
AMD-Like Substrate Causes Epithelial Mesenchymal Transition in iPSC-Derived Retinal Pigment Epithelial Cells Wild Type but Not C3-Knockout

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
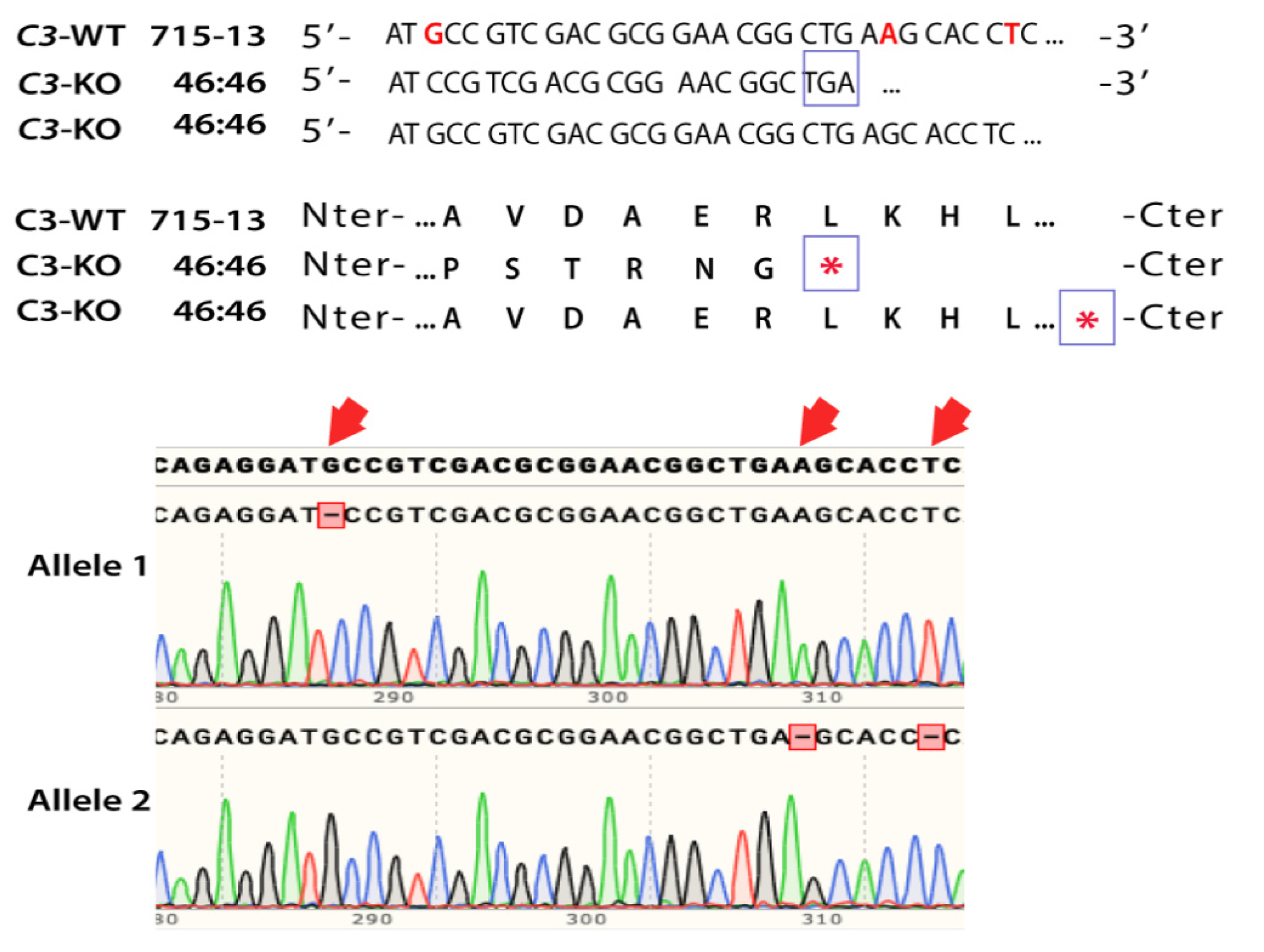
2.1. Generation of CRISPR-Edited iPSC-RPE Cells C3KO/KO
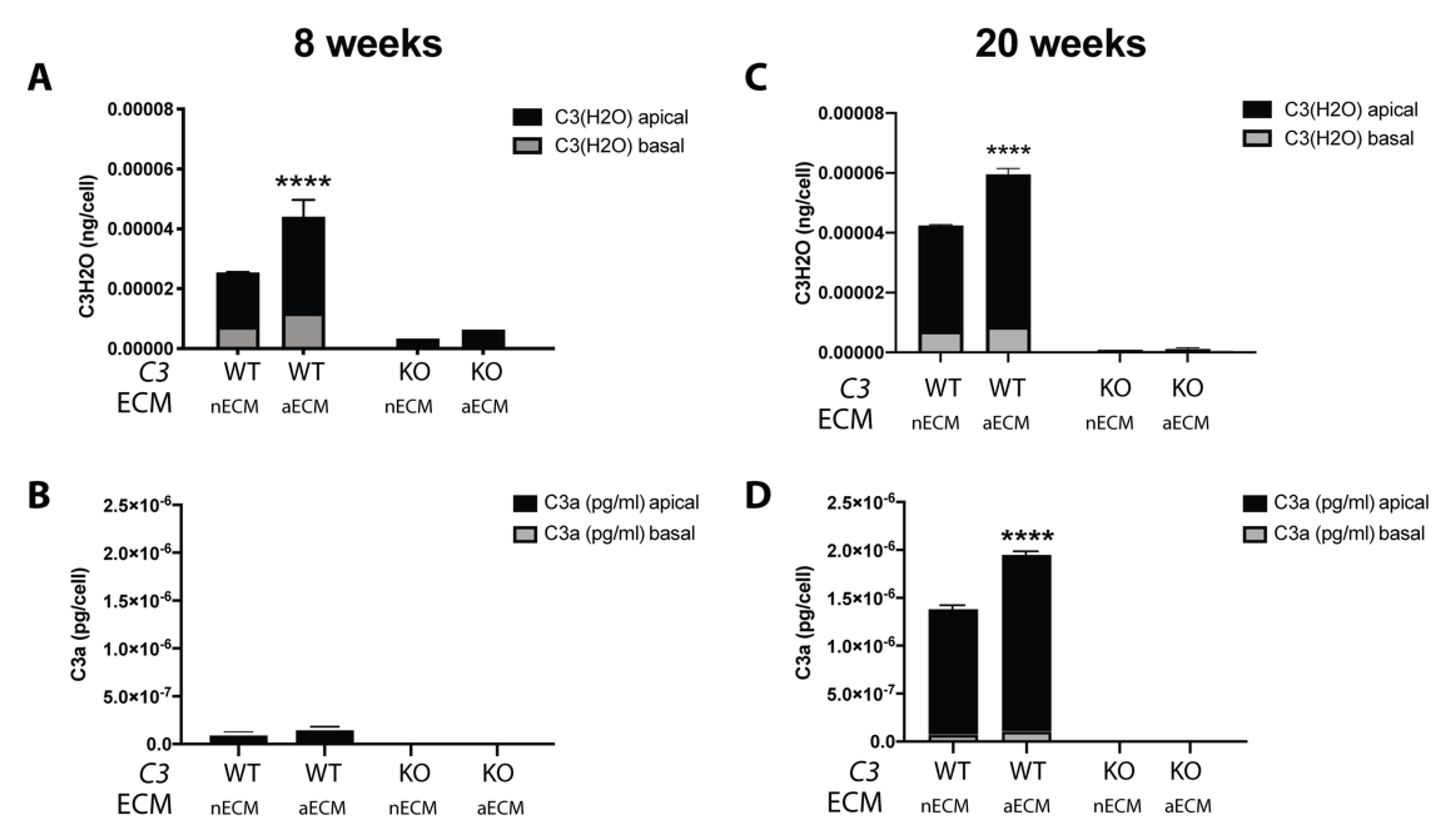
2.2. AMD-Like Substrate Increases Complement Activation by Tick-Over in iPSC-RPE Cell Cultures
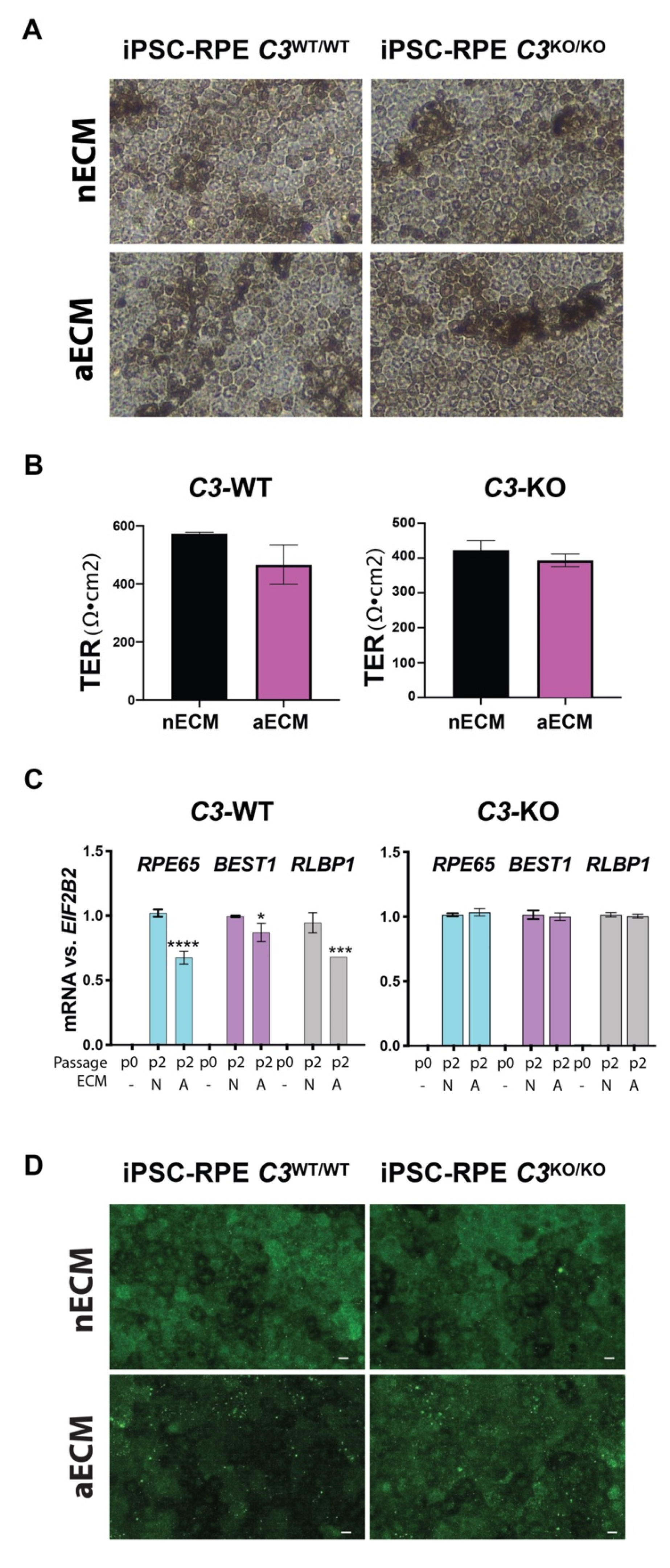
2.3. AMD-Like Substrate Causes Dedifferentiation of Mature iPSC-RPE Cells C3WT/WT but Not C3KO/KO
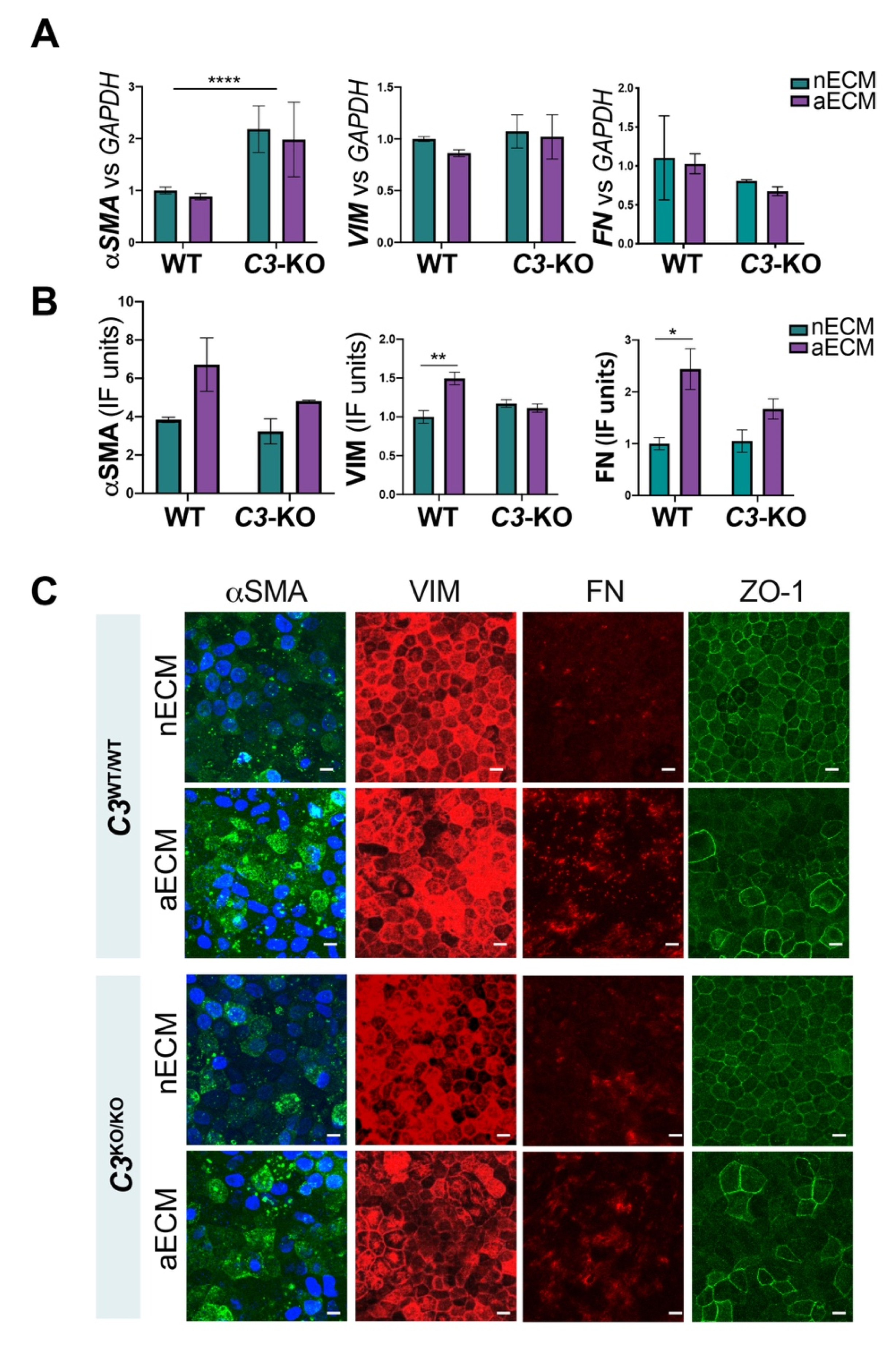
2.4. AMD-Like Substrate Triggers Epithelial Mesenchymal Transition (EMT) in iPSC-RPE Cell Cultures C3WT/WT but Not C3KO/KO
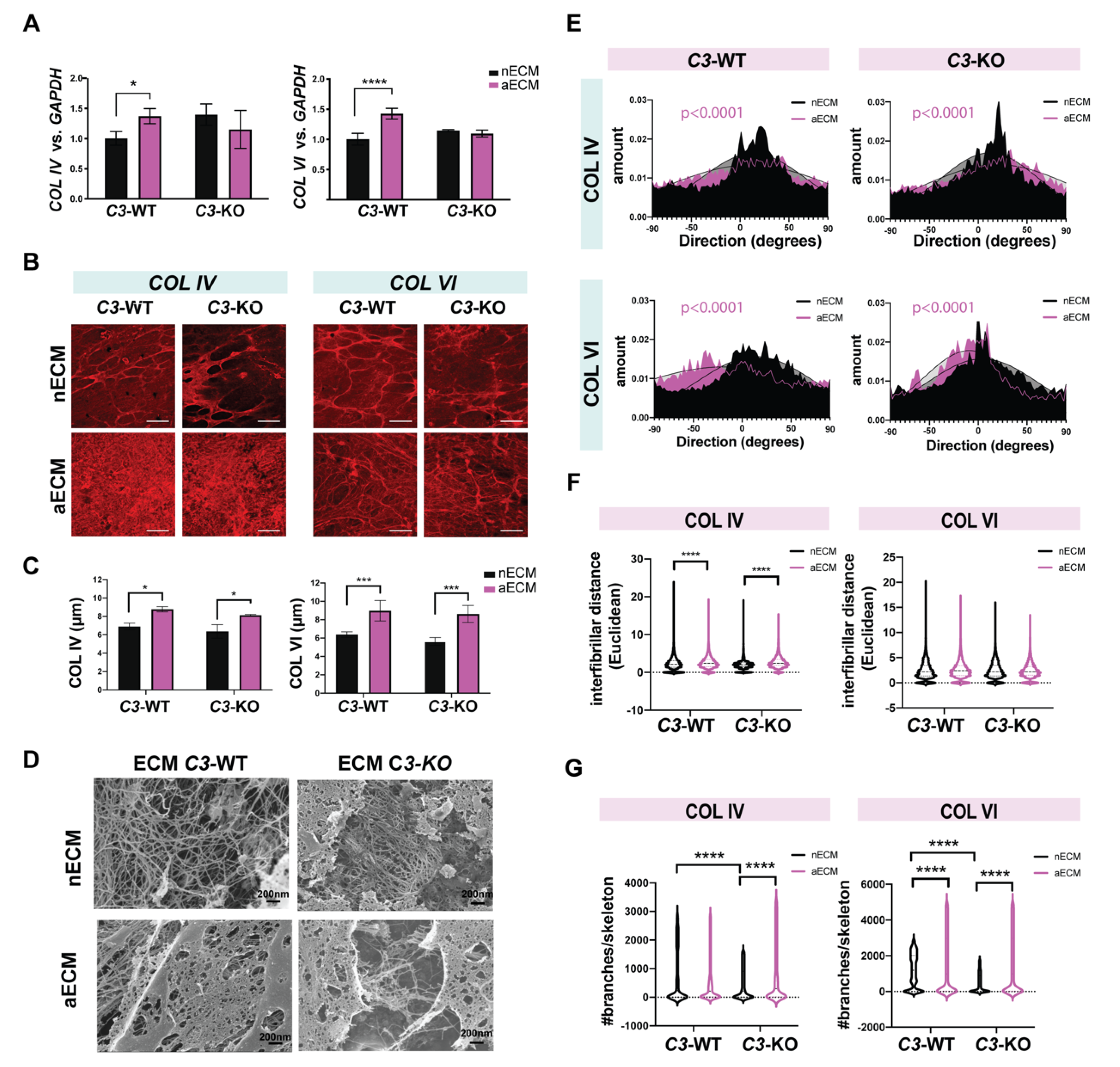
2.5. AMD-Like Substrate Enhances Deposition of Misaligned and Branched Collagen Fibers
2.6. AMD-Like Substrate Does Not Alter Collagens Turnover
3. Discussion
4. Materials and Methods
4.1. Clustered Regularly Interspaced Palindromic Repeats (CRISPR)-Cas9 Editing of iPSC-RPE Cells
4.2. Transepithelial Electrical Resistance (TER)
4.3. RNA Expression
4.4. ELISA
4.5. AMD-Like Substrate
4.6. Immunostaining
4.7. Transmission Electron Microscopy (TEM)
4.8. Scanning Electron Microscopy (SM)
4.9. Fiber Orientation
4.9.1. Directionality of Collagen Fibers
4.9.2. Interfibrillar Distance and Branch Information
4.10. Zymography
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Campochiaro, P.A.; Jerdon, J.A.; Glaser, B.M. The extracellular matrix of human retinal pigment epithelial cells in vivo and its synthesis in vitro. Investig. Ophthalmol. Vis. Sci. 1986, 27, 1615–1621. [Google Scholar]
- Curcio, C.A.; Johnson, M. Structure, function, and pathology of bruch’s membrane. In Retina, 5th ed.; Elsevier: Amsterdam, The Netherlands, 2012; Volume 1, pp. 465–481. [Google Scholar]
- Zarbin, M.A. Analysis of retinal pigment epithelium integrin expression and adhesion to aged submacular human Bruch’s membrane. Trans. Am. Ophthalmol. Soc. 2003, 101, 499–520. [Google Scholar] [PubMed]
- Ho, T.C.; Del Priore, L.V. Reattachment of cultured human retinal pigment epithelium to extracellular matrix and human Bruch’s membrane. Investig. Ophthalmol. Vis. Sci. 1997, 38, 1110–1118. [Google Scholar]
- Handa, J.T.; Verzijl, N.; Matsunaga, H.; Aotaki-Keen, A.; Lutty, G.A.; Koppele, J.M.T.; Miyata, T.; Hjelmeland, L.M. Increase in the advanced glycation end product pentosidine in Bruch’s membrane with age. Investig. Ophthalmol. Vis. Sci. 1999, 40, 775–779. [Google Scholar]
- Wagnera, D.R.; Reiser, K.M.; Lotz, J.C. Glycation increases human annulus fibrosus stiffness in both experimental measurements and theoretical predictions. J. Biomech. 2006, 39, 1021–1029. [Google Scholar] [CrossRef] [PubMed]
- Keenan, T.D.L.; Pickford, C.E.; Holley, R.J.; Clark, S.; Lin, W.; Dowsey, A.; Merry, C.; Day, A.; Bishop, P. Age-dependent changes in heparan sulfate in human Bruch’s membrane: Implications for age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2014, 55, 5370–5379. [Google Scholar] [CrossRef]
- Fritsche, L.G.; Fariss, R.N.; Stambolian, D.; Abecasis, G.R.; Curcio, C.A.; Swaroop, A. Age-related macular degeneration: Genetics and biology coming together. Annu. Rev. Genom. Hum. Genet. 2014, 15, 151–171. [Google Scholar] [CrossRef] [Green Version]
- Moore, D.J.; Clover, G.M. The effect of age on the macromolecular permeability of human Bruch’s membrane. Investig. Ophthalmol. Vis. Sci. 2001, 42, 2970–2975. [Google Scholar]
- Mishra, M.; Prasad, K. Do advanced glycation end products and its receptor play a role in pathophysiology of hypertension? Int. J. Angiol. 2017, 26, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Pastor-Pareja, J.C.; Xu, T. Shaping cells and organs in drosophila by opposing roles of fat body-secreted collagen IV and perlecan. Dev. Cell 2011, 21, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Chong, V.; Keonin, J.; Luthert, P.J.; Frennesson, C.I.; Weingeist, D.M.; Wolf, R.L.; Mullins, R.; Hageman, G.S. Decreased thickness and integrity of the macular elastic layer of Bruch’s membrane correspond to the distribution of lesions associated with age-related macular degeneration. Am. J. Pathol. 2005, 166, 241–251. [Google Scholar] [CrossRef]
- Gullapalli, V.K.; Sugino, I.K.; Van Patten, Y.; Shah, S.; Zarbin, M.A. Impaired RPE survival on aged submacular human Bruch’s membrane. Exp. Eye Res. 2005, 80, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Gullapalli, V.K.; Sugino, I.K.; Van Patten, Y.; Shah, S.; Zarbin, M.A. Retinal pigment epithelium resurfacing of aged submacular human Bruch’s membrane. Trans. Am. Ophthalmol. Soc. 2004, 102, 123. [Google Scholar]
- Fernandez-Godino, R.; Garland, D.L.; Pierce, E.A. A local complement response by RPE causes early-stage macular degeneration. Hum. Mol. Genet. 2015, 24, 5555–5569. [Google Scholar] [CrossRef] [Green Version]
- Godino, R.F.; Bujakowska, K.M.; Pierce, E.A. Changes in extracellular matrix cause RPE cells to make basal deposits and activate the alternative complement pathway. Hum. Mol. Genet. 2017, 27, 147–159. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Godino, R.; Pierce, E.A. C3a triggers formation of sub-retinal pigment epithelium deposits via the ubiquitin proteasome pathway. Sci. Rep. 2018, 8, 9679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, L.V.; Forest, D.L.; Banna, C.D.; Radeke, C.M.; Maloney, M.A.; Hu, J.; Spencer, C.N.; Walker, A.M.; Tsie, M.S.; Bok, D.; et al. Cell culture model that mimics drusen formation and triggers complement activation associated with age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2011, 108, 18277–18282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilgrim, M.; Lengyel, I.; Lanzirotti, A.; Newville, M.; Fearn, S.; Emri, E.; Knowles, J.C.; Messinger, J.D.; Read, R.W.; Guidry, C.; et al. Subretinal pigment epithelial deposition of drusen components including hydroxyapatite in a primary cell culture model. Investig. Ophthalmol. Vis. Sci. 2017, 58, 708–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, D.H.; Radeke, M.J.; Gallo, N.B.; Chapin, E.A.; Johnson, P.T.; Curletti, C.R.; Hancox, L.S.; Hu, J.; Ebright, J.N.; Malek, G.; et al. The pivotal role of the complement system in aging and age-related macular degeneration: Hypothesis re-visited. Prog. Retin. Eye Res. 2010, 29, 95–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hageman, G.S.; Luthert, P.J.; Chong, N.V.; Johnson, L.V.; Anderson, D.H.; Mullins, R.F. An integrated hypothesis that considers drusen as biomarkers of immune-mediated processes at the RPE-Bruch’s membrane interface in aging and age-related macular degeneration. Prog. Retin. Eye Res. 2001, 20, 705–732. [Google Scholar] [CrossRef]
- Nilsson, B.; Ekdahl, K.N. The tick-over theory revisited: Is C3 a contact-activated protein? Immunobiology 2012, 217, 1106–1110. [Google Scholar] [CrossRef]
- Chinchilla, B.; Foltopoulou, P.; Fernandez-Godino, R. Tick-over mediated complement activation is sufficient to cause basal deposit formation in cell-based models of macular degeneration. J. Pathol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A. Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J. Clin. Investig. 2007, 117, 524–529. [Google Scholar] [CrossRef] [Green Version]
- Wynn, T.A. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2008, 214, 199–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murtha, L.; Morten, M.; Schuliga, M.J.; Mabotuwana, N.S.; Hardy, S.A.; Waters, D.W.; Burgess, J.K.; Ngo, D.T.; Sverdlov, A.; Knight, D.A.; et al. The role of pathological aging in cardiac and pulmonary fibrosis. Aging Dis. 2019, 10, 419–428. [Google Scholar] [CrossRef] [Green Version]
- Sugino, I.K.; Sun, Q.; Wang, J.; Nunes, C.F.; Cheewatrakoolpong, N.; Rapista, A.; Johnson, A.C.; Malcuit, C.; Klimanskaya, I.; Lanza, R.; et al. Comparison of FRPE and human embryonic stem cell–derived RPE behavior on aged human Bruch’s membrane. Investig. Ophthalmol. Vis. Sci. 2011, 52, 4979–4997. [Google Scholar] [CrossRef]
- Tezel, T.H.; Kaplan, H.J.; Del Priore, L.V. Fate of human retinal pigment epithelial cells seeded onto layers of human Bruch’s membrane. Investig. Ophthalmol. Vis. Sci. 1999, 40, 467–476. [Google Scholar]
- Radeke, M.J.; Radeke, C.M.; Shih, Y.-H.; Hu, J.; Bok, D.; Johnson, L.V.; Coffey, P.J. Restoration of mesenchymal retinal pigmented epithelial cells by TGFβ pathway inhibitors: Implications for age-related macular degeneration. Genome Med. 2015, 7, 58. [Google Scholar] [CrossRef] [Green Version]
- Guidry, C.; Medeiros, N.E.; Curcio, C.A. Phenotypic variation of retinal pigment epithelium in age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2002, 43, 267–273. [Google Scholar]
- Acloque, H.; Adams, M.S.; Fishwick, K.; Bronner, M.; Nieto, M.A. Epithelial-mesenchymal transitions: The importance of changing cell state in development and disease. J. Clin. Investig. 2009, 119, 1438–1449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Tang, Z.; Lu, B.; Hatch, E.; Sacks, S.; Sheerin, N.S. C3a mediates epithelial-to-mesenchymal transition in proteinuric nephropathy. J. Am. Soc. Nephrol. 2009, 20, 593–603. [Google Scholar] [CrossRef] [Green Version]
- Cho, M.S.; Rupaimoole, R.; Choi, H.J.; Noh, K.; Chen, J.; Hu, Q.; Sood, A.K.; Afshar-Kharghan, V. Complement component 3 is regulated by TWIST1 and mediates epithelial–mesenchymal transition. J. Immunol. 2016, 196, 1412–1418. [Google Scholar] [CrossRef] [Green Version]
- Garland, D.L.; Fernandez-Godino, R.; Kaur, I.; Speicher, K.D.; Harnly, J.M.; Lambris, J.D.; Speicher, D.W.; Pierce, E.A. Mouse genetics and proteomic analyses demonstrate a critical role for complement in a model of DHRD/ML, an inherited macular degeneration. Hum. Mol. Genet. 2013, 23, 52–68. [Google Scholar] [CrossRef] [Green Version]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [Green Version]
- Brydon, E.M.; Bronstein, R.; Buskin, A.; Lako, M.; Pierce, E.A.; Fernandez-Godino, R. AAV-mediated gene augmentation therapy restores critical functions in mutant PRPF31+/− iPSC-derived RPE cells. Mol. Ther. Methods Clin. Dev. 2019, 15, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Foltz, L.P.; Clegg, D.O. Rapid, directed differentiation of retinal pigment epithelial cells from human embryonic or induced pluripotent stem cells. J. Vis. Exp. 2017, 128, e56274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leach, L.L.; Croze, R.H.; Hu, Q.; Nadar, V.P.; Clevenger, T.N.; Pennington, B.O.; Gamm, D.M.; Clegg, D.O. Induced pluripotent stem cell-derived retinal pigmented epithelium: A comparative study between cell lines and differentiation methods. J. Ocul. Pharmacol. Ther. 2016, 32, 317–330. [Google Scholar] [CrossRef] [Green Version]
- Stone, E.M.; Lotery, A.; Munier, F.L.; Héon, E.; Piguet, B.; Guymer, R.; VanDenburgh, K.; Cousin, P.; Nishimura, D.; Swiderski, R.E.; et al. A single EFEMP1 mutation associated with both Malattia Leventinese and Doyne honeycomb retinal dystrophy. Nat. Genet. 1999, 22, 199–202. [Google Scholar] [CrossRef]
- Elvington, M.; Liszewski, M.K.; Liszewski, A.R.; Kulkarni, H.S.; Hachem, R.R.; Mohanakumar, T.; Kim, A.; Atkinson, J.P. Development and optimization of an ELISA to quantitate C3(H2O) as a marker of human disease. Front. Immunol. 2019, 10, 703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galloway, C.A.; Dalvi, S.; Hung, S.S.; MacDonald, L.A.; Latchney, L.R.; Wong, R.C.; Guymer, R.H.; Mackey, D.A.; Williams, D.S.; Chung, M.M.; et al. Drusen in patient-derived hiPSC-RPE models of macular dystrophies. Proc. Natl. Acad. Sci. USA 2017, 114, E8214–E8223. [Google Scholar] [CrossRef] [Green Version]
- Zhou, M.; Geathers, J.S.; Grillo, S.L.; Weber, S.R.; Wang, W.; Zhao, Y.; Sundstrom, J.M. Role of epithelial-mesenchymal transition in retinal pigment epithelium dysfunction. Front. Cell Dev. Biol. 2020, 8, 501. [Google Scholar] [CrossRef]
- Shu, D.Y.; Butcher, E.; Saint-Geniez, M. EMT and EndMT: Emerging roles in age-related macular degeneration. Int. J. Mol. Sci. 2020, 21, 4271. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, M.; Neilson, E.G. Biomarkers for epithelial-mesenchymal transitions. J. Clin. Investig. 2009, 119, 1429–1437. [Google Scholar] [CrossRef] [Green Version]
- Georgiadis, A.; Tschernutter, M.; Bainbridge, J.W.B.; Balaggan, K.S.; Mowat, F.; West, E.; Munro, P.M.G.; Thrasher, A.J.; Matter, K.; Balda, M.S.; et al. The tight junction associated signalling proteins ZO-1 and ZONAB regulate retinal pigment epithelium homeostasis in mice. PLoS ONE 2010, 5, e15730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, N.; Ishii, M.; Brandon, C.; Ablonczy, Z.; Cai, J.; Liu, Y.; Chou, C.J.; Rohrer, B. Extracellular vesicle-mediated long-range communication in stressed retinal pigment epithelial cell monolayers. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2018, 1864, 2610–2622. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.L.; Lukas, T.J.; Yuan, M.; Du, N.; Tso, M.O.; Neufeld, A.H. Autophagy and exomoses in the aged retinal pigment epithelium: Possible relevance to drusen formation and age-related macular degeneration. PLoS ONE 2009, 4, e4160. [Google Scholar]
- Kalluri, R.; Neilson, E.G. Epithelial-mesenchymal transition and its implications for fibrosis. J. Clin. Invest. 2003, 112, 1776–1784. [Google Scholar] [CrossRef]
- Knupp, C.; Chong, V.; Munro, P.M.; Luthert, P.; Squire, J. Analysis of the collagen VI assemblies associated with Sorsby’s fundus dystrophy. J. Struct. Biol. 2002, 137, 31–40. [Google Scholar] [CrossRef] [Green Version]
- Theocharidis, G.; Drymoussi, Z.; Kao, A.P.; Barber, A.; Lee, D.; Braun, K.M.; Connelly, J.T. Type VI collagen regulates dermal matrix assembly and fibroblast motility. J. Investig. Dermatol. 2016, 136, 74–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arganda-Carreras, I.; Fernández-González, R.; Muñoz-Barrutia, A.; de Solórzano, C.O. 3D reconstruction of histological sections: Application to mammary gland tissue. Microsc. Res. Tech. 2010, 73, 1019–1029. [Google Scholar] [CrossRef] [PubMed]
- Romaniuk, W.; Nita, M.; Strzalka-Mrozik, B.; Grzybowski, A.; Mazurek, U. Age-related macular degeneration and changes in the extracellular matrix. Med. Sci. Monit. 2014, 20, 1003–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Godino, R.; Pierce, E.A.; Garland, D.L. Extracellular matrix alterations and deposit formation in AMD. In Advances in Experimental Medicine and Biology; Springer Science and Business Media LLC: Berlin, Germany, 2016; Volume 854, pp. 53–58. [Google Scholar]
- Pauleikhoff, D.; Harper, C.A.; Marshall, J.; Bird, A.C. Aging changes in Bruch’s membrane. A histochemical and morphologic study. Ophthalmology 1990, 97, 171–178. [Google Scholar] [CrossRef]
- Fields, M.A.; Bowrey, H.E.; Gong, J.; Moreira, E.F.; Cai, H.; Del Priore, L.V. Extracellular matrix nitration alters growth factor release and activates bioactive complement in human retinal pigment epithelial cells. PLoS ONE 2017, 12, e0177763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balaratnasingam, C.; Yannuzzi, L.A.; Curcio, C.A.; Morgan, W.H.; Querques, G.; Capuano, V.; Souied, E.; Jung, J.; Freund, K.B. Associations between retinal pigment epithelium and drusen volume changes during the lifecycle of large drusenoid pigment epithelial detachments. Investig. Ophthalmol. Vis. Sci. 2016, 57, 5479–5489. [Google Scholar] [CrossRef]
- Ghosh, S.; Shang, P.; Terasaki, H.; Stepicheva, N.; Hose, S.; Yazdankhah, M.; Weiss, J.; Sakamoto, T.; Bhutto, I.A.; Xia, S.; et al. A role for βA3/A1-crystallin in type 2 EMT of RPE cells occurring in dry age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2018, 59, AMD104–AMD113. [Google Scholar] [CrossRef] [Green Version]
- Sarks, J.P.; Sarks, S.H.; Killingsworth, M.C. Evolution of geographic atrophy of the retinal pigment epithelium. Eye 1988, 2, 552–577. [Google Scholar] [CrossRef]
- Wan, J.-X.; Fukuda, N.; Endo, M.; Tahira, Y.; Yao, E.-H.; Matsuda, H.; Ueno, T.; Matsumoto, K. Complement 3 is involved in changing the phenotype of human glomerular mesangial cells. J. Cell. Physiol. 2007, 213, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Zeisberg, M.; Lively, J.C.; Nyberg, P.; Afdhal, N.; Kalluri, R. Integrin α1β1 and α2β1 are the key regulators of hepatocarcinoma cell invasion across the fibrotic matrix microenvironment. Cancer Res. 2003, 63, 8312–8317. [Google Scholar] [PubMed]
- Das, A.; Puklin, J.E.; Frank, R.N.; Zhang, N.L. Ulrastructural immunocytochemistry of subretinal neovascular membranes in age-related macular degeneration. Ophthalmology 1992, 99, 1368–1376. [Google Scholar] [CrossRef]
- Alavi, M.V.; Mao, M.; Pawlikowski, B.T.; Kvezereli, M.; Duncan, J.L.; Libby, R.T.; John, S.W.; Gould, D.B. Col4a1 mutations cause progressive retinal neovascular defects and retinopathy. Sci. Rep. 2016, 6, 18602. [Google Scholar] [CrossRef]
- Karwatowski, W.S.; Jeffries, T.E.; Duance, V.C.; Albon, J.; Bailey, A.J.; Easty, D.L. Preparation of Bruch’s membrane and analysis of the age-related changes in the structural collagens. Br. J. Ophthalmol. 1995, 79, 944–952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishikawa, K.; Kannan, R.; Hinton, D.R. Molecular mechanisms of subretinal fibrosis in age-related macular degeneration. Exp. Eye Res. 2016, 142, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef]
- Chen, G.; Gulbranson, D.R.; Hou, Z.; Bolin, J.M.; Ruotti, V.; Probasco, M.D.; Smuga-Otto, K.; Howden, S.; Diol, N.R.; Propson, N.E.; et al. Chemically defined conditions for human iPSC derivation and culture. Nat. Methods 2011, 8, 424–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, K.; Yu, J.; Suknuntha, K.; Tian, S.; Montgomery, K.; Choi, K.-D.; Stewart, R.; Thomson, J.A.; Slukvin, I.I. Efficient generation of transgene-free induced pluripotent stem cells from normal and neoplastic bone marrow and cord blood mononuclear cells. Blood 2011, 117, e109–e119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maminishkis, A.; Chen, S.; Jalickee, S.; Banzon, T.; Shi, G.; Wang, F.E.; Ehalt, T.; Hammer, J.A.; Miller, S.S. Confluent monolayers of cultured human fetal retinal pigment epithelium exhibit morphology and physiology of native tissue. Investig. Ophthalmol. Vis. Sci. 2006, 47, 3612–3624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godino, R.F.; Garland, D.L.; Pierce, E. Isolation, culture and characterization of primary mouse RPE cells. Nat. Protoc. 2016, 11, 1206–1218. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chinchilla, B.; Fernandez-Godino, R. AMD-Like Substrate Causes Epithelial Mesenchymal Transition in iPSC-Derived Retinal Pigment Epithelial Cells Wild Type but Not C3-Knockout. Int. J. Mol. Sci. 2021, 22, 8183. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22158183
Chinchilla B, Fernandez-Godino R. AMD-Like Substrate Causes Epithelial Mesenchymal Transition in iPSC-Derived Retinal Pigment Epithelial Cells Wild Type but Not C3-Knockout. International Journal of Molecular Sciences. 2021; 22(15):8183. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22158183
Chicago/Turabian StyleChinchilla, Blanca, and Rosario Fernandez-Godino. 2021. "AMD-Like Substrate Causes Epithelial Mesenchymal Transition in iPSC-Derived Retinal Pigment Epithelial Cells Wild Type but Not C3-Knockout" International Journal of Molecular Sciences 22, no. 15: 8183. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22158183