
Exploration of Novel Xanthine Oxidase Inhibitors Based on 1,6-Dihydropyrimidine-5-Carboxylic Acids by an Integrated in Silico Study

Abstract
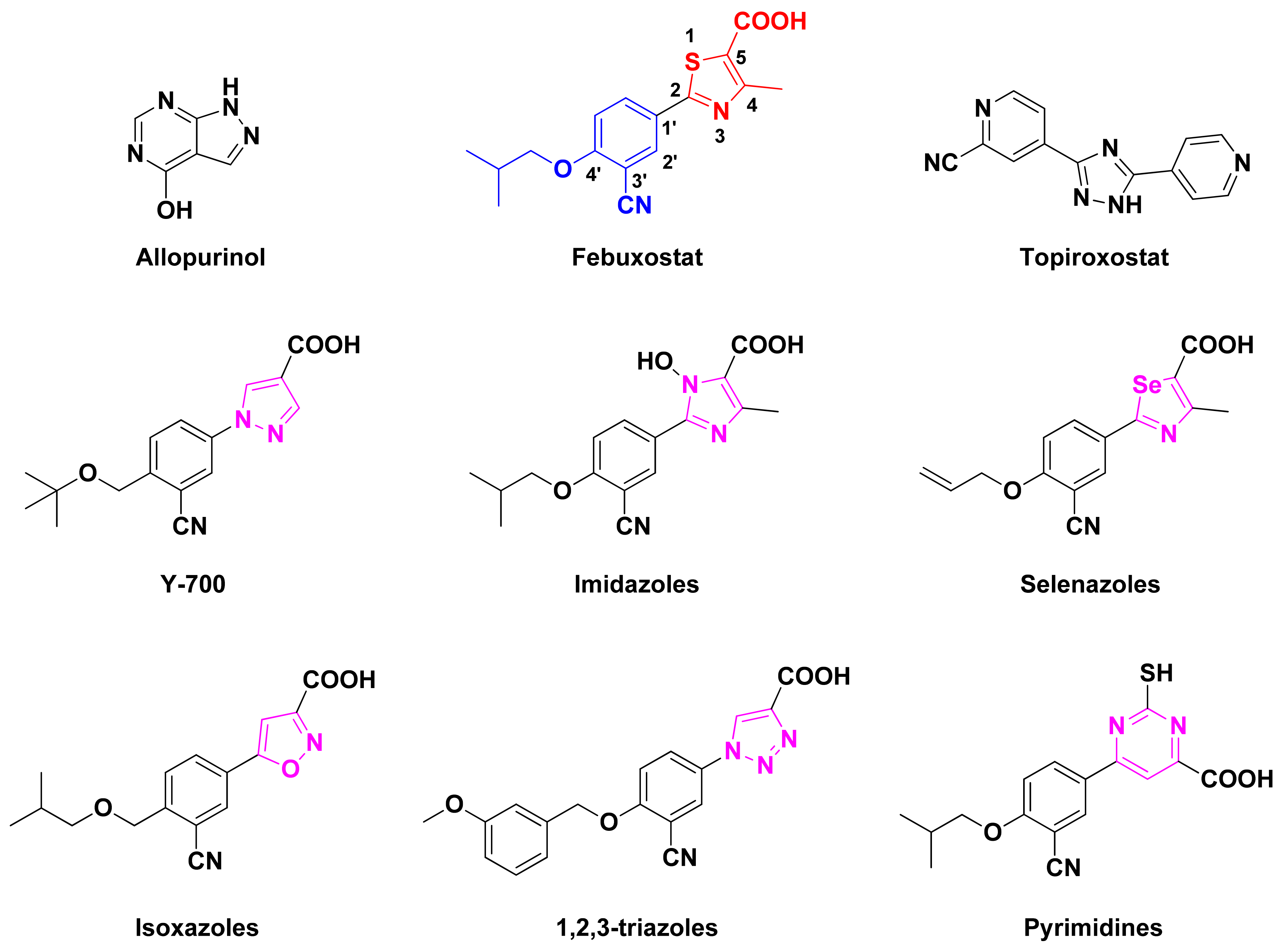
:1. Introduction
2. Results and Discussion
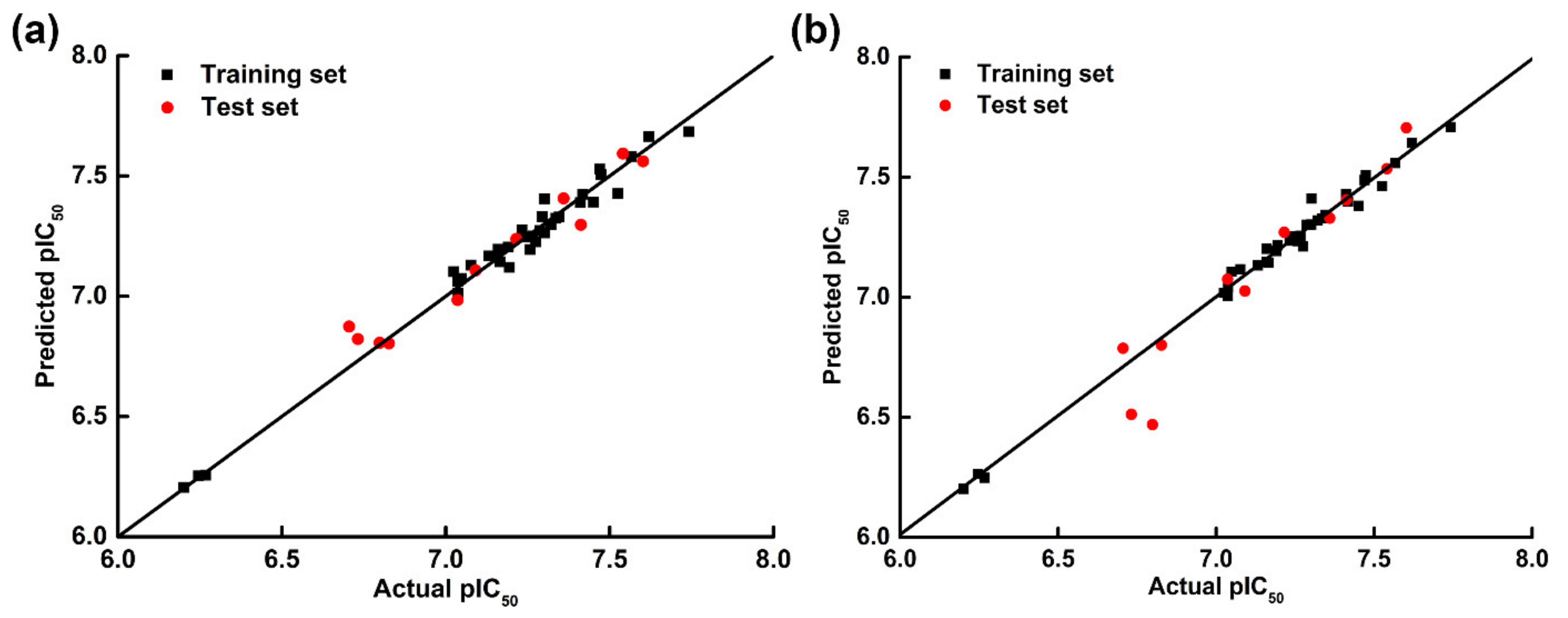
2.1. CoMFA and CoMSIA Statistical Results
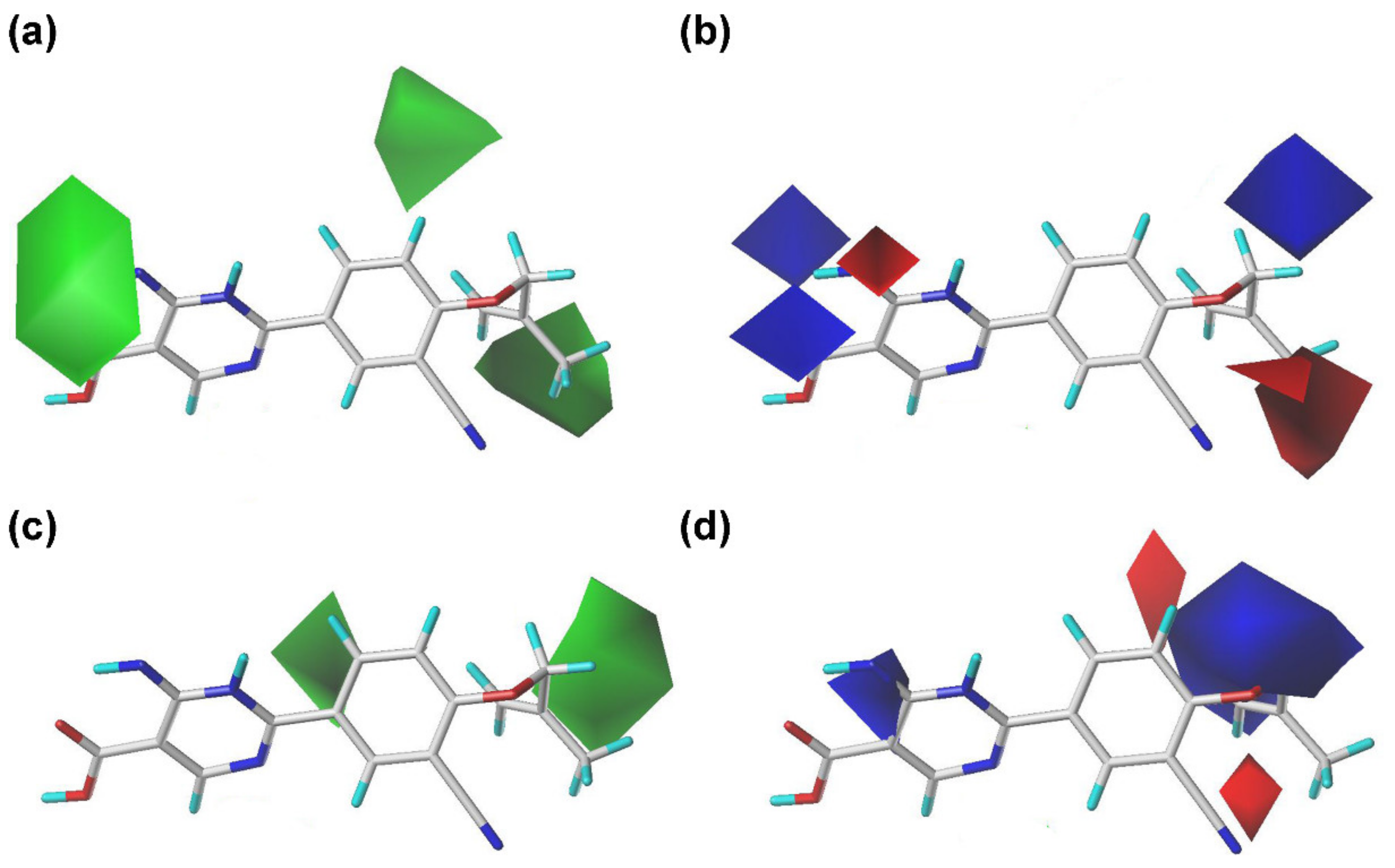
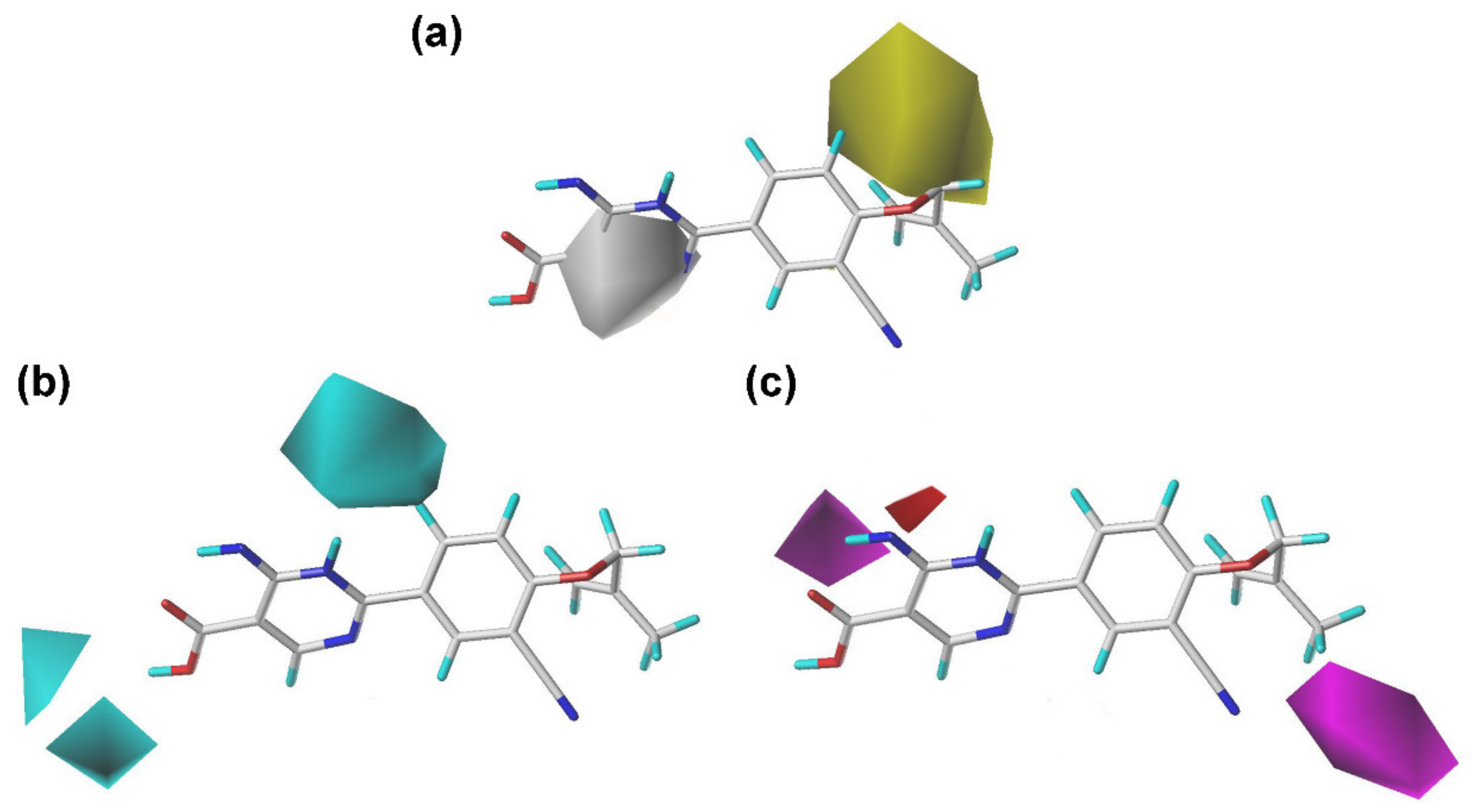
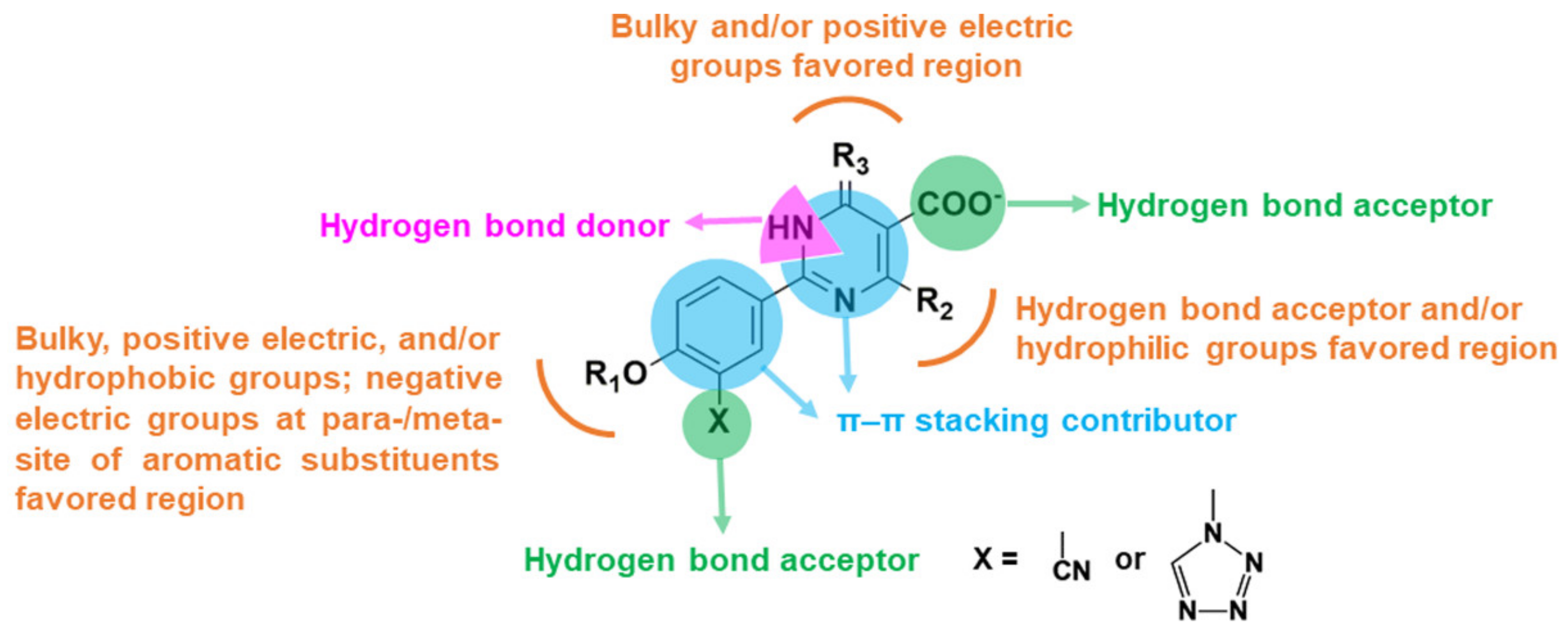
2.2. Contour Maps of the CoMFA and CoMSIA Models
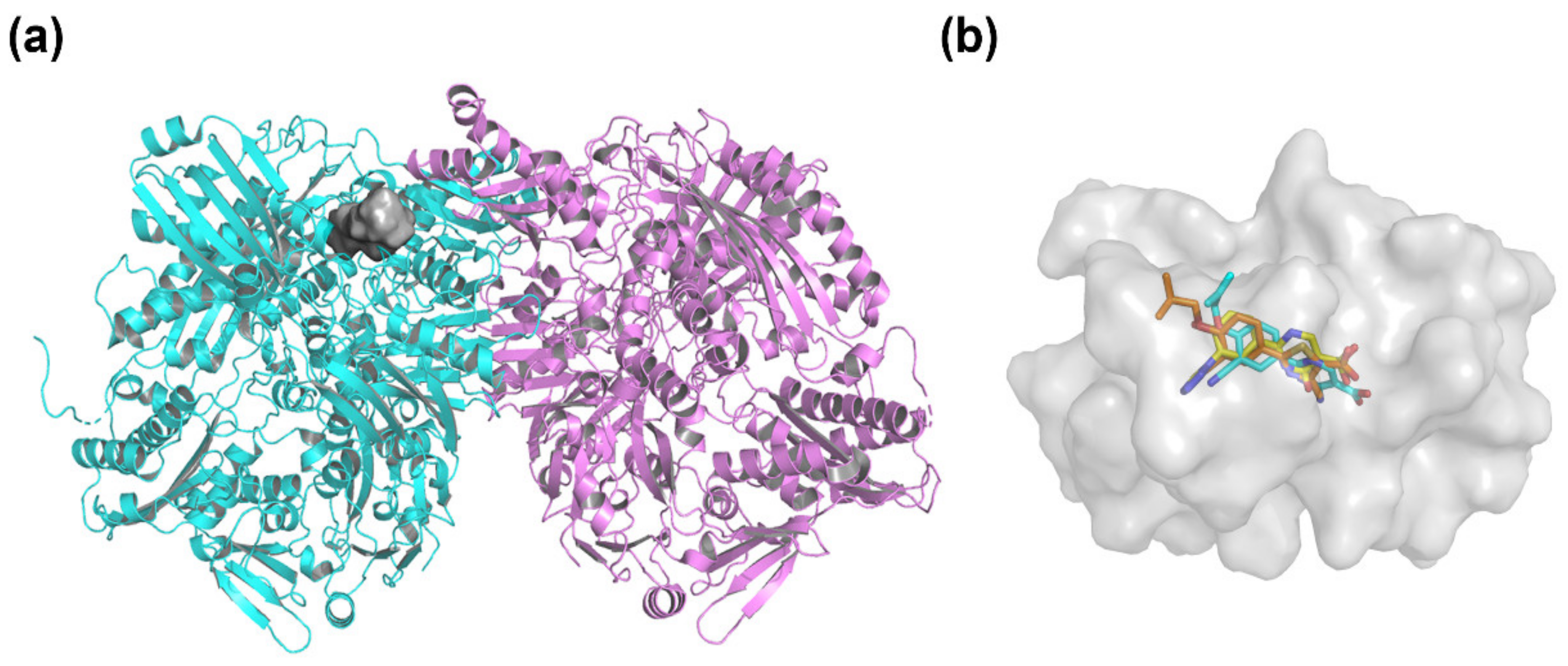
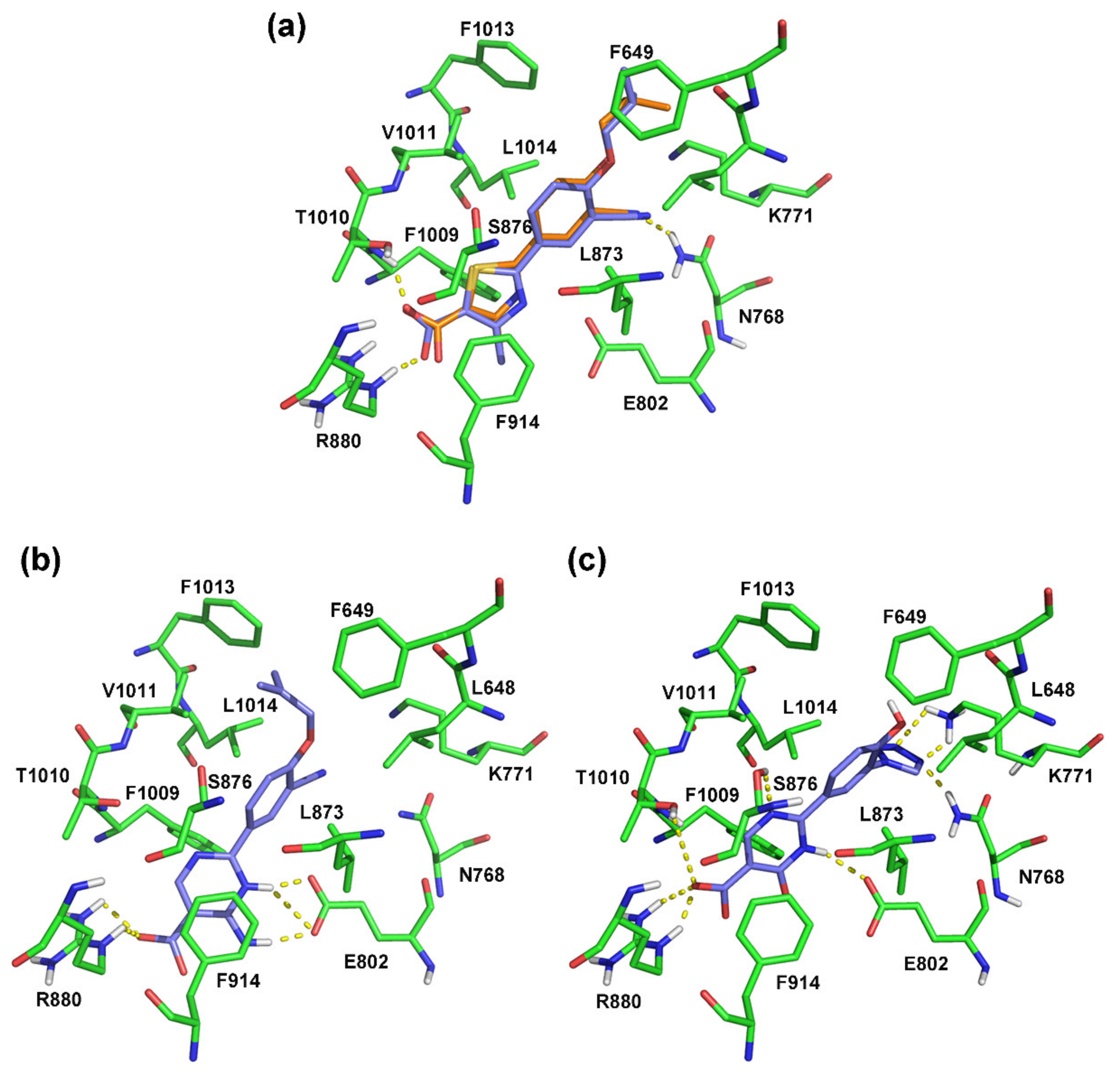
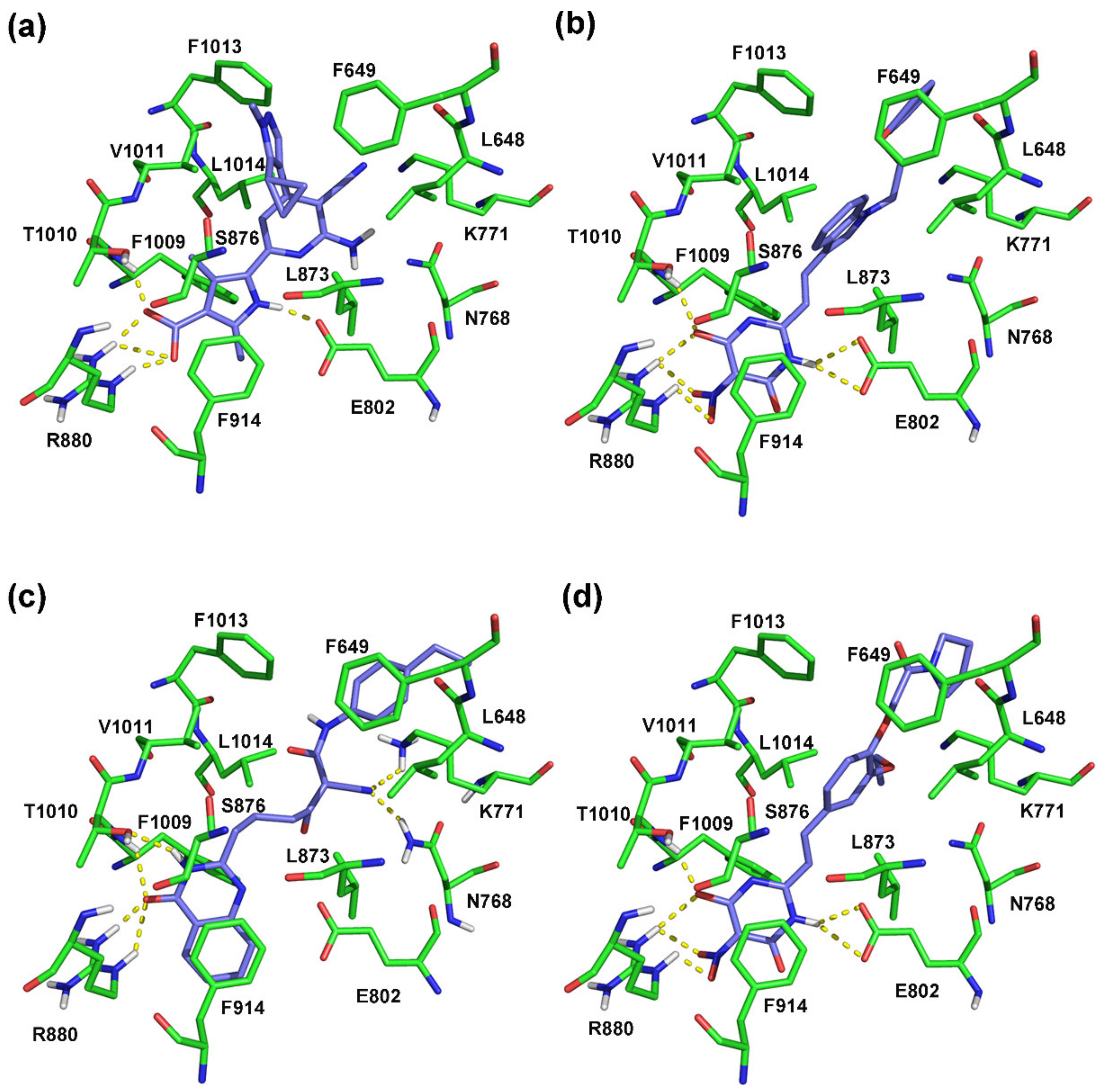
2.3. Molecular Docking
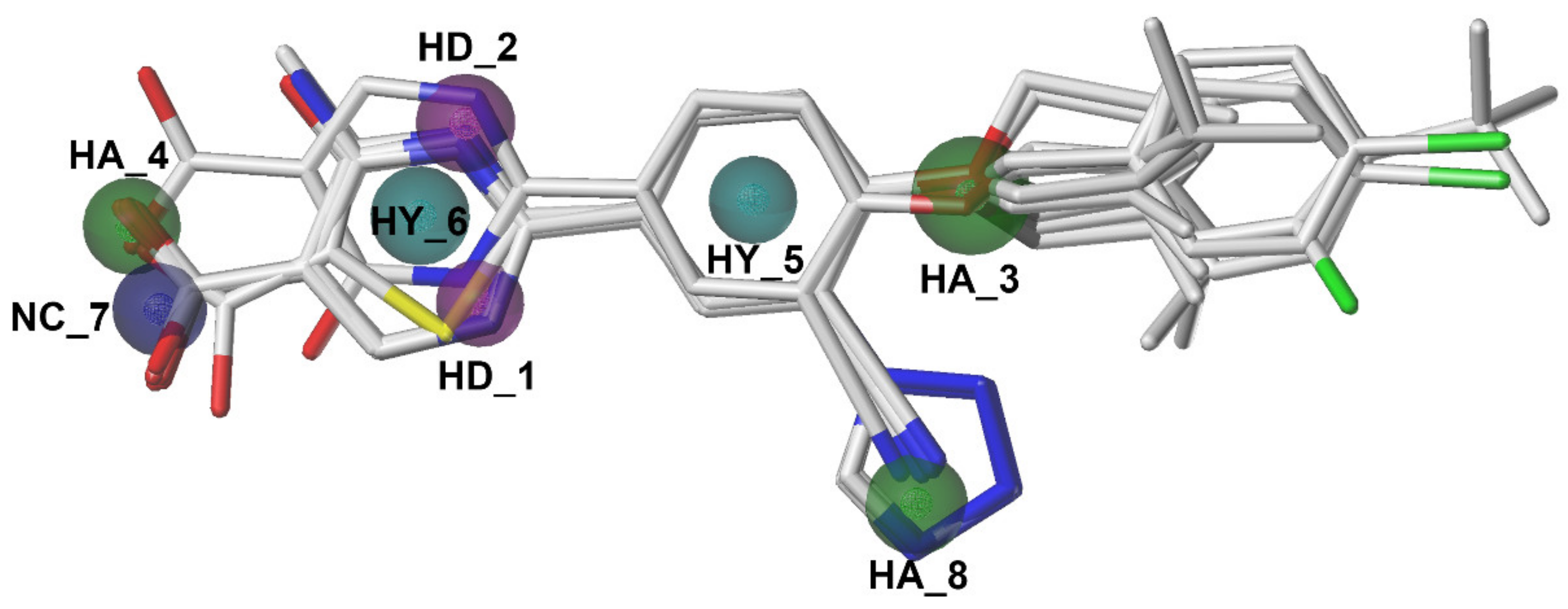
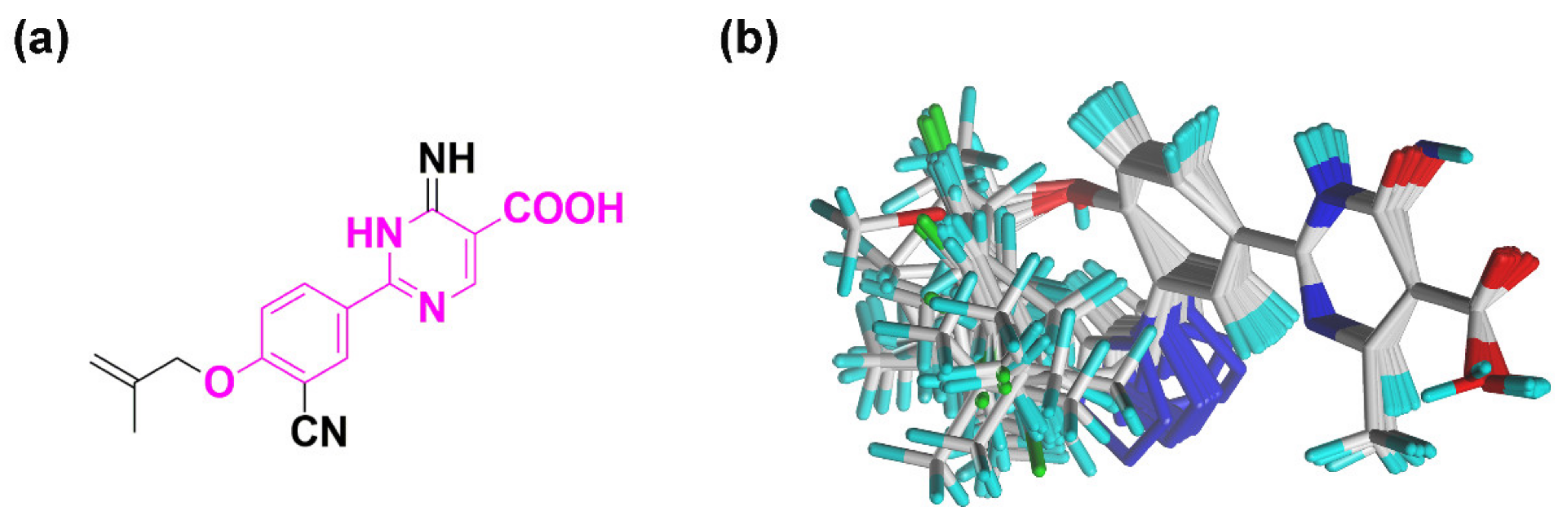
2.4. Pharmacophore Model
2.5. Virtual Screening and Docking Analysis
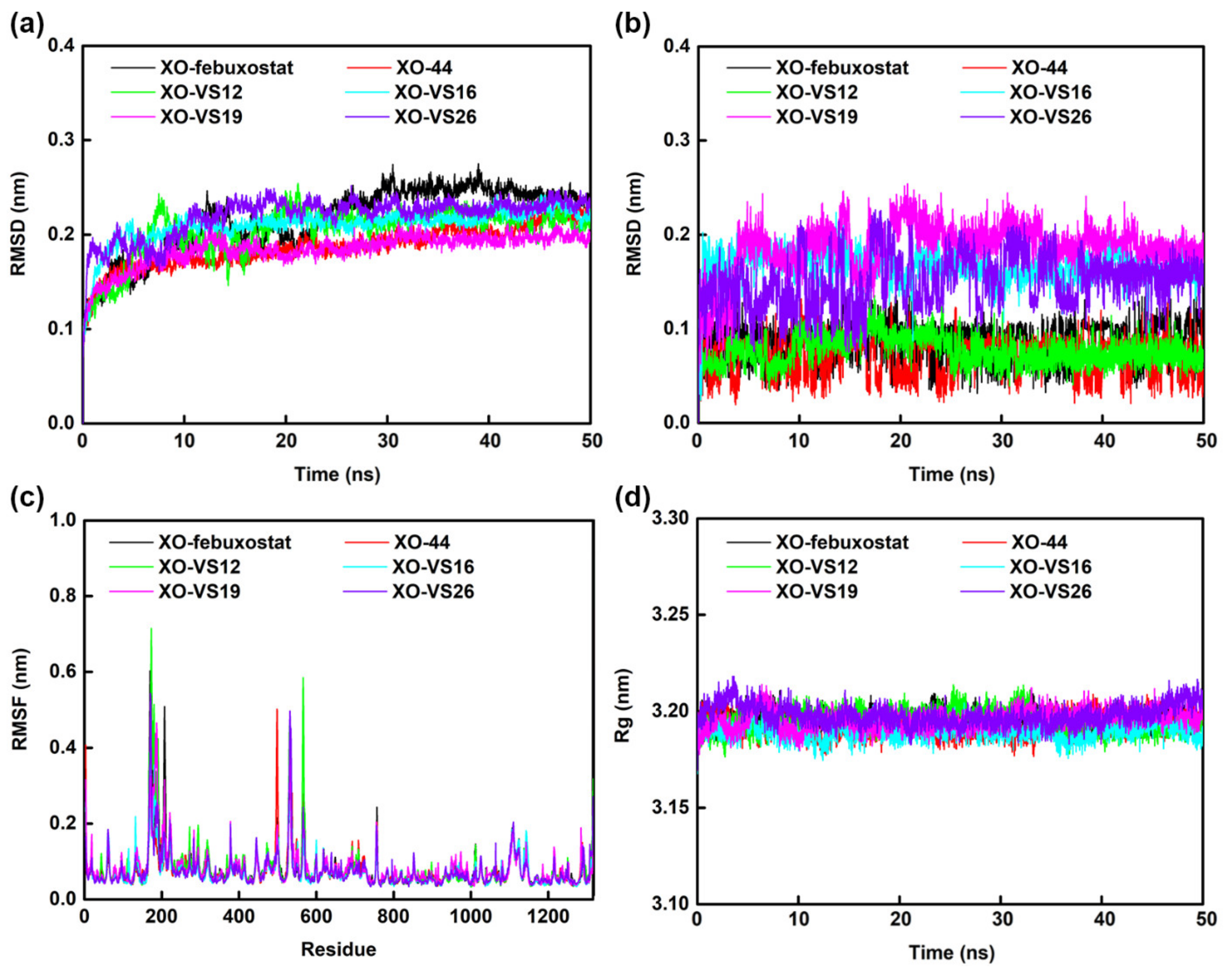
2.6. Molecular Dynamics Simulations
3. Materials and Methods
3.1. Molecules Set and Optimization
3.2. Molecular Modeling and Molecular Alignment
3.3. Model Validation
3.4. Molecular Docking
3.5. Pharmacophore Model
3.6. Virtual Screening
3.7. Molecular Dynamics Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, Y.; Gao, Y.; Wu, F.; Luo, X.; Ju, X.; Liu, G. Computationally exploring novel xanthine oxidase inhibitors using docking-based 3D-QSAR, molecular dynamics, and virtual screening. N. J. Chem. 2020, 44, 19276–19287. [Google Scholar] [CrossRef]
- Burns, C.M.; Wortmann, R.L. Gout therapeutics: New drugs for an old disease. Lancet 2011, 377, 165–177. [Google Scholar] [CrossRef]
- Zhang, B.; Dai, X.; Bao, Z.; Mao, Q.; Duan, Y.; Yang, Y.; Wang, S. Targeting the subpocket in xanthine oxidase: Design, synthesis, and biological evaluation of 2-[4-alkoxy-3-(1H-tetrazol-1-yl)phenyl]-6-oxo-1,6-dihydropyrimidine-5-carboxylic acid derivatives. Eur. J. Med. Chem. 2019, 181, 111559. [Google Scholar] [CrossRef]
- Mao, Q.; Dai, X.; Xu, G.; Su, Y.; Zhang, B.; Liu, D.; Wang, S. Design, synthesis and biological evaluation of 2-(4-alkoxy-3-cyano)phenyl-6-oxo-1,6-dihydropyrimidine-5-carboxylic acid derivatives as novel xanthine oxidase inhibitors. Eur. J. Med. Chem. 2019, 181, 111558. [Google Scholar] [CrossRef]
- Guan, Q.; Cheng, Z.; Ma, X.; Wang, L.; Feng, D.; Cui, Y.; Bao, K.; Wu, L.; Zhang, W. Synthesis and bioevaluation of 2-phenyl-4-methyl-1,3-selenazole-5-carboxylic acids as potent xanthine oxidase inhibitors. Eur. J. Med. Chem. 2014, 85, 508–516. [Google Scholar] [CrossRef]
- Tang, H.; Zhao, D. Studies of febuxostat analogues as xanthine oxidase inhibitors through 3D-QSAR, Topomer CoMFA and molecular modeling. J. Iran. Chem. Soc. 2019, 16, 2659–2671. [Google Scholar] [CrossRef]
- Luna, G.; Dolzhenko, A.V.; Mancera, R.L. Inhibitors of xanthine oxidase: Scaffold diversity and structure-based drug design. ChemMedChem 2019, 14, 714–743. [Google Scholar] [CrossRef] [Green Version]
- Mehmood, A.; Ishaq, M.; Zhao, L.; Safdar, B.; Rehman, A.U.; Munir, M.; Raza, A.; Nadeem, M.; Iqbal, W.; Wang, C. Natural compounds with xanthine oxidase inhibitory activity: A review. Chem. Biol. Drug Des. 2019, 93, 387–418. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhang, T.; Wu, Q.; Olounfeh, K.M.; Zhang, Y.; Meng, F. Synthesis and biological evaluation of 5-benzyl-3-pyridyl-1H-1,2,4-triazole derivatives as xanthine oxidase inhibitors. Med. Chem. 2020, 16, 119–127. [Google Scholar] [CrossRef]
- Casas, A.I.; Nogales, C.; Mucke, H.A.M.; Petraina, A.; Cuadrado, A.; Rojo, A.I.; Ghezzi, P.; Jaquet, V.; Augsburger, F.; Dufrasne, F.; et al. On the clinical pharmacology of reactive oxygen species. Pharm. Rev. 2020, 72, 801–828. [Google Scholar] [CrossRef] [PubMed]
- Dhiman, R.; Sharma, S.; Singh, G.; Nepali, K.; Singh Bedi, P.M. Design and synthesis of aza-flavones as a new class of xanthine oxidase inhibitors. Arch. Pharm. 2013, 346, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Smelcerovic, A.; Tomovic, K.; Smelcerovic, Z.; Petronijevic, Z.; Kocic, G.; Tomasic, T.; Jakopin, Z.; Anderluh, M. Xanthine oxidase inhibitors beyond allopurinol and febuxostat; an overview and selection of potential leads based on in silico calculated physico-chemical properties, predicted pharmacokinetics and toxicity. Eur. J. Med. Chem. 2017, 135, 491–516. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Zhang, Y.; Tu, S.; Wu, Y.; Zhang, Z.; Meng, F. Design, synthesis and biological evaluation of N-(3-(1H-tetrazol-1-yl)phenyl)isonicotinamide derivatives as novel xanthine oxidase inhibitors. Eur. J. Med. Chem. 2019, 183, 111717. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhou, H.; Mo, X.; Zhang, L.; Li, J. In silico study of febuxostat analogs as inhibitors of xanthine oxidoreductase: A combined 3D-QSAR and molecular docking study. J. Mol. Struct. 2019, 1181, 428–435. [Google Scholar] [CrossRef]
- Okamoto, K.; Eger, B.T.; Nishino, T.; Kondo, S.; Pai, E.F.; Nishino, T. An extremely potent inhibitor of xanthine oxidoreductase. Crystal structure of the enzyme-inhibitor complex and mechanism of inhibition. J. Biol. Chem. 2003, 278, 1848–1855. [Google Scholar] [CrossRef] [Green Version]
- Okafor, O.N.; Farrington, K.; Gorog, D.A. Allopurinol as a therapeutic option in cardiovascular disease. Pharmacol. Ther. 2017, 172, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Malik, N.; Dhiman, P.; Khatkar, A. In silico and 3D QSAR studies of natural based derivatives as xanthine oxidase inhibitors. Curr. Top. Med. Chem. 2019, 19, 123–138. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Lv, Y.; Lei, Y.; Liu, D.; Feng, Y.; Zhao, J.; Chen, S.; Meng, F.; Wang, S. Design, synthesis and biological evaluation of 1-hydroxy-2-phenyl-4-pyridyl-1H-imidazole derivatives as xanthine oxidase inhibitors. Eur. J. Med. Chem. 2018, 146, 668–677. [Google Scholar] [CrossRef]
- Xu, X.; Deng, L.; Nie, L.; Chen, Y.; Liu, Y.; Xie, R.; Li, Z. Discovery of 2-phenylthiazole-4-carboxylic acid, a novel and potent scaffold as xanthine oxidase inhibitors. Bioorg. Med. Chem. Lett. 2019, 29, 525–528. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.V.; Mal, G.; Kaur, G.; Gupta, M.K.; Singh, A.; Nepali, K.; Singh, H.; Sharma, S.; PM, S.B. Benzoflavone derivatives as potent antihyperuricemic agents. Medchemcomm 2019, 10, 128–147. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, L.; Tian, J.; Ye, F.; Xiao, Z. Identification of xanthine oxidase inhibitors through hierarchical virtual screening. RSC Adv. 2020, 10, 27752–27763. [Google Scholar] [CrossRef]
- Singh, J.V.; Bedi, P.M.S.; Singh, H.; Sharma, S. Xanthine oxidase inhibitors: Patent landscape and clinical development (2015–2020). Expert Opin. Ther. Pat. 2020, 30, 769–780. [Google Scholar] [CrossRef]
- Peng, J.; Li, Y.; Zhou, Y.; Zhang, L.; Liu, X.; Zuo, Z. Pharmacophore modeling, molecular docking and molecular dynamics studies on natural products database to discover novel skeleton as non-purine xanthine oxidase inhibitors. J. Recept. Signal Transduct. Res. 2018, 38, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, J.; Ma, L.; Fu, P. Pharmacological urate-lowering approaches in chronic kidney disease. Eur. J. Med. Chem. 2019, 166, 186–196. [Google Scholar] [CrossRef]
- Kayikci, M.; Venkatakrishnan, A.J.; Scott-Brown, J.; Ravarani, C.N.J.; Flock, T.; Babu, M.M. Visualization and analysis of non-covalent contacts using the Protein Contacts Atlas. Nat. Struct. Mol. Biol. 2018, 25, 185–194. [Google Scholar] [CrossRef]
- Gao, Y.; Wang, H.; Wang, J.; Cheng, M. In silico studies on p21-activated kinase 4 inhibitors: Comprehensive application of 3D-QSAR analysis, molecular docking, molecular dynamics simulations, and MM-GBSA calculation. J. Biomol. Struct. Dyn. 2020, 38, 4119–4133. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Wan, Y.; Wang, W.; Fang, S.; Gu, S.; Ju, X. Docking-based 3D-QSAR and pharmacophore studies on diarylpyrimidines as non-nucleoside inhibitors of HIV-1 reverse transcriptase. Mol. Divers. 2019, 23, 107–121. [Google Scholar] [CrossRef]
- Gao, Y.; Chen, Y.; Tian, Y.; Zhao, Y.; Wu, F.; Luo, X.; Ju, X.; Liu, G. In silico study of 3-hydroxypyrimidine-2,4-diones as inhibitors of HIV RT-associated RNase H using molecular docking, molecular dynamics, 3D-QSAR, and pharmacophore models. N. J. Chem. 2019, 43, 17004–17017. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Shaaban, S.; Vervandier-Fasseur, D.; Andreoletti, P.; Zarrouk, A.; Richard, P.; Negm, A.; Manolikakes, G.; Jacob, C.; Cherkaoui-Malki, M. Cytoprotective and antioxidant properties of organic selenides for the myelin-forming cells, oligodendrocytes. Bioorg. Chem. 2018, 80, 43–56. [Google Scholar] [CrossRef]
- Li, X.; Xu, Y.; Lai, L.; Pei, J. Prediction of human cytochrome P450 inhibition using a multitask deep autoencoder neural network. Mol. Pharm. 2018, 15, 4336–4345. [Google Scholar] [CrossRef]
- Gao, Y.; Zhang, Y.; Wu, F.; Pei, J.; Luo, X.; Ju, X.; Zhao, C.; Liu, G. Exploring the interaction mechanism of desmethyl-broflanilide in insect GABA receptors and screening potential antagonists by in silico simulations. J. Agric. Food Chem. 2020, 68, 14768–14780. [Google Scholar] [CrossRef]
- Liu, W.; Wang, R.; Sun, Y.; Li, W.; Li, H.; Liu, C.; Ma, Y.; Wang, R. Exploring the effect of inhibitor AKB-9778 on VE-PTP by molecular docking and molecular dynamics simulation. J. Cell Biochem. 2019, 120, 17015–17029. [Google Scholar] [CrossRef]
- Gerlt, J.A.; Kreevoy, M.M.; Cleland, W.W.; Frey, P.A. Understanding enzymic catalysis: The importance of short, strong hydrogen bonds. Chem. Biol. 1997, 4, 259–267. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Gao, Y.; Chen, Y.; Liu, G.; Ju, X. Identification of the fipronil resistance associated mutations in Nilaparvata lugens GABA receptors by molecular modeling. Molecules 2019, 24, 4116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, Y.; Jung, H.; Kim, H.; Baek, J.; Jun, J.; Cho, H.; Im, D.; Hah, J. Design and synthesis of a novel PLK1 inhibitor scaffold using a hybridized 3D-QSAR model. Int. J. Mol. Sci. 2021, 22, 3865. [Google Scholar] [CrossRef] [PubMed]
- Cramer, R.D.; Patterson, D.E.; Bunce, J.D. Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J. Am. Chem. Soc. 1988, 110, 5959–5967. [Google Scholar] [CrossRef]
- Klebe, G. Comparative molecular similarity indices: CoMSIA. Perspect. Drug Discov. Des. 1998, 12, 87–104. [Google Scholar] [CrossRef]
- Wang, W.; Tian, Y.; Wan, Y.; Gu, S.; Ju, X.; Luo, X.; Liu, G. Insights into the key structural features of N1-ary-benzimidazols as HIV-1 NNRTIs using molecular docking, molecular dynamics, 3D-QSAR, and pharmacophore modeling. Struct. Chem. 2018, 30, 385–397. [Google Scholar] [CrossRef]
- Liu, G.; Wang, W.; Wan, Y.; Ju, X.; Gu, S. Application of 3D-QSAR, pharmacophore, and molecular docking in the molecular design of diarylpyrimidine derivatives as HIV-1 nonnucleoside reverse transcriptase inhibitors. Int. J. Mol. Sci. 2018, 19, 1436. [Google Scholar] [CrossRef] [Green Version]
- Wan, Y.; Tian, Y.; Wang, W.; Gu, S.; Ju, X.; Liu, G. In silico studies of diarylpyridine derivatives as novel HIV-1 NNRTIs using docking-based 3D-QSAR, molecular dynamics, and pharmacophore modeling approaches. RSC Adv. 2018, 8, 40529–40543. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Peng, J.; Wang, J.; Geng, Y.; Zuo, Z.; Hua, Y. Structure-activity relationship of xanthones as inhibitors of xanthine oxidase. Molecules 2018, 23, 365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Tian, Y.; Gao, Y.; Wu, F.; Luo, X.; Ju, X.; Liu, G. In silico design of novel HIV-1 NNRTIs based on combined modeling studies of dihydrofuro[3,4-d]pyrimidines. Front. Chem. 2020, 8, 164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sterling, T.; Irwin, J.J. ZINC 15—Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| No. | R1 | R2 | R3 | IC50 (µM) | pIC50 | CoMFA | CoMSIA | ||
|---|---|---|---|---|---|---|---|---|---|
| Predicted pIC50 | Residuals | Predicted pIC50 | Residuals | ||||||
| 01 * | methyl | H | O | 0.0920 | 7.0362 | 6.984 | 0.0522 | 7.075 | 0.0388 |
| 02 | iso-propyl | H | O | 0.0737 | 7.1325 | 7.167 | 0.0345 | 7.132 | 0.0005 |
| 03 | iso-butyl | H | O | 0.0644 | 7.1911 | 7.204 | 0.0129 | 7.192 | 0.0009 |
| 04 | iso-pentyl | H | O | 0.0541 | 7.2668 | 7.25 | 0.0168 | 7.255 | 0.0118 |
| 05 *,# | allyl | H | O | 0.0437 | 7.3595 | 7.407 | 0.0475 | 7.329 | 0.0305 |
| 06 | iso-butenyl | H | O | 0.0569 | 7.2449 | 7.246 | 0.0011 | 7.253 | 0.0081 |
| 07 | iso-pentenyl | H | O | 0.0692 | 7.1599 | 7.163 | 0.0031 | 7.146 | 0.0139 |
| 08# | propinyl | H | O | 0.0500 | 7.301 | 7.263 | 0.038 | 7.302 | 0.001 |
| 09# | methylene cyclopropane | H | O | 0.0461 | 7.3363 | 7.324 | 0.0123 | 7.328 | 0.0083 |
| 10 | cyclopentyl | H | O | 0.0585 | 7.2328 | 7.275 | 0.0422 | 7.235 | 0.0022 |
| 11 | methylene cyclohexane | H | O | 0.0683 | 7.1656 | 7.143 | 0.0226 | 7.144 | 0.0216 |
| 12 | benzyl | H | O | 0.0945 | 7.0246 | 7.102 | 0.0774 | 7.018 | 0.0066 |
| 13 | p-methylbenzyl | H | O | 0.0894 | 7.0487 | 7.074 | 0.0253 | 7.106 | 0.0573 |
| 14 * | p-tert-butylbenzyl | H | O | 0.1490 | 6.8268 | 6.804 | 0.0228 | 6.801 | 0.0258 |
| 15 | p-methoxylbenzyl | H | O | 0.0507 | 7.295 | 7.330 | 0.035 | 7.301 | 0.006 |
| 16 | p-fluorobenzyl | H | O | 0.0531 | 7.2749 | 7.226 | 0.0489 | 7.211 | 0.0639 |
| 17 | p-chlorobenzyl | H | O | 0.0691 | 7.1605 | 7.195 | 0.0345 | 7.201 | 0.0405 |
| 18 | p-bromobenzyl | H | O | 0.0552 | 7.2581 | 7.194 | 0.0641 | 7.233 | 0.0251 |
| 19 | m-methoxylbenzyl | H | O | 0.0516 | 7.2874 | 7.273 | 0.0144 | 7.301 | 0.0136 |
| 20 | m-fluorobenzyl | H | O | 0.0477 | 7.3215 | 7.298 | 0.0235 | 7.319 | 0.0025 |
| 21 *,# | m-chlorobenzyl | H | O | 0.0288 | 7.5406 | 7.593 | 0.0524 | 7.535 | 0.0056 |
| 22 | m-bromobenzyl | H | O | 0.0450 | 7.3468 | 7.329 | 0.0178 | 7.342 | 0.0048 |
| 23 | o-chlorobenzyl | H | O | 0.0917 | 7.0376 | 7.062 | 0.0244 | 7.004 | 0.0336 |
| 24 | 2,5-dichlorobenzyl | H | O | 0.0639 | 7.1945 | 7.120 | 0.0745 | 7.215 | 0.0205 |
| 25 | 2,4-dichlorobenzyl | H | O | 0.0838 | 7.0768 | 7.129 | 0.0522 | 7.116 | 0.0392 |
| 26 | hydrogen | H | O | 0.6290 | 6.2013 | 6.205 | 0.0313 | 6.201 | 0.0003 |
| 27 | iso-propyl | H | O | 0.0916 | 7.0381 | 7.013 | 0.0222 | 7.042 | 0.0039 |
| 28 * | iso-butyl | H | O | 0.0609 | 7.2154 | 7.238 | 0.0226 | 7.270 | 0.0546 |
| 29 *,# | iso-pentyl | H | O | 0.0250 | 7.6021 | 7.561 | 0.0411 | 7.705 | 0.1029 |
| 30 * | allyl | H | O | 0.0811 | 7.091 | 7.107 | 0.016 | 7.026 | 0.065 |
| 31 # | iso-butenyl | H | O | 0.0336 | 7.4737 | 7.505 | 0.0061 | 7.507 | 0.0333 |
| 32 | iso-pentenyl | H | O | 0.0388 | 7.4112 | 7.389 | 0.1031 | 7.429 | 0.0178 |
| 33 * | benzyl | H | O | 0.0387 | 7.4123 | 7.297 | 0.1153 | 7.405 | 0.0073 |
| 34 | p-fluorobenzyl | H | O | 0.0382 | 7.4179 | 7.424 | 0.0998 | 7.398 | 0.0199 |
| 35 | p-chlorobenzyl | H | O | 0.0499 | 7.3019 | 7.405 | 0.06 | 7.410 | 0.1081 |
| 36 # | p-bromobenzyl | H | O | 0.0298 | 7.5258 | 7.426 | 0.0116 | 7.462 | 0.0638 |
| 37 *,# | p-tert-butylbenzyl | H | O | 0.1970 | 6.7055 | 6.874 | 0.1685 | 6.787 | 0.0815 |
| 38 | p-methylbenzyl | H | O | 0.0354 | 7.451 | 7.391 | 0.06 | 7.380 | 0.071 |
| 39 | iso-pentyl | CH3 | O | 0.5400 | 6.2676 | 6.256 | 0.0116 | 6.248 | 0.0196 |
| 40 | iso-butenyl | CH3 | O | 0.5677 | 6.2459 | 6.253 | 0.0071 | 6.263 | 0.0171 |
| 41 * | p-bromobenzyl | CH3 | O | 0.1854 | 6.7319 | 6.822 | 0.0901 | 6.512 | 0.2199 |
| 42 * | p-methylbenzyl | CH3 | O | 0.1590 | 6.7986 | 6.807 | 0.0084 | 6.469 | 0.3296 |
| 43 # | iso-pentyl | H | NH | 0.0240 | 7.6198 | 7.664 | 0.0442 | 7.642 | 0.0222 |
| 44 # | iso-butenyl | H | NH | 0.0181 | 7.7423 | 7.684 | 0.0583 | 7.708 | 0.0343 |
| 45 # | p-bromobenzyl | H | NH | 0.0271 | 7.567 | 7.58 | 0.013 | 7.558 | 0.009 |
| 46 | p-methylbenzyl | H | NH | 0.0339 | 7.4698 | 7.528 | 0.0582 | 7.487 | 0.0172 |
| Febuxostat # | - | - | - | 0.0236 | 7.6271 | - | - | - | - |
| Model | q2 | ONC | SEE | R2 | F | rpred2 | Field Contribution (%) | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| S | E | H | D | A | ||||||||
| CoMFA | S + E | 0.897 | 7 | 0.050 | 0.983 | 229.50 | 0.948 | 0.773 | 0.227 | |||
| CoMSIA | S + E + H + D + A | 0.922 | 11 | 0.041 | 0.990 | 212.26 | 0.840 | 0.105 | 0.248 | 0.372 | 0.193 | 0.082 |
| H + D | 0.907 | 12 | 0.048 | 0.987 | 141.51 | 0.670 | 0.763 | 0.237 | ||||
| S + E + D | 0.931 | 7 | 0.055 | 0.980 | 189.71 | 0.665 | 0.175 | 0.404 | 0.421 | |||
| E + H + A | 0.875 | 11 | 0.043 | 0.989 | 195.52 | 0.871 | 0.331 | 0.528 | 0.141 | |||
| S + E + H + D | 0.926 | 10 | 0.041 | 0.990 | 232.65 | 0.825 | 0.107 | 0.257 | 0.422 | 0.214 | ||
| E + H + D + A | 0.921 | 11 | 0.042 | 0.990 | 205.711 | 0.848 | 0.271 | 0.436 | 0.204 | 0.089 | ||
| Validation Parameters | RMSE | r2 | r02 | r’02 | (r2 − r’02)/r2 | k | k’ | rm2 | r’m2 | Δrm2 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| CoMFA | 0.074 | 0.950 | 0.946 | 0.936 | 0.0147 | 0.998 | 1.002 | 0.890 | 0.838 | 0.052 | 0.864 |
| CoMSIA (S + E + H + D + A) | 0.130 | 0.922 | 0.840 | 0.897 | 0.027 | 1.005 | 0.995 | 0.658 | 0.776 | 0.118 | 0.717 |
| Name | SPECIFICITY | N_HITS | FEATS | PARETO | ENERGY | STERICS | HBOND | MOL_QRY |
|---|---|---|---|---|---|---|---|---|
| Model_1 | 4.629 | 11 | 8 | 0 | 8.76 | 2052.10 | 497.60 | 84.69 |
| Model_2 | 4.630 | 11 | 8 | 0 | 9.53 | 2064.40 | 498.40 | 81.58 |
| Model_3 | 2.330 | 9 | 10 | 0 | 8.47 | 2023.00 | 499.90 | 65.32 |
| Model_4 | 4.651 | 11 | 8 | 0 | 11.39 | 1986.30 | 494.50 | 87.28 |
| Model_5 | 4.629 | 11 | 8 | 0 | 10.10 | 1911.90 | 492.00 | 91.15 |
| Model_6 | 5.709 | 11 | 8 | 0 | 9.41 | 1864.20 | 486.00 | 104.79 |
| Model_7 | 3.440 | 12 | 10 | 0 | 11.68 | 1834.50 | 495.80 | 117.25 |
| Model_8 | 4.630 | 11 | 8 | 0 | 10.80 | 1840.80 | 503.10 | 85.90 |
| Model_9 | 3.440 | 12 | 10 | 0 | 12.21 | 1835.30 | 494.70 | 117.25 |
| Model_10 | 4.629 | 11 | 8 | 0 | 22.58 | 2149.70 | 496.10 | 85.45 |
| Model_11 | 4.628 | 11 | 8 | 0 | 10.47 | 1965.60 | 490.20 | 85.45 |
| Model_12 | 4.649 | 11 | 8 | 0 | 12.18 | 1996.40 | 487.40 | 93.3 |
| Model_13 | 4.630 | 11 | 8 | 0 | 1240.81 | 2059.40 | 497.30 | 85.90 |
| Model_14 | 4.630 | 11 | 8 | 0 | 10.80 | 1786.20 | 499.50 | 85.90 |
| Model_15 | 3.440 | 12 | 10 | 0 | 9.89 | 1780.70 | 484.10 | 141.11 |
| Model_16 | 3.440 | 12 | 10 | 0 | 14.36 | 1750.50 | 492.50 | 152.73 |
| Model_17 | 3.439 | 12 | 10 | 0 | 10.26 | 1929.00 | 470.00 | 129.80 |
| Model_18 | 4.637 | 11 | 8 | 0 | 10.82 | 1830.30 | 487.90 | 97.00 |
| Model_19 | 3.441 | 12 | 10 | 0 | 10.19 | 1723.60 | 485.60 | 130.83 |
| Model_20 | 4.636 | 11 | 8 | 0 | 1385.32 | 2189.60 | 481.20 | 97.00 |
| Parameter | Compound | |||||
|---|---|---|---|---|---|---|
| VS12 | VS16 | VS19 | VS26 | Febuxostat | 44 | |
| MW (g/mol) | 375.4 | 417.39 | 401.44 | 429.4 | 316.37 | 310.31 |
| Fraction Csp3 | 0.3 | 0.09 | 0.22 | 0.35 | 0.31 | 0.12 |
| Rotatable bonds | 4 | 7 | 8 | 9 | 5 | 5 |
| TPSA (Å2) | 113.4 | 105.7 | 98.6 | 130.3 | 111.5 | 122.9 |
| GI absorption | High | High | High | High | High | High |
| BBB permeant | No | No | No | No | No | No |
| CYP1A2 inhibitor | Yes | No | Yes | No | Yes | No |
| CYP2C19 inhibitor | No | Yes | No | Yes | Yes | No |
| CYP2C9 inhibitor | No | No | No | No | Yes | No |
| CYP2D6 inhibitor | No | No | No | No | No | No |
| CYP3A4 inhibitor | No | No | No | No | No | No |
| Lipinski violations | 0 | 0 | 0 | 0 | 0 | 0 |
| SA score | 3.4 | 3.32 | 3.25 | 3.34 | 3.12 | 2.64 |
| Hit Compound | VS12 | VS16 | VS19 | VS26 |
|---|---|---|---|---|
| Structure |  |  |  |  |
| Docking score | 10.62 | 11.87 | 10.76 | 11.03 |
| Complex | ΔEvdW | ΔEele | ΔGPB | ΔGSA | ΔGbinding |
|---|---|---|---|---|---|
| XO-febuxostat | −175.32 ± 8.81 | −39.95 ± 6.37 | 136.43 ± 11.05 | −18.20 ± 0.73 | −97.04 ± 9.45 |
| XO-44 | −160.03 ± 10.29 | −107.25 ± 10.15 | 189.25 ± 11.17 | −17.27 ± 0.78 | −95.30 ± 8.32 |
| XO-VS12 | −195.31 ± 9.21 | −69.99 ± 13.91 | 197.26 ± 13.45 | −20.27 ± 0.81 | −88.31 ± 10.40 |
| XO-VS16 | −220.50 ± 9.93 | −28.10 ± 8.40 | 109.67 ± 10.71 | −20.78 ± 0.86 | −159.71 ± 11.41 |
| XO-VS19 | −152.19 ± 8.97 | −49.01 ± 6.76 | 124.32 ± 11.20 | −19.03 ± 0.86 | −95.91 ± 10.52 |
| XO-VS26 | −216.89 ± 9.57 | −40.34 ± 8.46 | 127.33 ± 14.15 | −20.94 ± 0.91 | −150.84 ± 9.61 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhai, N.; Wang, C.; Wu, F.; Xiong, L.; Luo, X.; Ju, X.; Liu, G. Exploration of Novel Xanthine Oxidase Inhibitors Based on 1,6-Dihydropyrimidine-5-Carboxylic Acids by an Integrated in Silico Study. Int. J. Mol. Sci. 2021, 22, 8122. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22158122
Zhai N, Wang C, Wu F, Xiong L, Luo X, Ju X, Liu G. Exploration of Novel Xanthine Oxidase Inhibitors Based on 1,6-Dihydropyrimidine-5-Carboxylic Acids by an Integrated in Silico Study. International Journal of Molecular Sciences. 2021; 22(15):8122. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22158122
Chicago/Turabian StyleZhai, Na, Chenchen Wang, Fengshou Wu, Liwei Xiong, Xiaogang Luo, Xiulian Ju, and Genyan Liu. 2021. "Exploration of Novel Xanthine Oxidase Inhibitors Based on 1,6-Dihydropyrimidine-5-Carboxylic Acids by an Integrated in Silico Study" International Journal of Molecular Sciences 22, no. 15: 8122. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22158122