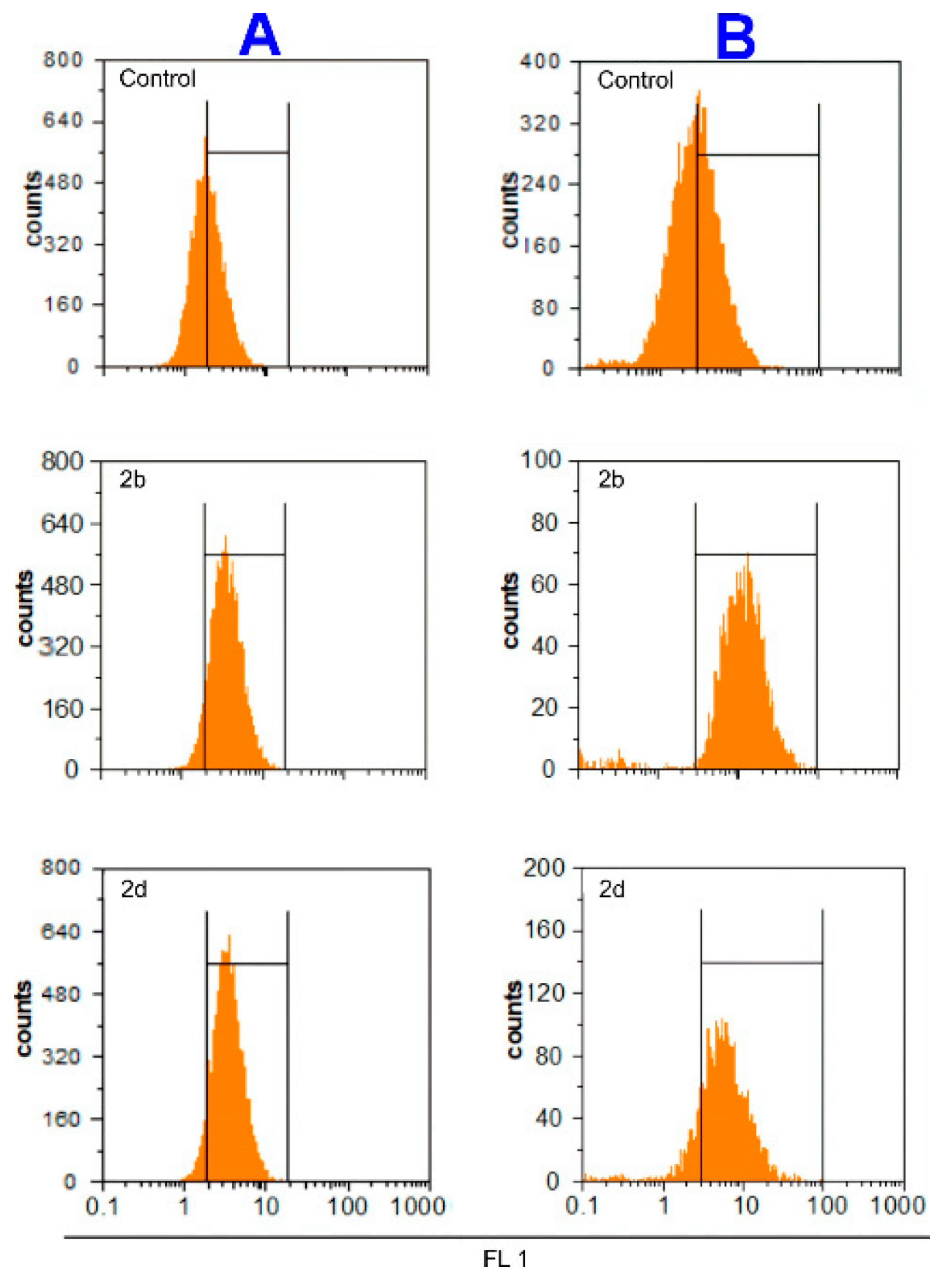
2.3. Mechanism of Action
3. Conclusions
4. Experimental
4.1. Materials and Methods
4.2. Synthesis and Characterization of Compounds
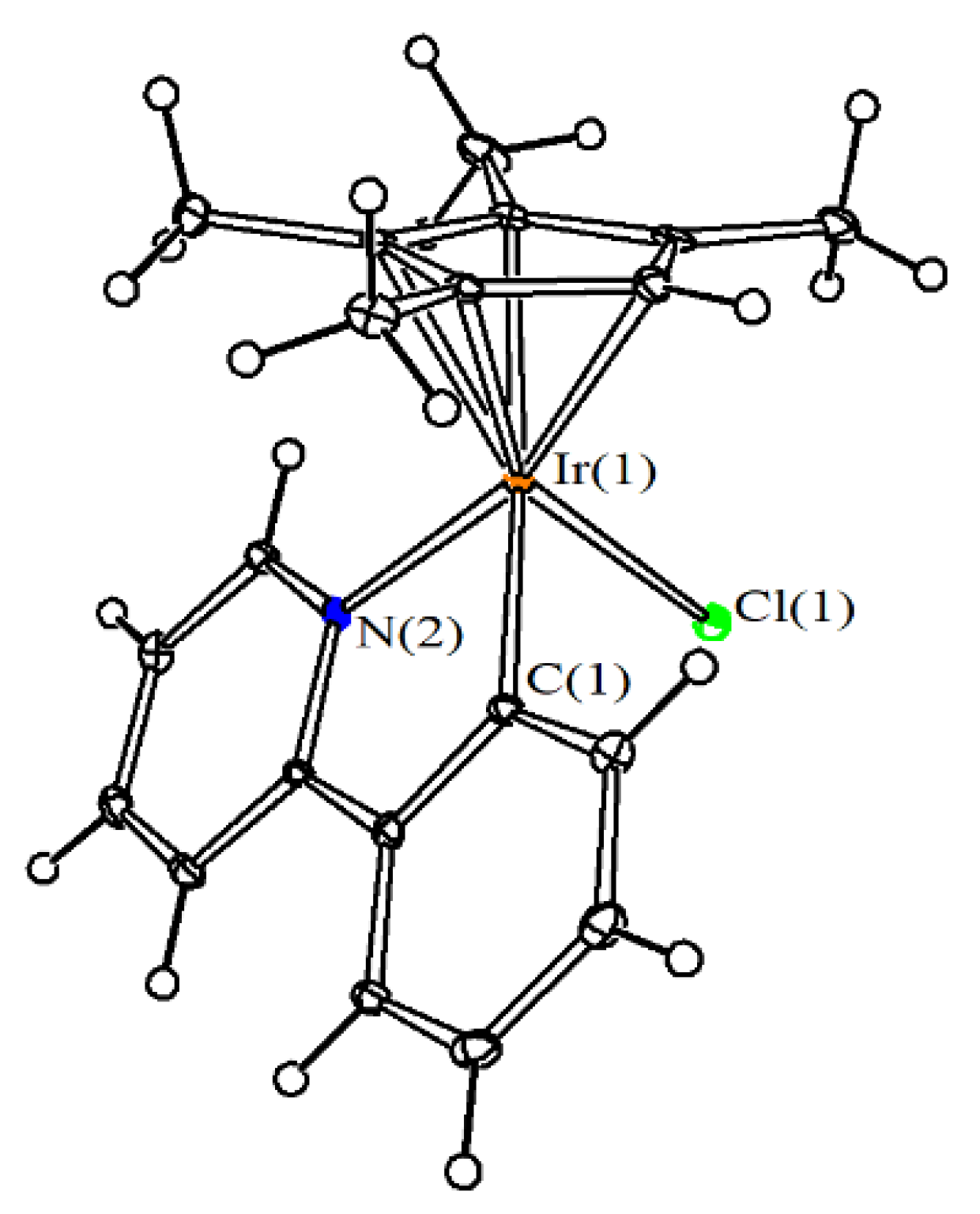
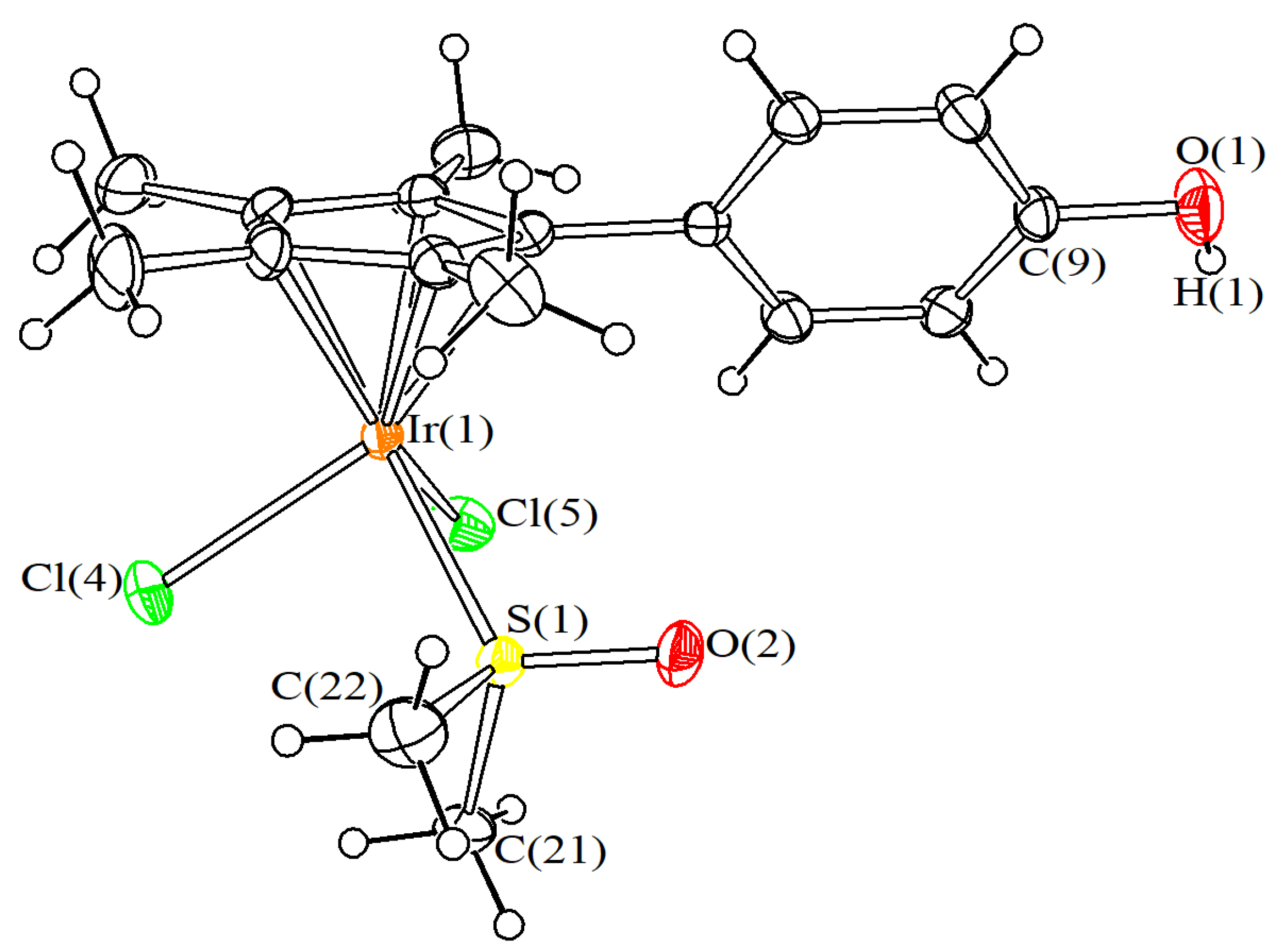
4.3. X-ray Crystallography
4.4. Behavior in Aqueous Solutions
4.5. Biological Studies
4.5.1. Reagents and Cells
4.5.2. Preparation of Drug Solutions
4.5.3. Colorimetric Assays for Cellular Viability
4.5.4. Flow Cytometry
4.5.5. Fluorescence Microscopy
4.5.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Webpage
References
- Apps, M.G.; Choi, E.H.Y.; Wheate, N.J. The state-of-play and future of platinum drugs. Endocr. Relat. Cancer 2015, 22, R219–R233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oun, R.; Moussa, Y.E.; Wheate, N.J. The side effects of platinum-based chemotherapy drugs: A review for chemists. Dalton Trans. 2018, 47, 6645–6653. [Google Scholar] [CrossRef]
- Hanif, M.; Hartinger, C.G. Anticancer metallodrugs: Where is the next cisplatin? Fut. Med. Chem. 2018, 10, 615–617. [Google Scholar] [CrossRef]
- Anthony, E.J.; Bolitho, E.M.; Bridgewater, H.E.; Carter, O.W.L.; Donnelly, J.M.; Imberti, C.E.; Lant, C.; Lermyte, F.; Needham, R.J.; Palau, M.; et al. Metallodrugs are unique: Opportunities and challenges of discovery and development. Chem. Sci. 2020, 11, 12888–12917. [Google Scholar] [CrossRef]
- Murray, B.S.; Dyson, P.J. Recent progress in the development of organometallics for the treatment of cancer. Curr. Opinion Chem. Biol. 2020, 56, 28–34. [Google Scholar] [CrossRef]
- Liu, Z.; Sadler, P.J. Organoiridium Complexes: Anticancer Agents and Catalysts. Acc. Chem. Res. 2014, 47, 1174–1185. [Google Scholar] [CrossRef]
- Lord, R.M.; Zegke, M.; Henderson, I.R.; Pask, C.M.; Shepherd, H.J.; McGowan, P.C. β-Ketoiminato Iridium (III) Organometallic Complexes: Selective Cytotoxicity towards Colorectal Cancer Cells HCT116 p53-l-. Chem. Eur. J. 2019, 25, 495–500. [Google Scholar] [CrossRef]
- Yang, Y.; Guo, L.; Ge, X.; Shi, S.; Gong, Y.; Xu, Z.; Zheng, X.; Liu, Z. Structure-activity relationships for highly potent half-sandwich organoiridium(III) anticancer complexes with C^N-chelated ligands. J. Inorg. Biochem. 2019, 191, 1–7. [Google Scholar] [CrossRef]
- Liu, Z.; Habtemariam, A.; Pizarro, A.M.; Fletcher, S.A.; Kisova, A.; Vrana, O.; Salassa, L.; Bruijnincx, P.C.; Clarkson, G.J.; Brabec, V.; et al. Organometallic Half-Sandwich Iridium Anticancer Complexes. J. Med. Chem. 2011, 54, 3011–3026. [Google Scholar] [CrossRef]
- Liu, X.; Chen, S.; Ge, X.; Zhang, Y.; Xie, Y.; Hao, Y.; Wu, D.; Zhao, J.; Yuan, X.-A.; Tian, L.; et al. Dual functions of iridium(III) 2-phenylpyridine complexes: Metastasis inhibition and lysosomal damage. J. Inorg. Biochem. 2020, 205, 110983. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Romero-Canelon, I.; Qamar, B.; Hearn, J.M.; Habtemariam, A.; Barry, N.; Pizarro, A.M.; Clarkson, G.J.; Sadler, P.J. The Potent Oxidant Anticancer Activity of Organoiridium Catalysts. Angew. Chem. Int. Ed. 2014, 53, 3941–3946. [Google Scholar] [CrossRef] [Green Version]
- Carrasco, A.C.; Rodríguez-Fanjul, V.; Habtemariam, A.; Pizarro, A.M. Structurally Strained Half-Sandwich Iridium(III) Complexes As Highly Potent Anticancer Agents. J. Med. Chem. 2020, 63, 4005–4021. [Google Scholar] [CrossRef] [PubMed]
- Lucas, S.J.; Lord, R.M.; Basri, A.M.; Allison, S.J.; Phillips, R.M.; McGowan, P.C.; Blacker, A.J. Increasing anti-cancer activity with longer tether lengths of group 9 Cp* complexes. Dalton Trans. 2016, 45, 6812–6815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatto, G.; De Palo, A.; Carrasco, A.C.; Pizarro, A.M.; Zacchini, S.; Pampaloni, G.; Marchetti, F.; Macchioni, A. Modulating the water oxidation catalytic activity of iridium complexes by functionalizing the Cp*-ancillary ligand: Hints on the nature of the active species. Catal. Sci. Technol. 2021, 11, 2885–2895. [Google Scholar] [CrossRef]
- Fooladi, E.; Graham, T.; Turner, M.L.; Dalhus, B.; Maitlis, P.M.; Tilset, M. Oxidatively induced M–C bond cleavage reactions of Cp*Ir(Me2SO)Me2 and Cp*Rh(Me2SO)Me2 (Cp* = η5-C5Me5). J. Chem. Soc. Dalton Trans. 2002, 975–982. [Google Scholar] [CrossRef]
- Frasco, D.A.; Lilly, C.P.; Boyle, P.D.; Ison, E.A. Cp*IrIII-Catalyzed Oxidative Coupling of Benzoic Acids with Alkynes. ACS Catal. 2013, 3, 2421–2429. [Google Scholar] [CrossRef]
- Krämer, R.; Polborn, K.; Beck, W. Metallkomplexe mit biologisch wichtigen Liganden: LIX. Darstellung und Struktur des chiralen Chloro(dimethylsulfoxid)(η5-pentamethylcyclopentadienyl)-thyminatoiridium (III). J. Organomet. Chem. 1991, 410, 111–116. [Google Scholar] [CrossRef]
- Frasco, D.A.; Mukherjee, S.; Sommer, R.D.; Perry, C.M.; Lambic, N.S.; Abboud, K.A.; Jakubikova, E.; Ison, E.A. Nondirected C–H Activation of Arenes with Cp*Ir(III) Acetate Complexes: An Experimental and Computational Study. Organometallics 2016, 35, 2435–2445. [Google Scholar] [CrossRef]
- Blakemore, J.D.; Schley, N.D.; Balcells, D.; Hull, J.F.; Olack, G.W.; Incarvito, C.D.; Eisenstein, O.; Brudvig, G.W.; Crabtree, R.H. Half-Sandwich Iridium Complexes for Homogeneous Water-Oxidation Catalysis. J. Am. Chem. Soc. 2010, 132, 16017–16029. [Google Scholar] [CrossRef] [PubMed]
- Boutadla, Y.; Al-Duaij, O.; Davies, D.; Griffith, G.A.; Singh, K. Mechanistic Study of Acetate-Assisted C−H Activation of 2-Substituted Pyridines with [MCl2Cp*]2 (M = Rh, Ir) and [RuCl2(p-cymene)]2. Organometallics 2009, 28, 433–440. [Google Scholar] [CrossRef]
- Liu, Z.; Habtemariam, A.; Pizarro, A.M.; Clarkson, G.J.; Sadler, P.J. Organometallic Iridium(III) Cyclopentadienyl Anticancer Complexes Containing C,N-Chelating Ligands. Organometallics 2011, 30, 4702–4710. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Na, T.; Wang, L.; Gao, Q.; Yin, W.; Wang, J.; Yuan, B.-Z. Human diploid MRC-5 cells exhibit several critical properties of human umbilical cord-derived mesenchymal stem cells. Vaccine 2014, 32, 6820–6827. [Google Scholar] [CrossRef] [PubMed]
- Paglin, S.; Lee, N.-Y.; Nakar, C.; Fitzgerald, M.; Plotkin, J.; Deuel, B.; Hackett, N.; McMahill, M.; Sphicas, E.; Lampen, N.; et al. Rapamycin-Sensitive Pathway Regulates Mitochondrial Membrane Potential, Autophagy, and Survival in Irradiated MCF-7 Cells. Cancer Res. 2005, 65, 11061–11070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mijatović, S.; Bulatović, M.; Mojić, M.; Stosic-Grujicic, S.; Miljković, Đ.; Maksimović-Ivanić, D.; Gómez-Ruiz, S.; Pinkas, J.; Horáček, M.; Kaluđerović, G.N. Study of the anticancer properties of methyl- and phenyl-substituted carbon- and silicon-bridged ansa-titanocene complexes. J. Organomet. Chem. 2014, 751, 361–367. [Google Scholar] [CrossRef]
- Kroemer, G.; Galluzzi, L.; Vandenabeele, P.; Abrams, J.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; El-Deiry, W.S.; Golstein, P.; Green, D.R.; et al. Classification of cell death: Recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009, 16, 3–11. [Google Scholar] [CrossRef]
- Ludwig, G.; Ranđelović, I.; Maksimović-Ivanić, D.; Mijatović, S.; Bulatović, M.Z.; Miljković, D.; Korb, M.; Lang, H.; Steinborn, D.; Kaluđerović, G.N. Anticancer Potential of (Pentamethylcyclopentadienyl)chloridoiridium(III) Complexes Bearing κP and κP,κS-Coordinated Ph2PCH2CH2CH2S(O)xPh (x=0–2) Ligands. ChemMedChem 2014, 9, 1586–1593. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, G.; Miljković, S.; Ranđelović, I.; Bulatovićb, M.; Miljković, D.; Maksimović-Ivanić, D.; Korb, M.; Lang, H.; Steinborn, D.; Kaluđerović, G.N. Biological activity of neutral and cationic iridium(III) complexes with κP and κP,κS coordinated Ph2PCH2S(O)xPh (x = 0–2) ligands. Eur. J. Med. Chem. 2013, 69, 216–222. [Google Scholar] [CrossRef]
- Herranz, N.; Gil, J. Mechanisms and functions of cellular senescence. J. Clin. Invest. 2018, 128, 1238–1246. [Google Scholar] [CrossRef]
- Paskaš, S.; Krajnović, T.; Basile, M.S.; Dunđerović, D.; Cavalli, E.; Mangano, K.; Mammana, S.; Al-Abed, Y.; Nicoletti, F.; Mijatović, S.; et al. Senescence as a main mechanism of Ritonavir and Ritonavir-NO action against melanoma. Mol. Carcinog. 2019, 58, 1362–1375. [Google Scholar] [CrossRef]
- Wang, X.; Wong, S.C.; Pan, J.; Tsao, S.W.; Fung, K.H.; Kwong, D.L.; Sham, J.S.; Nicholls, J.M. Evidence of cisplatin-induced senescent-like growth arrest in nasopharyngeal carcinoma cells. Cancer Res. 1998, 58, 5019–5022. [Google Scholar]
- Bulatović, M.Z.; Maksimović-Ivanić, D.; Bensing, C.; Gómez-Ruiz, S.; Steinborn, D.; Schmidt, H.; Mojić, M.; Korać, A.; Golić, I.; Pérez-Quintanilla, D.; et al. Organotin (IV)-loaded mesoporous silica as a biocompatible strategy in cancer treatment. Angew. Chem. Int. Ed. 2014, 53, 5982–5987. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.-J.; Gu, D.-N.; Dai, J.-J.; Huang, Q.; Tian, L. Dark Side of Cytotoxic Therapy: Chemoradiation-Induced Cell Death and Tumor Repopulation. Trends Cancer 2020, 6, 419–431. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.-Y.; Banerjee, S.; Hughes, G.M.; Bridgewater, H.E.; Song, J.-I.; Breeze, B.G.; Clarkson, G.J.; Coverdale, J.P.C.; Sanchez-Cano, C.; Ponte, F.; et al. Ligand-centred redox activation of inert organoiridium anticancer catalysts. Chem. Sci. 2020, 11, 5466–5480. [Google Scholar] [CrossRef] [PubMed]
- White, C.; Yates, A.; Maitlis, P.M.; Henekey, D.M. (η5-Pentamethylcyclopentadienyl)Rhodium and -Iridium Compounds. Inorg. Synth. 1992, 29, 228–234. [Google Scholar]
- Willker, W.; Leibfritz, D.; Kerssebaum, R.; Bermel, W. Gradient selection in inverse heteronuclear correlation spectroscopy. Magn. Reson. Chem. 1993, 31, 287–292. [Google Scholar] [CrossRef]
- Li, L.; Brennessel, W.; Jones, W.D. An Efficient Low-Temperature Route to Polycyclic Isoquinoline Salt Synthesis via C−H Activation with [Cp*MCl2]2 (M = Rh, Ir). J. Am. Chem. Soc. 2008, 130, 12414–12419. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SADABS-2008/1—Bruker AXS Area Detector Scaling and Absorption Correction; Bruker AXS: Madison, WI, USA, 2008. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Rice, N.M.; Irving, H.M.N.H.; Leonard, M.A. Nomenclature for Liquid-Liquid Distribution (Solvent Extraction). IUPAC Standards Online 2016, 65, 2373–2396. [Google Scholar] [CrossRef]
- Agonigi, G.; Biancalana, L.; Lupo, M.G.; Montopoli, M.; Ferri, N.; Zacchini, S.; Binacchi, F.; Biver, T.; Campanella, B.; Pampaloni, G.; et al. Exploring the Anticancer Potential of Diiron Bis-cyclopentadienyl Complexes with Bridging Hydrocarbyl Ligands: Behavior in Aqueous Media and In Vitro Cytotoxicity. Organometallics 2020, 39, 645–657. [Google Scholar] [CrossRef]
- Liu, L.; Salassa, Z.; Habtemariam, A.; Pizarro, A.M.; Clarkson, G.J.; Sadler, P.J. Contrasting Reactivity and Cancer Cell Cytotoxicity of Isoelectronic Organometallic Iridium(III) Complexes. Inorg. Chem. 2011, 50, 5777–5783. [Google Scholar] [CrossRef] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 2b | 3 | |
|---|---|---|
| Formula | C20H21ClIrN | C17H23Cl2IrO2S |
| FW | 503.03 | 554.51 |
| T, K | 100(2) | 293(2) |
| λ, Å | 0.71073 | 0.71073 |
| Crystal system | Monoclinic | Monoclinic |
| Space group | P21/n | P21/c |
| a, Å | 14.4923(5) | 13.9835(5) |
| b, Å | 7.3769(3) | 8.8692(3) |
| c, Å | 15.5178(5) | 15.3611(6) |
| β,° | 95.9050(10) | 96.317(2) |
| Cell volume, Å3 | 1650.18(10) | 1893.55(12) |
| Z | 4 | 4 |
| Dc, g∙cm−3 | 2.025 | 1.945 |
| μ,mm−1 | 8.251 | 7.449 |
| F(000) | 968 | 1072 |
| Crystal size, mm | 0.16 × 0.13 × 0.10 | 0.18 × 0.13 × 0.10 |
| θ limits,° | 1.831–25.999 | 1.465–25.992 |
| Reflections collected | 21,455 | 28,963 |
| Independent reflections | 3237 [Rint = 0.0255] | 3715 [Rint = 0.0394] |
| Data/restraints/parameters | 3237/36/213 | 3715/1/217 |
| Goodness on fit on F2 | 1.390 | 1.068 |
| R1 (I > 2σ(I)) | 0.0275 | 0.0193 |
| wR2 (all data) | 0.0702 | 0.0487 |
| Largest diff. peak and hole, e Å−3 | 1.648/–2.034 | 0.497/–1.017 |
| 1b | 1c | 1d | 1e | 2a | 2b | 2c | 2d | 3 | Cisplatin | |
|---|---|---|---|---|---|---|---|---|---|---|
| B16 | >50 | 28 ± 4 | 20.4 ± 0.1 | >100 | 5.1 ± 0.1 | 2.5 ± 0.2 | 6.7 ± 0.6 | 1.2 ± 0.2 | >100 | 5.6 ± 0.2 |
| B16 | >50 | 44 ± 8 | 28.1 ± 0.1 | >100 | 6 ± 2 | 3.0 ± 0.1 | 8 ± 2 | 1.7 ± 0.4 | >100 | 11.6 ± 0.3 |
| SW620 | >50 | 27 ± 2 | 8 ± 2 | >100 | 7.4 ± 0.9 | 2.6 ± 0.1 | 10.4 ± 0.4 | 2.0 ± 0.1 | >100 | 11.6 ± 0.1 |
| SW620 | >50 | 45 ± 8 | 18.5 ± 0.4 | >100 | 5.3 ± 0.2 | 2.9 ± 0.5 | 12.4 ± 1.5 | 2.0 ± 0.2 | >100 | 11 ± 3 |
| C6 | >50 | 27 ± 3 | 34 ± 2 | 90 ± 14 | 4.9 ± 0.8 | 2.4 ± 0.3 | 4.0 ± 0.3 | 2.00 ± 0.01 | >100 | 0.6 ± 0.2 |
| C6 | >50 | 54 ± 5 | 49 ± 1 | >100 | 4.7 ± 0.5 | 2.6 ± 0.1 | 5.9 ± 0.5 | 2.1 ± 0.1 | >100 | 1.2 ± 0.1 |
| MCF-7 | >50 | 28 ± 5 | 9.2 ± 0.5 | 88 ± 12 | 15 ± 5 | 8.6 ± 0.2 | 12.0 ± 1.3 | 2.5 ± 0.2 | >100 | 0.8 ± 0.2 |
| MCF-7 | >50 | 55 ± 8 | 30.0 ± 0.4 | 94 ± 8 | 13.3 ± 0.6 | 37 ± 3 | 17.2 ± 0.1 | 6.8 ± 0.7 | >100 | 1.2 ± 0.3 |
| HCT116 | >50 | 30 ± 4 | 8.1 ± 0.7 | 80 ± 3 | 4.8 ± 0.9 | 8.0 ± 0.2 | 8.0 ± 0.6 | 1.3 ± 0.3 | >100 | 1.5 ± 0.1 |
| HCT116 | >50 | 38 ± 3 | 14.5 ± 1.5 | 79 ± 8 | 24.6 ± 0.5 | 7 ± 2 | 13.0 ± 1.1 | 5.3 ± 1.3 | >100 | 8.0 ± 0.7 |
| A2780 | >50 | 13 ± 1 | 16.0 ± 0.3 | N.A. | 2.9 ± 0.1 | 5.5 ± 0.5 | 7.3 ± 0.6 | 1.3 ± 0.1 | N.A. | 1.9 ± 0.2 |
| A2780 | >50 | 39 ± 7 | 20.2 ± 0.3 | N.A. | 12.3 ± 0.3 | 4.4 ± 0.6 | 7.2 ± 0.8 | 1.8 ± 0.1 | N.A. | 3.8 ± 0.5 |
| MRC5 | >50 | 36 ± 2 | 19.1 ± 0.2 | >100 | 2.5 ± 0.5 | 6.7 ± 0.2 | 3.7 ± 0.2 | 2.4 ± 0.3 | >100 | 0.6 ± 0.2 |
| MRC5 | >50 | >50 | 22.2 ± 0.1 | >100 | 5.8 ± 0.1 | 6 ± 2 | 2.7 ± 0.1 | 2.1 ± 0.6 | >100 | 0.8 ± 0.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Palo, A.; Draca, D.; Murrali, M.G.; Zacchini, S.; Pampaloni, G.; Mijatovic, S.; Maksimovic-Ivanic, D.; Marchetti, F. A Comparative Analysis of the In Vitro Anticancer Activity of Iridium(III) {η5-C5Me4R} Complexes with Variable R Groups. Int. J. Mol. Sci. 2021, 22, 7422. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147422
De Palo A, Draca D, Murrali MG, Zacchini S, Pampaloni G, Mijatovic S, Maksimovic-Ivanic D, Marchetti F. A Comparative Analysis of the In Vitro Anticancer Activity of Iridium(III) {η5-C5Me4R} Complexes with Variable R Groups. International Journal of Molecular Sciences. 2021; 22(14):7422. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147422
Chicago/Turabian StyleDe Palo, Alice, Dijana Draca, Maria Grazia Murrali, Stefano Zacchini, Guido Pampaloni, Sanja Mijatovic, Danijela Maksimovic-Ivanic, and Fabio Marchetti. 2021. "A Comparative Analysis of the In Vitro Anticancer Activity of Iridium(III) {η5-C5Me4R} Complexes with Variable R Groups" International Journal of Molecular Sciences 22, no. 14: 7422. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147422