
Costunolide Induces Apoptosis via the Reactive Oxygen Species and Protein Kinase B Pathway in Oral Cancer Cells

 ,
,  , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
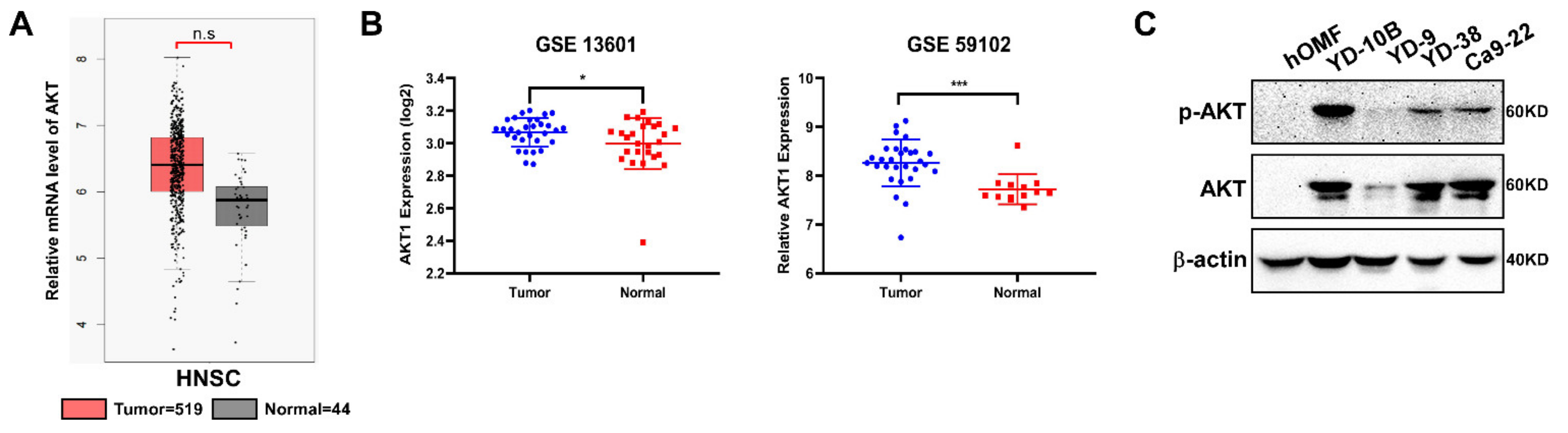
2.1. AKT Expresses in OSCC Patients Tissue
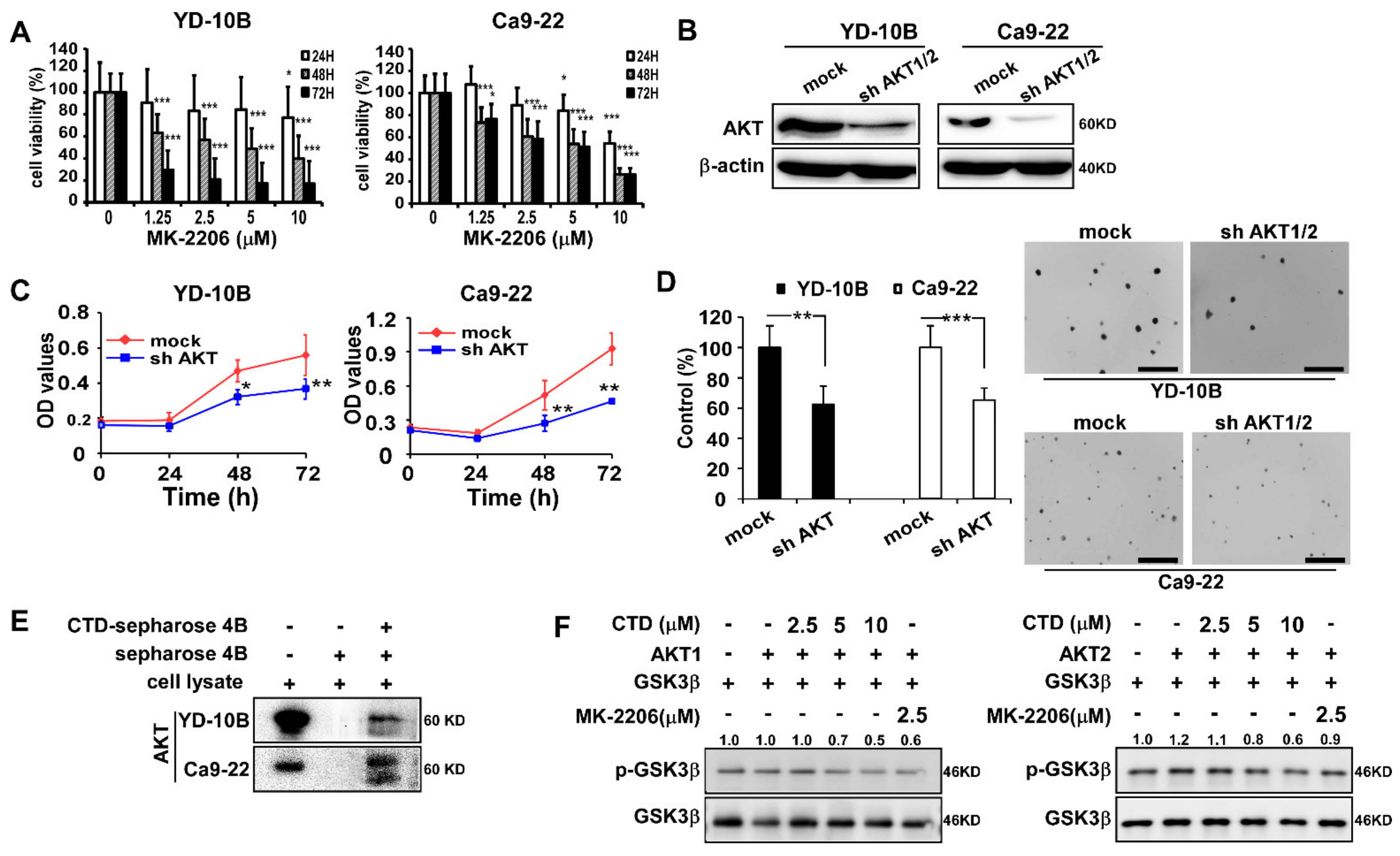
2.2. The Target of Costunolide Is AKT
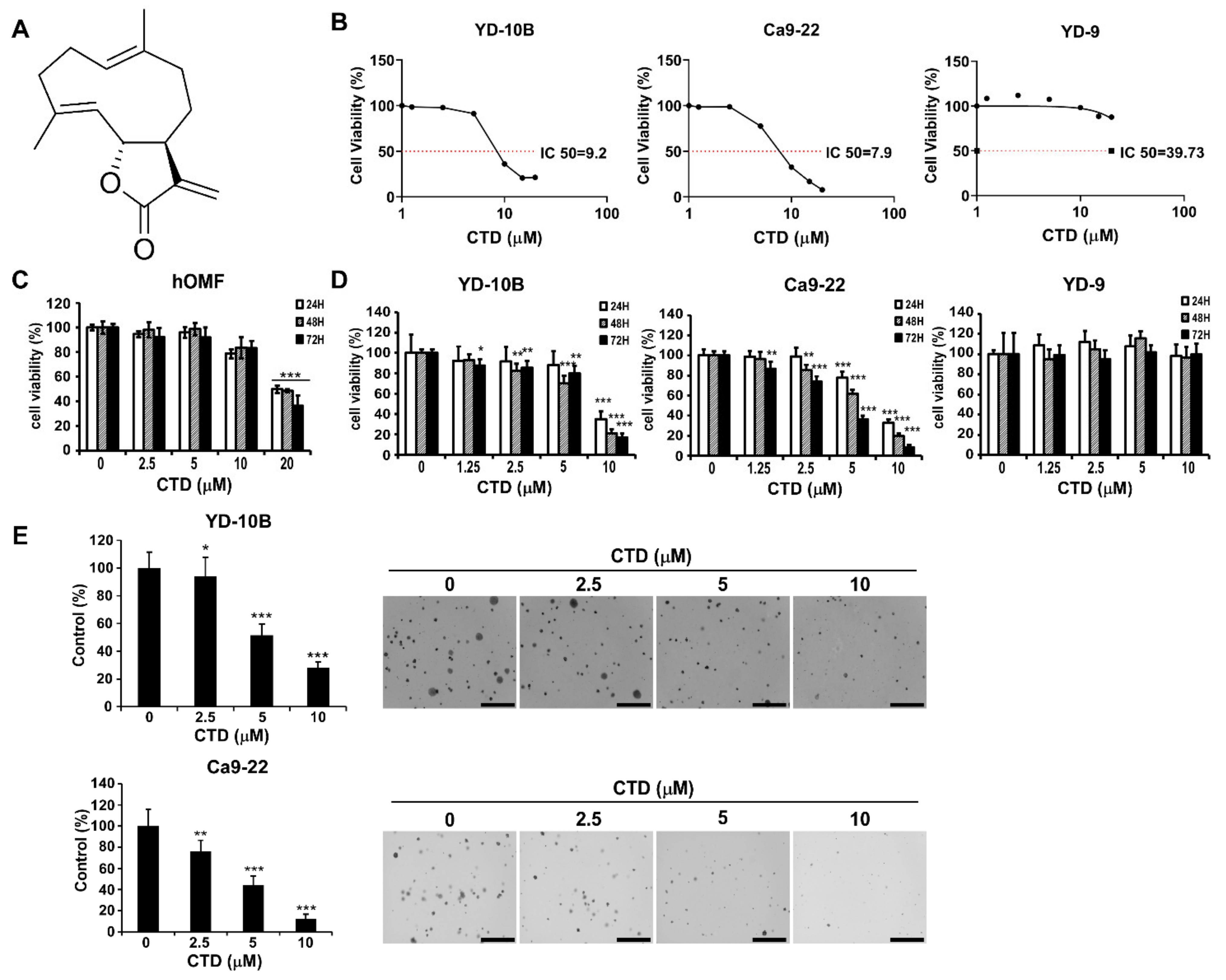
2.3. The Effect of Costunolide on the Viability of Oral Cancer Cells
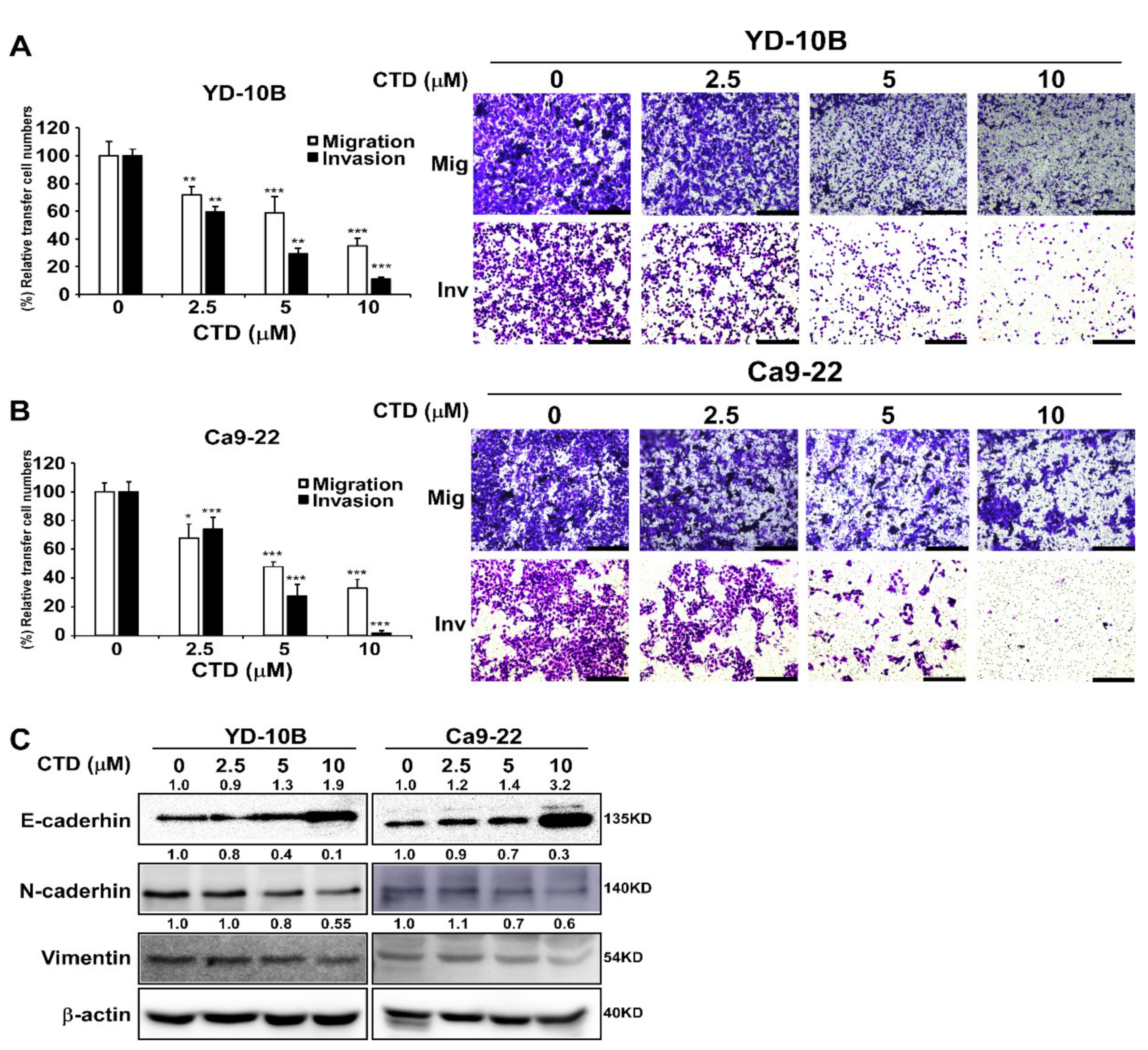
2.4. Costunolide Efficiently Inhibits Cell Migration and Invasion
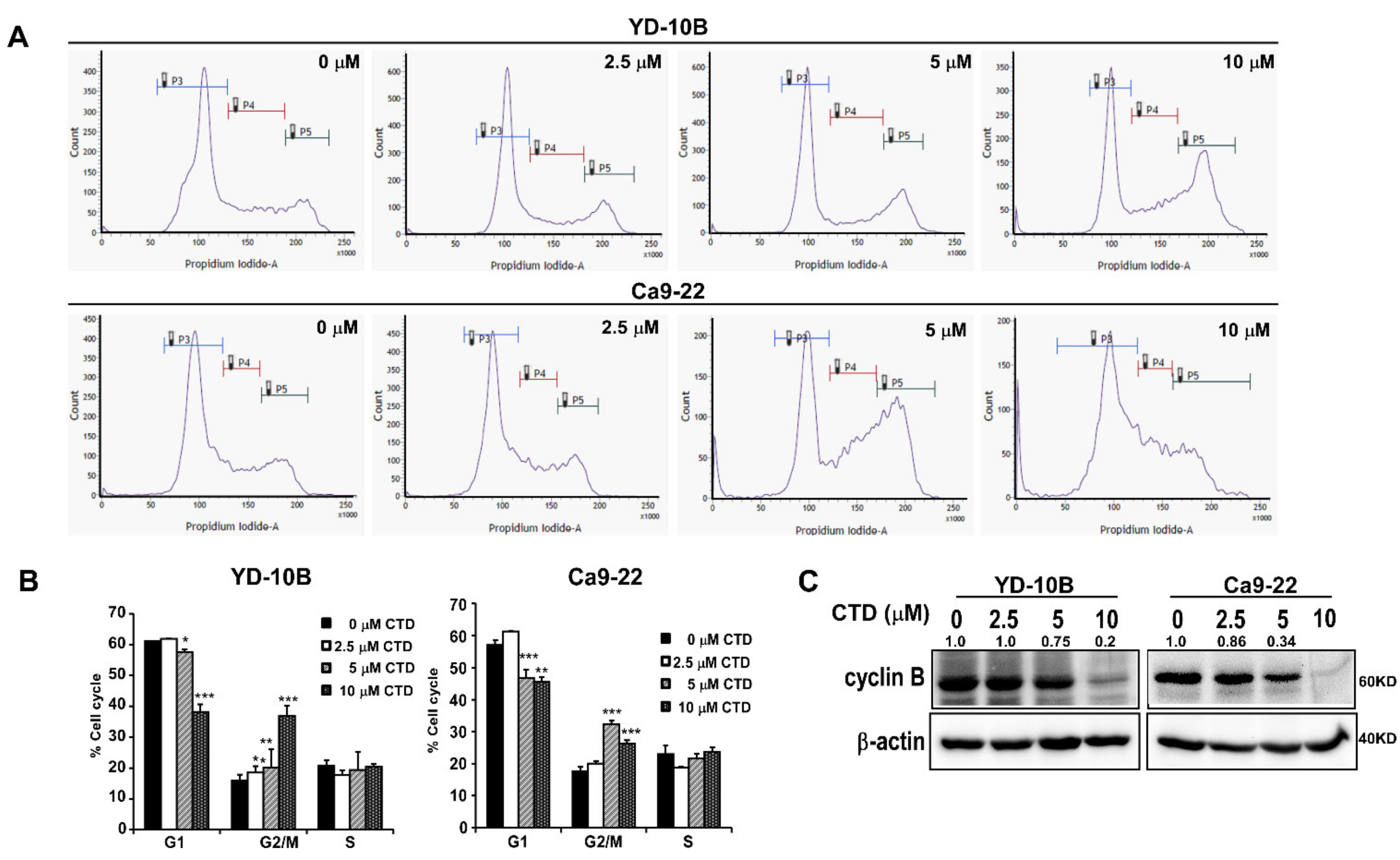
2.5. Costunolide Induces Cell Cycle Arrest in the G2/M Phase
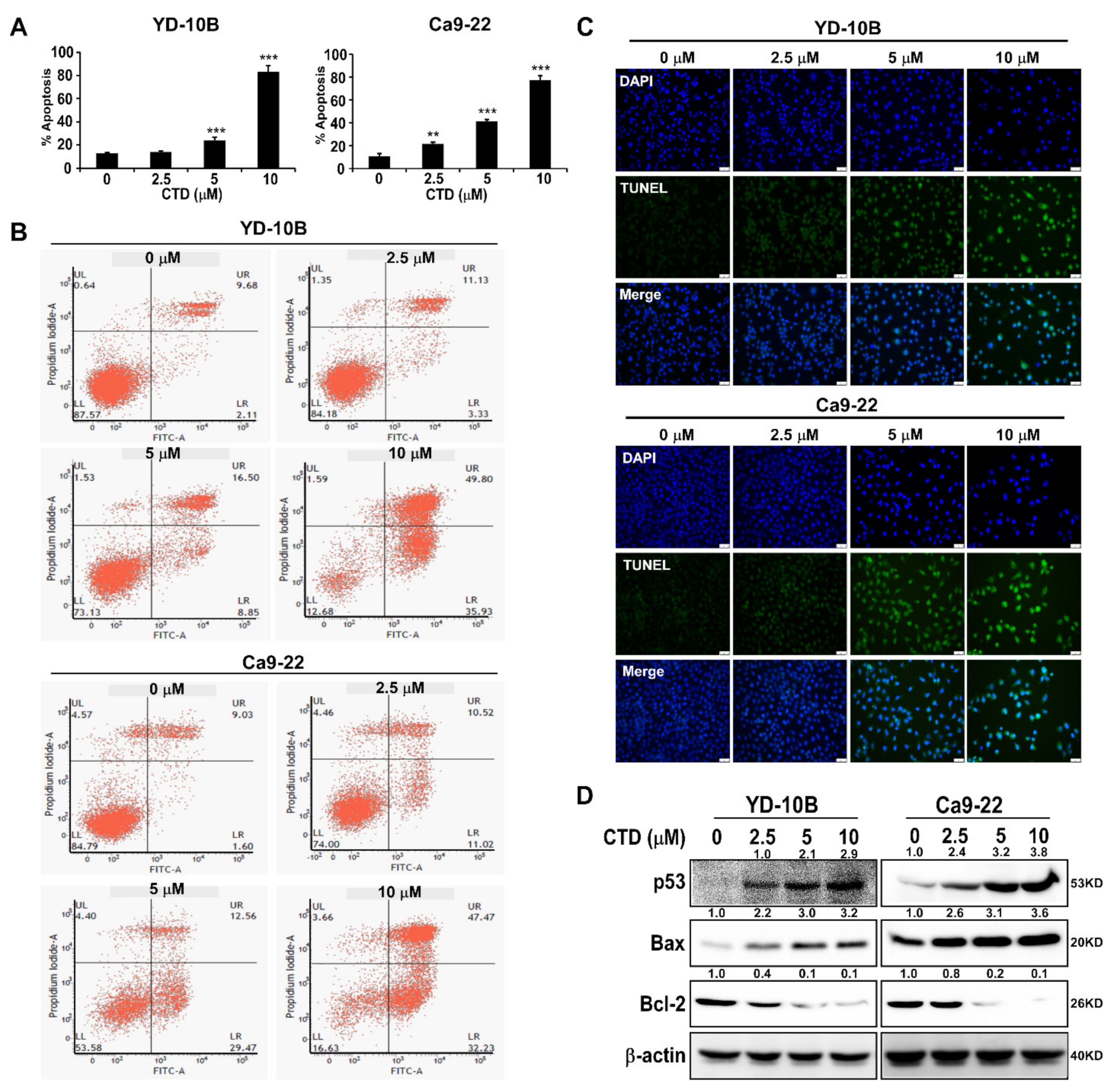
2.6. Costunolide Triggers Apoptosis of Oral Cancer Cells
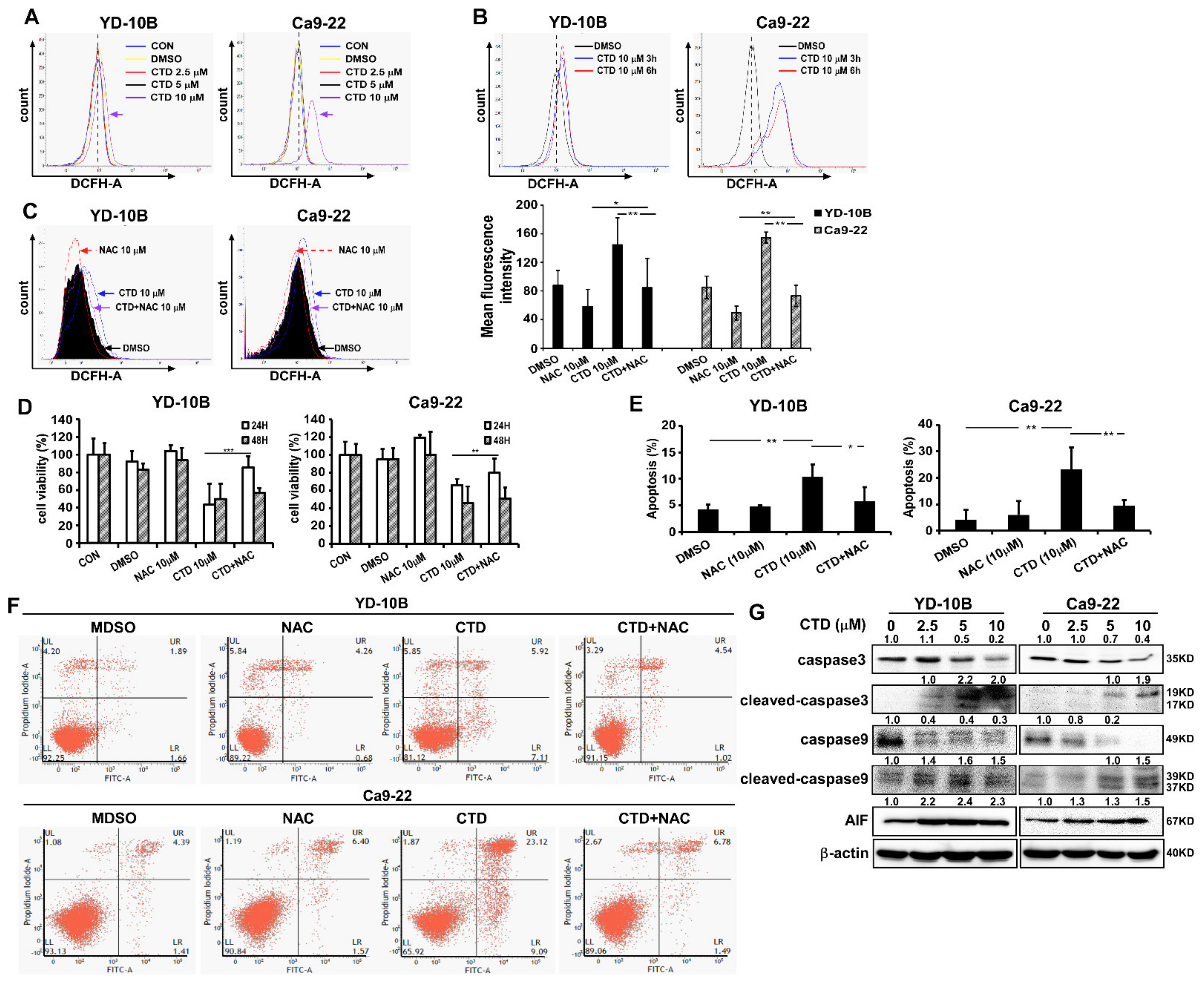
2.7. ROS Induction in Costunolide-Treated Oral Cancer Cells
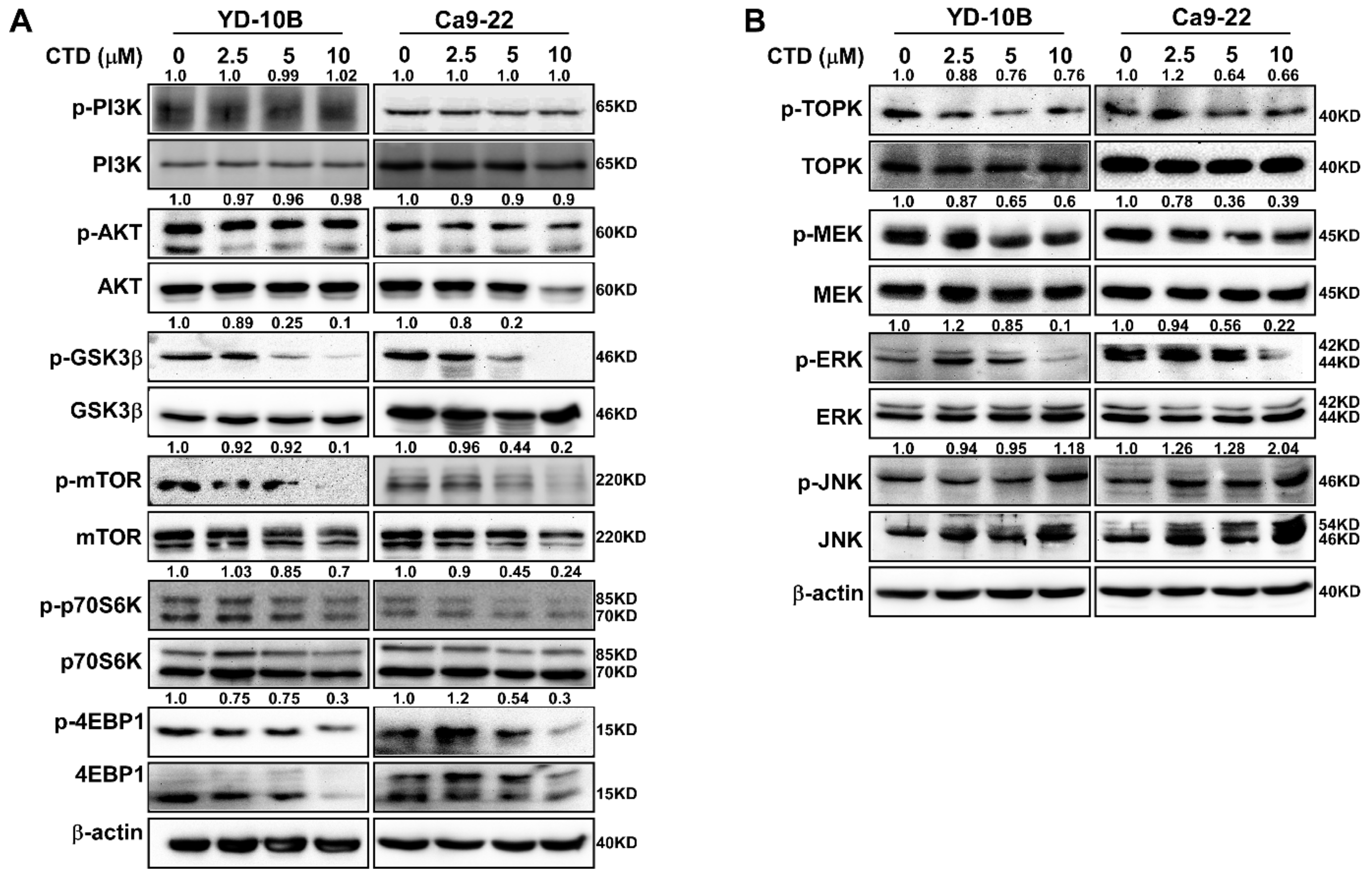
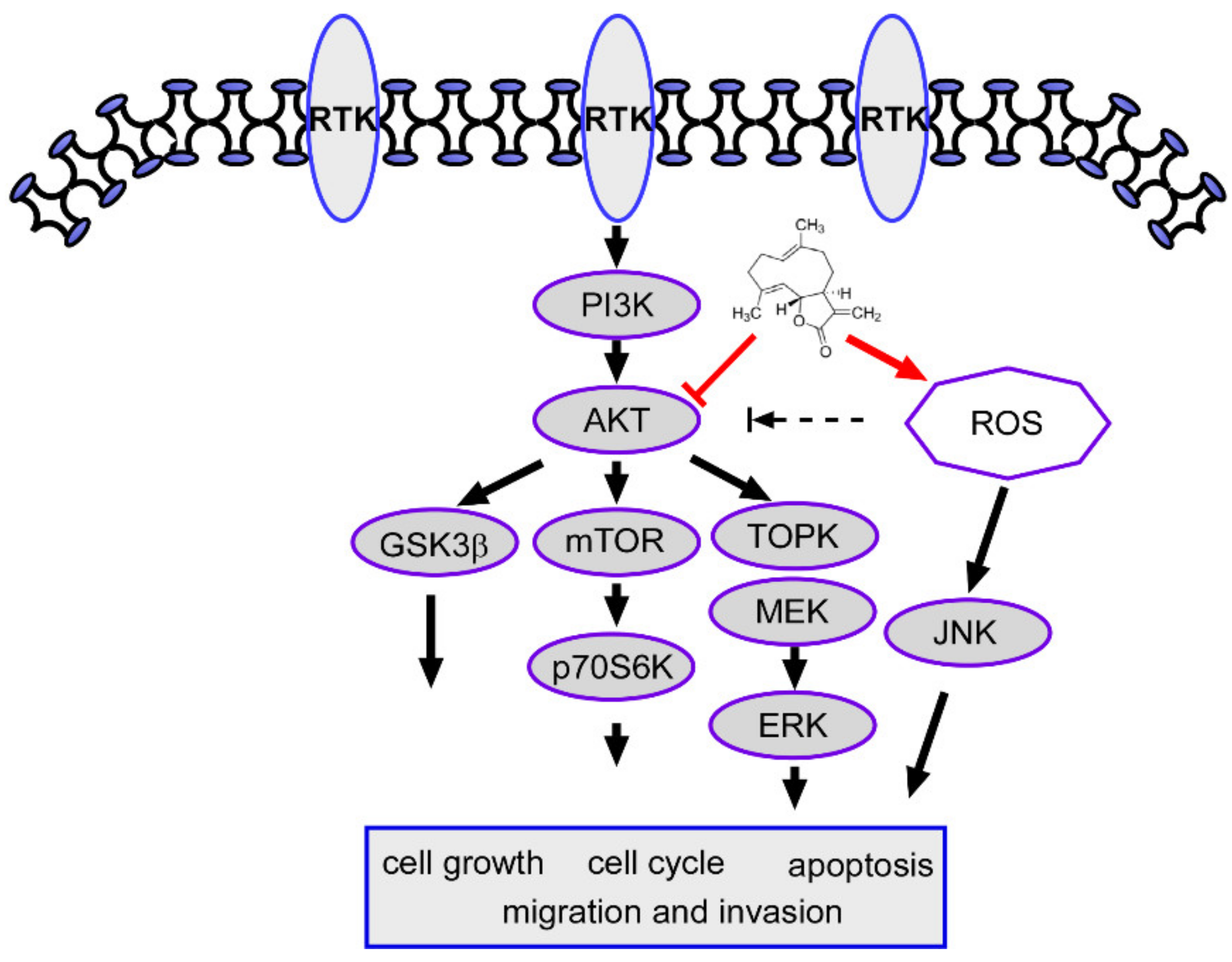
2.8. Costunolide Inhibits Oral Cancer Cell Growth through the AKT Pathway
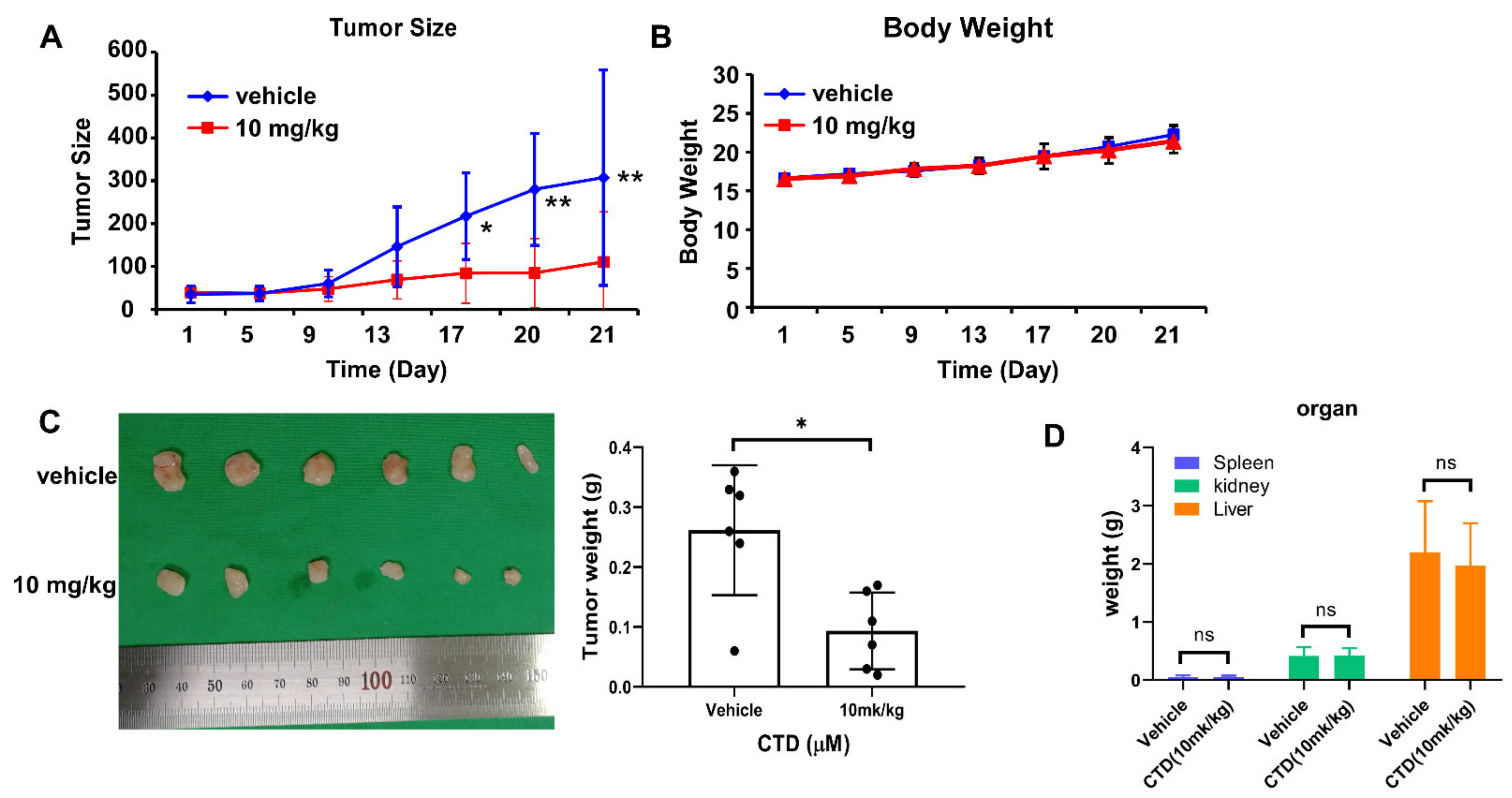
2.9. Costunolide Inhibits Tumor Growth of Cell-Derived-Xenograft (CDX)
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
4.3. Analysis of AKT Expression
4.4. Cell Viability Assay
4.5. Cell Cycle and Apoptosis Analysis
4.6. TUNEL Assay
4.7. Migration and Invasion Assays
4.8. Measurement of Reactive Oxygen Species Generation
4.9. Western Blot Assay
4.10. AKT Kinase Analysis In Vitro
4.11. Binding Assay
4.12. Lentiviral Infection
4.13. In Vivo Analysis
4.14. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Khurshid, Z.; Zafar, M.S.; Khan, R.S.; Najeeb, S.; Slowey, P.D.; Rehman, I.U. Role of Salivary Biomarkers in Oral Cancer Detection. Adv. Clin. Chem. 2018, 86, 23–70. [Google Scholar] [CrossRef]
- Kumar, M.; Nanavati, R.; Modi, T.G.; Dobariya, C. Oral cancer: Etiology and risk factors: A review. J. Cancer Res. Ther. 2016, 12, 458–463. [Google Scholar] [CrossRef] [PubMed]
- Deschler, D.G.; Richmon, J.D.; Khariwala, S.S.; Ferris, R.L.; Wang, M.B. The “New” Head and Neck Cancer Patient-Young, Nonsmoker, Nondrinker, and HPV Positive: Evaluation. Otolaryngol. Head Neck Surg. 2014, 151, 375–380. [Google Scholar] [CrossRef] [Green Version]
- Chai, A.W.Y.; Lim, K.P.; Cheong, S.C. Translational genomics and recent advances in oral squamous cell carcinoma. Semin. Cancer Biol. 2020, 61, 71–83. [Google Scholar] [CrossRef]
- Jitender, S.; Sarika, G.; Varada, H.R.; Omprakash, Y.; Mohsin, K. Screening for oral cancer. J. Exp. Ther. Oncol. 2016, 11, 303–307. [Google Scholar] [PubMed]
- Chu, C.; Shang, W.; Sun, Y.; Zhang, X. Anlotinib is effective in patients with advanced oral cancer? Med. Hypotheses 2020, 137, 109578. [Google Scholar] [CrossRef]
- Yang, H.; Mo, C.; Xun, Y.; Liu, L.G.; Li, W.; Guan, J.; Liu, J.; Wu, J.; Yang, A.; Zheng, S.; et al. Combination of cetuximab with met inhibitor in control of cetuximab-resistant oral squamous cell carcinoma. Am. J. Transl. Res. 2019, 11, 2370–2381. [Google Scholar] [PubMed]
- Qiao, X.W.; Jiang, J.; Pang, X.; Huang, M.C.; Tang, Y.J.; Liang, X.H.; Tang, Y.L. The Evolving Landscape of PD-1/PD-L1 Pathway in Head and Neck Cancer. Front. Immunol. 2020, 11, 1721. [Google Scholar] [CrossRef]
- Nandini, D.B.; Rao, R.S.; Hosmani, J.; Khan, S.; Patil, S.; Awan, K.H. Novel therapies in the management of oral cancer: An update. Dis. Mon. 2020, 101036. [Google Scholar] [CrossRef] [PubMed]
- Mazumdar, T.; Byers, L.A.; Ng, P.K.S.; Mills, G.B.; Peng, S.H.; Diao, L.X.; Fan, Y.H.; Stemke-Hale, K.; Heymach, J.V.; Myers, J.N.; et al. A Comprehensive Evaluation of Biomarkers Predictive of Response to PI3K Inhibitors and of Resistance Mechanisms in Head and Neck Squamous Cell Carcinoma. Mol. Cancer Ther. 2014, 13, 2738–2750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harsha, C.; Banik, K.; Ang, H.L.; Girisa, S.; Vikkurthi, R.; Parama, D.; Rana, V.; Shabnam, B.; Khatoon, E.; Kumar, A.P.; et al. Targeting AKT/mTOR in Oral Cancer: Mechanisms and Advances in Clinical Trials. Int. J. Mol. Sci. 2020, 21, 3285. [Google Scholar] [CrossRef]
- Marquard, F.E.; Jucker, M. PI3K/AKT/mTOR signaling as a molecular target in head and neck cancer. Biochem. Pharmacol. 2020, 172. [Google Scholar] [CrossRef] [PubMed]
- Chang, F.; Lee, J.T.; Navolanic, P.M.; Steelman, L.S.; Shelton, J.G.; Blalock, W.L.; Franklin, R.A.; McCubrey, J.A. Involvement of PI3K/Akt pathway in cell cycle progression, apoptosis, and neoplastic transformation: A target for cancer chemotherapy. Leukemia 2003, 17, 590–603. [Google Scholar] [CrossRef] [Green Version]
- Song, M.Q.; Liu, X.J.; Liu, K.D.; Zhao, R.; Huang, H.; Shi, Y.Y.; Zhang, M.; Zhou, S.L.; Xie, H.; Chen, H.Y.; et al. Targeting AKT with Oridonin Inhibits Growth of Esophageal Squamous Cell Carcinoma In Vitro and Patient-Derived Xenografts In Vivo. Mol. Cancer Ther. 2018, 17, 1540–1553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, M.Q.; Bode, A.M.; Dong, Z.G.; Lee, M.H. AKT as a Therapeutic Target for Cancer. Cancer Res. 2019, 79, 1019–1031. [Google Scholar] [CrossRef] [Green Version]
- Moloney, J.N.; Cotter, T.G. ROS signalling in the biology of cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, V.; Hay, N. Molecular Pathways: Reactive Oxygen Species Homeostasis in Cancer Cells and Implications for Cancer Therapy. Clin. Cancer Res. 2013, 19, 4309–4314. [Google Scholar] [CrossRef] [Green Version]
- Teppo, H.R.; Soini, Y.; Karihtala, P. Reactive Oxygen Species-Mediated Mechanisms of Action of Targeted Cancer Therapy. Oxidative Med. Cell. Longev. 2017, 2017. [Google Scholar] [CrossRef]
- Wang, J.; Li, X.M.; Bai, Z.; Chi, B.X.; Wei, Y.; Chen, X. Curcumol induces cell cycle arrest in colon cancer cells via reactive oxygen species and Akt/GSK3 beta/cyclin D1 pathway. J. Ethnopharmacol. 2018, 210, 1–9. [Google Scholar] [CrossRef]
- Hong, Y.N.; Fan, D.D. Ginsenoside Rk1 induces cell death through ROS-mediated PTEN/PI3K/Akt/mTOR signaling pathway in MCF-7 cells. J. Funct. Foods 2019, 57, 255–265. [Google Scholar] [CrossRef]
- Zhuge, W.; Chen, R.; Vladimir, K.; Dong, X.; Zia, K.; Sun, X.; Dai, X.; Bao, M.; Shen, X.; Liang, G. Costunolide specifically binds and inhibits thioredoxin reductase 1 to induce apoptosis in colon cancer. Cancer Lett. 2018, 412, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Nobili, S.; Lippi, D.; Witort, E.; Donnini, M.; Bausi, L.; Mini, E.; Capaccioli, S. Natural compounds for cancer treatment and prevention. Pharmacol. Res. 2009, 59, 365–378. [Google Scholar] [CrossRef]
- Vidoni, C.; Ferraresi, A.; Secomandi, E.; Vallino, L.; Dhanasekaran, D.N.; Isidoro, C. Epigenetic targeting of autophagy for cancer prevention and treatment by natural compounds. Semin. Cancer Biol. 2020, 66, 34–44. [Google Scholar] [CrossRef]
- Surh, Y.J. Cancer chemoprevention with dietary phytochemicals. Nat. Rev. Cancer 2003, 3, 768–780. [Google Scholar] [CrossRef]
- Hua, P.Y.; Sun, M.; Zhang, G.X.; Zhang, Y.F.; Song, G.; Liu, Z.Y.; Li, X.; Zhang, X.Y.; Li, B.J. Costunolide Induces Apoptosis through Generation of ROS and Activation of P53 in Human Esophageal Cancer Eca-109 Cells. J. Biochem. Mol. Toxicol. 2016, 30, 462–469. [Google Scholar] [CrossRef]
- Chen, J.S.; Chen, B.S.; Zou, Z.H.; Li, W.; Zhang, Y.M.; Xie, J.L.; Liu, C.X. Costunolide enhances doxorubicin-induced apoptosis in prostate cancer cells via activated mitogen-activated protein kinases and generation of reactive oxygen species. Oncotarget 2017, 8, 107701–107715. [Google Scholar] [CrossRef] [Green Version]
- Peng, Z.X.; Wang, Y.; Fan, J.H.; Lin, X.J.; Liu, C.Y.; Xu, Y.; Ji, W.D.; Yan, C.; Su, C.Q. Costunolide and dehydrocostuslactone combination treatment inhibit breast cancer by inducing cell cycle arrest and apoptosis through c-Myc/p53 and AKT/14-3-3 pathway. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.Y.; Choi, B.Y. Costunolide-A Bioactive Sesquiterpene Lactone with Diverse Therapeutic Potential. Int. J. Mol. Sci. 2019, 20, 2926. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.C.; Li, J.; Wu, Y.N.; Gui, L.; Shen, Y. Costunolide Inhibits the Growth of OAW42-A Multidrug-Resistant Human Ovarian Cancer Cells by Activating Apoptotic and Autophagic Pathways, Production of Reactive Oxygen Species (ROS), Cleaved Caspase-3 and Cleaved Caspase-9. Med. Sci. Monit. 2019, 25, 3231–3237. [Google Scholar] [CrossRef]
- Liu, X.; Song, M.; Wang, P.; Zhao, R.; Chen, H.; Zhang, M.; Shi, Y.; Liu, K.; Liu, F.; Yang, R.; et al. Targeted therapy of the AKT kinase inhibits esophageal squamous cell carcinoma growth in vitro and in vivo. Int. J. Cancer 2019. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Yi, J.; Park, S.; Zhang, H.; Kim, E.; Park, S.; Kwon, W.; Jang, S.; Zhang, X.; Chen, H.; et al. Costunolide suppresses melanoma growth via the AKT/mTOR pathway in vitro and in vivo. Am. J. Cancer Res. 2021, 11, 1410–1427. [Google Scholar]
- Ye, Q.; Cai, W.J.; Zheng, Y.B.; Evers, B.M.; She, Q.B. ERK and AKT signaling cooperate to translationally regulate survivin expression for promotion of cell motility and metastasis in colorectal cancer. Cancer Res. 2013, 73. [Google Scholar] [CrossRef]
- Choi, Y.K.; Seo, H.S.; Choi, H.S.; Choi, H.S.; Kim, S.R.; Shin, Y.C.; Ko, S.G. Induction of Fas-mediated extrinsic apoptosis, p21WAF1-related G2/M cell cycle arrest and ROS generation by costunolide in estrogen receptor-negative breast cancer cells, MDA-MB-231. Mol. Cell Biochem. 2012, 363, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Avan, A.; Narayan, R.; Giovannetti, E.; Peters, G.J. Role of Akt signaling in resistance to DNA-targeted therapy. World J. Clin. Oncol. 2016, 7, 352–369. [Google Scholar] [CrossRef] [PubMed]
- Alzahrani, A.S. PI3K/Akt/mTOR inhibitors in cancer: At the bench and bedside. Semin. Cancer Biol. 2019, 59, 125–132. [Google Scholar] [CrossRef]
- Liu, C.Y.; Chang, H.S.; Chen, I.S.; Chen, C.J.; Hsu, M.L.; Fu, S.L.; Chen, Y.J. Costunolide causes mitotic arrest and enhances radiosensitivity in human hepatocellular carcinoma cells. Radiat Oncol. 2011, 6, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, P.Y.; Zhang, G.X.; Zhang, Y.F.; Sun, M.; Cui, R.J.; Li, X.; Li, B.J.; Zhang, X.Y. Costunolide induces G1/S phase arrest and activates mitochondrial-mediated apoptotic pathways in SK-MES 1 human lung squamous carcinoma cells. Oncol. Lett. 2016, 11, 2780–2786. [Google Scholar] [CrossRef] [Green Version]
- Hu, M.; Liu, L.; Yao, W. Activation of p53 by costunolide blocks glutaminolysis and inhibits proliferation in human colorectal cancer cells. Gene 2018, 678, 261–269. [Google Scholar] [CrossRef]
- Yan, Z.; Xu, T.; An, Z.; Hu, Y.; Chen, W.; Ma, J.; Shao, C.; Zhu, F. Costunolide induces mitochondria-mediated apoptosis in human gastric adenocarcinoma BGC-823 cells. BMC Complement. Altern Med. 2019, 19, 151. [Google Scholar] [CrossRef] [Green Version]
- Ge, M.X.; Liu, H.T.; Zhang, N.; Niu, W.X.; Lu, Z.N.; Bao, Y.Y.; Huang, R.; Yu, D.K.; Shao, R.G.; He, H.W. Costunolide represses hepatic fibrosis through WW domain-containing protein 2-mediated Notch3 degradation. Br. J. Pharmacol. 2020, 177, 372–387. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Li, L.; Jiang, J.; Zhao, C.; Yang, C. Costunolide enhances sensitivity of K562/ADR chronic myeloid leukemia cells to doxorubicin through PI3K/Akt pathway. Phytother Res. 2019. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Park, S.; Zhang, H.; Park, S.; Kwon, W.; Kim, E.; Zhang, X.; Jang, S.; Yoon, D.; Choi, S.K.; et al. Targeting AKT with costunolide suppresses the growth of colorectal cancer cells and induces apoptosis in vitro and in vivo. J. Exp. Clin. Cancer Res. 2021, 40, 114. [Google Scholar] [CrossRef]
- Alkhadar, H.; Macluskey, M.; White, S.; Ellis, I. Nerve growth factor-induced migration in oral and salivary gland tumour cells utilises the PI3K/Akt signalling pathway: Is there a link to perineural invasion? J. Oral Pathol. Med. 2020, 49, 227–234. [Google Scholar] [CrossRef]
- Fulda, S. Modulation of Apoptosis by Natural Products for Cancer Therapy. Planta Med. 2010, 76, 1075–1079. [Google Scholar] [CrossRef] [Green Version]
- Fu, D.A.; Wu, D.; Cheng, W.; Gao, J.P.; Zhang, Z.Y.; Ge, J.P.; Zhou, W.Q.; Xu, Z.Y. Costunolide Induces Autophagy and Apoptosis by Activating ROS/MAPK Signaling Pathways in Renal Cell Carcinoma. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef]
- Vo, T.T.T.; Liu, J.F.; Wu, C.Z.; Lin, W.N.; Chen, Y.L.; Lee, I.T. Surfactin from Bacillus subtilis induces apoptosis in human oral squamous cell carcinoma through ROS-regulated mitochondrial pathway. J. Cancer 2020, 11, 7253–7263. [Google Scholar] [CrossRef]
- Lee, N.P.; Chan, C.M.; Tung, L.N.; Wang, H.K.; Law, S. Tumor xenograft animal models for esophageal squamous cell carcinoma. J. Biomed. Sci. 2018, 25, 66. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, H.; Yi, J.-K.; Lim, S.-G.; Park, S.; Zhang, H.; Kim, E.; Jang, S.; Lee, M.-H.; Liu, K.; Kim, K.-R.; et al. Costunolide Induces Apoptosis via the Reactive Oxygen Species and Protein Kinase B Pathway in Oral Cancer Cells. Int. J. Mol. Sci. 2021, 22, 7509. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147509
Huang H, Yi J-K, Lim S-G, Park S, Zhang H, Kim E, Jang S, Lee M-H, Liu K, Kim K-R, et al. Costunolide Induces Apoptosis via the Reactive Oxygen Species and Protein Kinase B Pathway in Oral Cancer Cells. International Journal of Molecular Sciences. 2021; 22(14):7509. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147509
Chicago/Turabian StyleHuang, Hai, Jun-Koo Yi, Su-Geun Lim, Sijun Park, Haibo Zhang, Eungyung Kim, Soyoung Jang, Mee-Hyun Lee, Kangdong Liu, Ki-Rim Kim, and et al. 2021. "Costunolide Induces Apoptosis via the Reactive Oxygen Species and Protein Kinase B Pathway in Oral Cancer Cells" International Journal of Molecular Sciences 22, no. 14: 7509. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22147509