
High Concentration of an ISS-N1-Targeting Antisense Oligonucleotide Causes Massive Perturbation of the Transcriptome


Abstract
:1. Introduction
2. Results
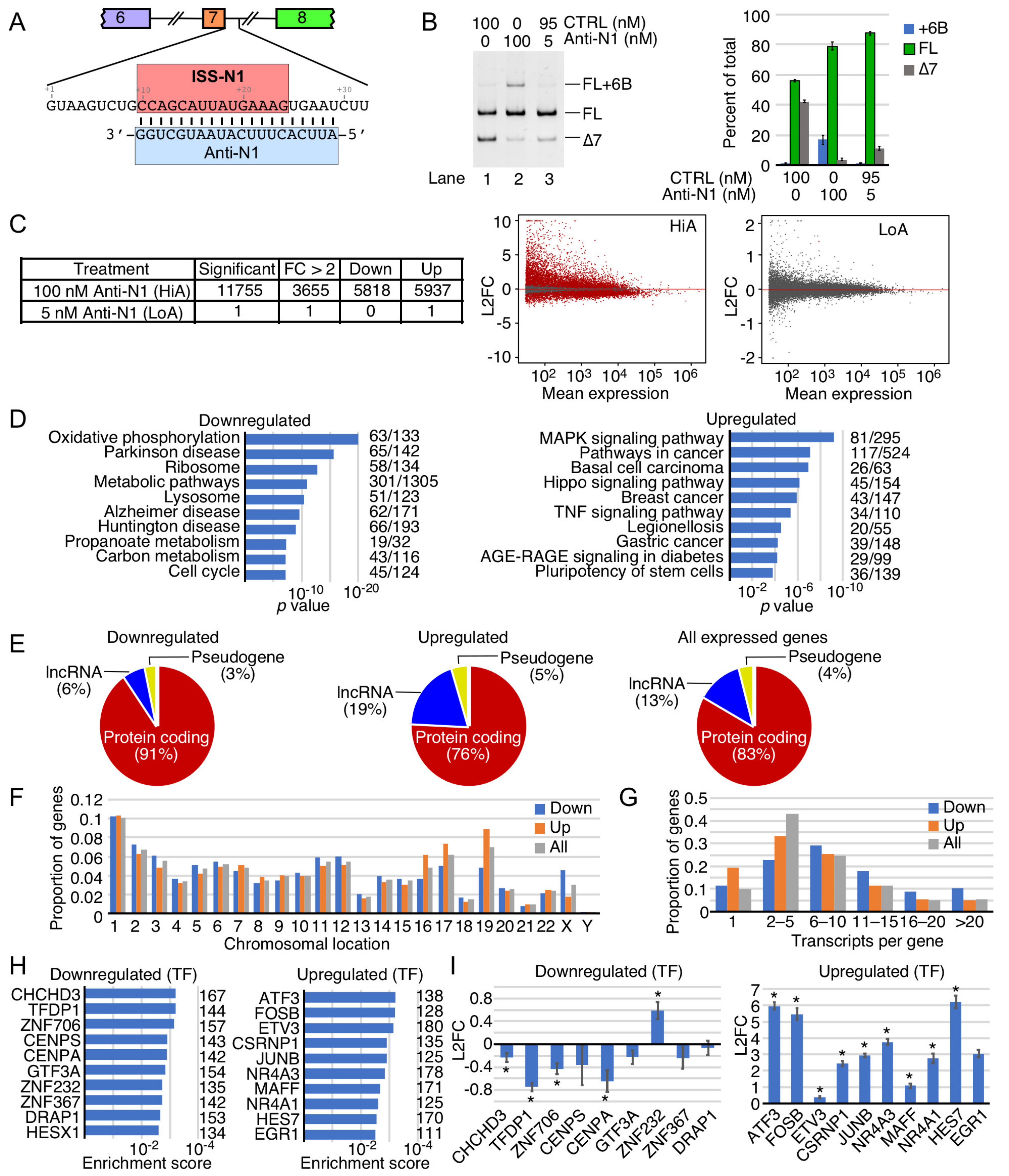
2.1. Transcriptome-Wide Effect of Anti-N1
2.2. Nature of Splicing Events Impacted by Anti-N1
2.3. Analysis of Exons Undergoing Enhanced Inclusion Caused by Anti-N1
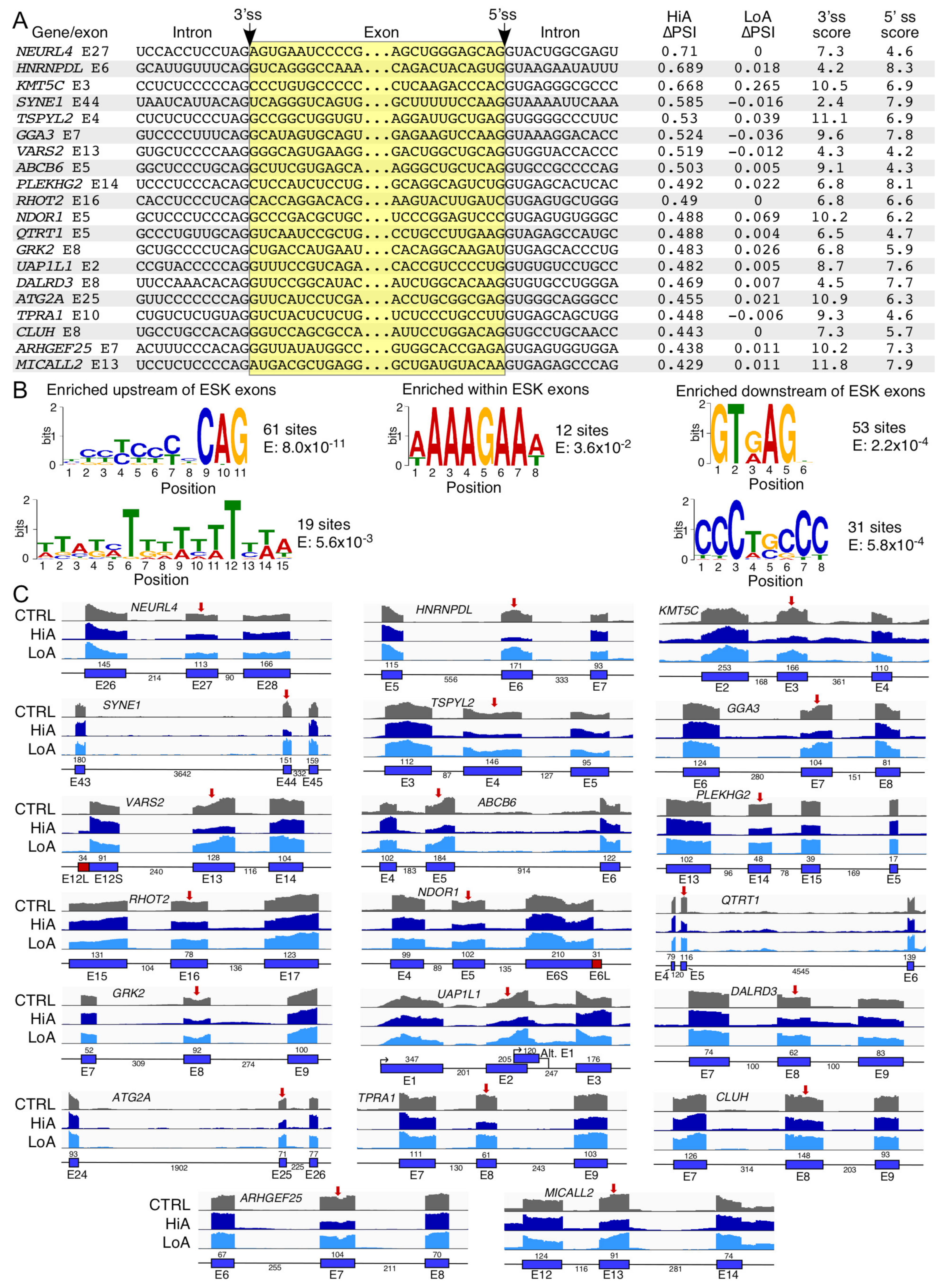
2.4. Analysis of Exons Undergoing Enhanced Skipping Caused by Anti-N1
2.5. Analysis of Intron Retention Events Induced by Anti-N1 Treatment
2.6. Analysis of Intron Removal Events Enhanced by Anti-N1
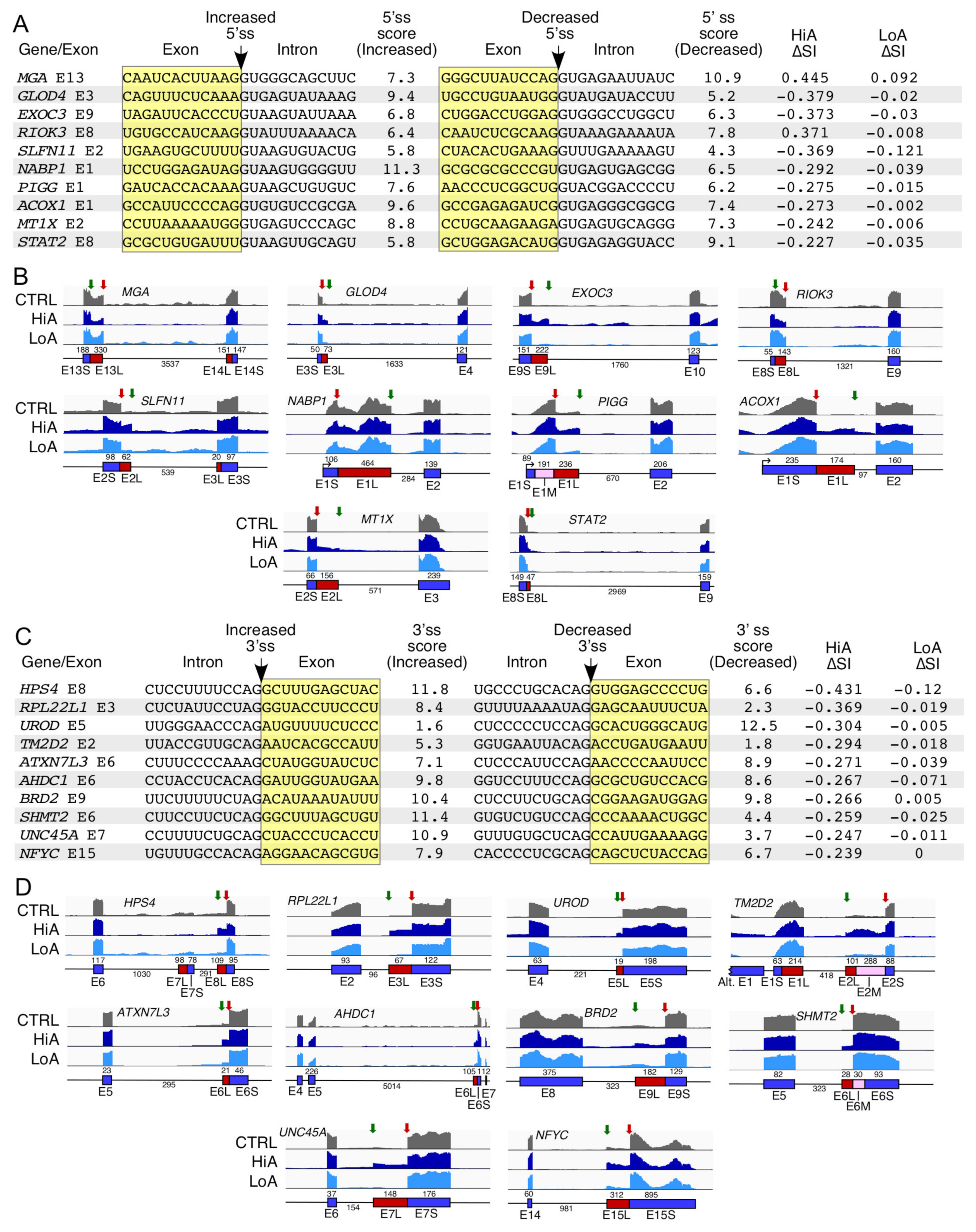
2.7. Analysis of Alternative Splice Usage Enhanced by Anti-N1
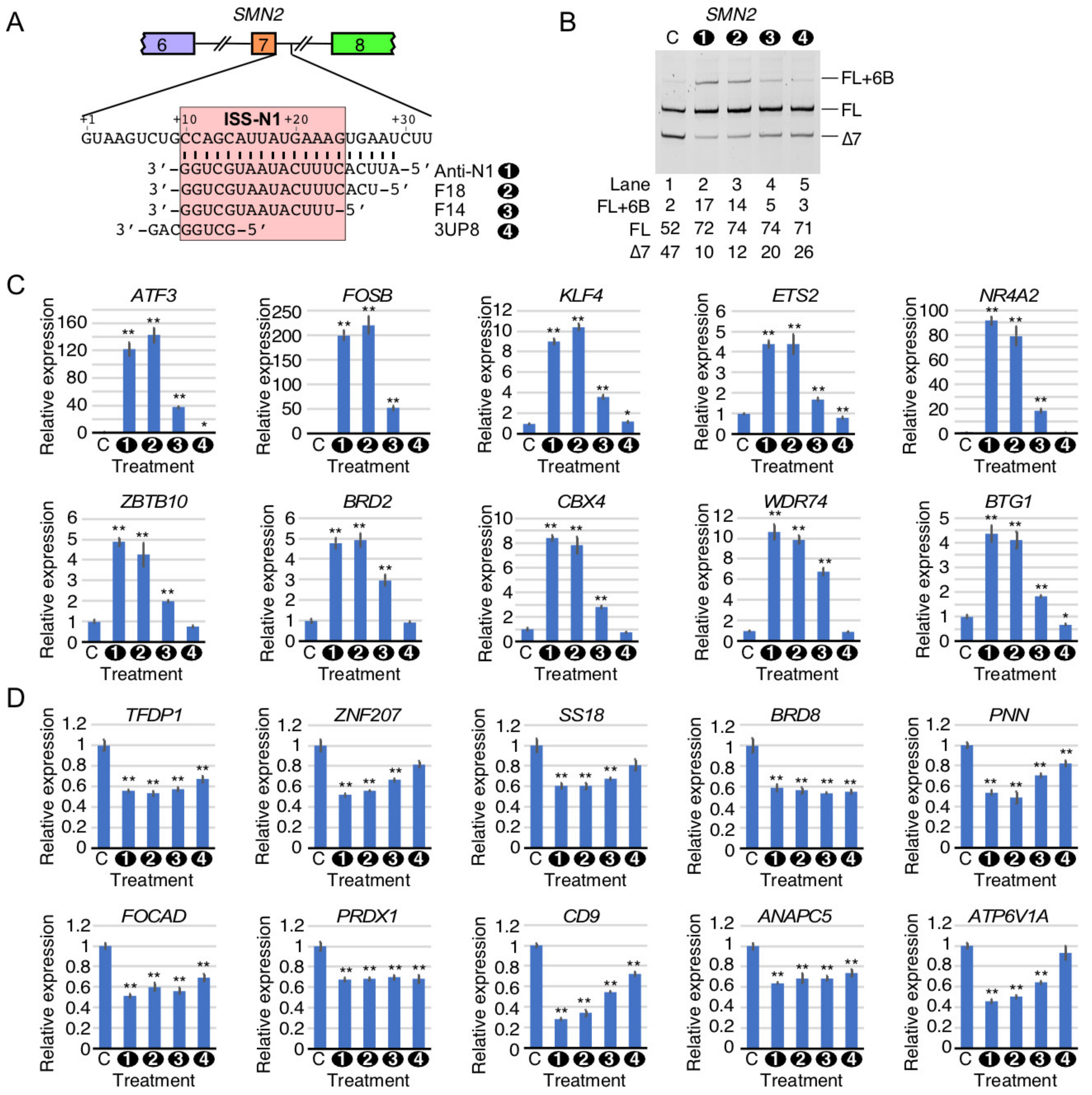
2.8. Comparison of Perturbed Expression of Genes by ISS-N1-Targeting Splice-Switching Oligonucleotides
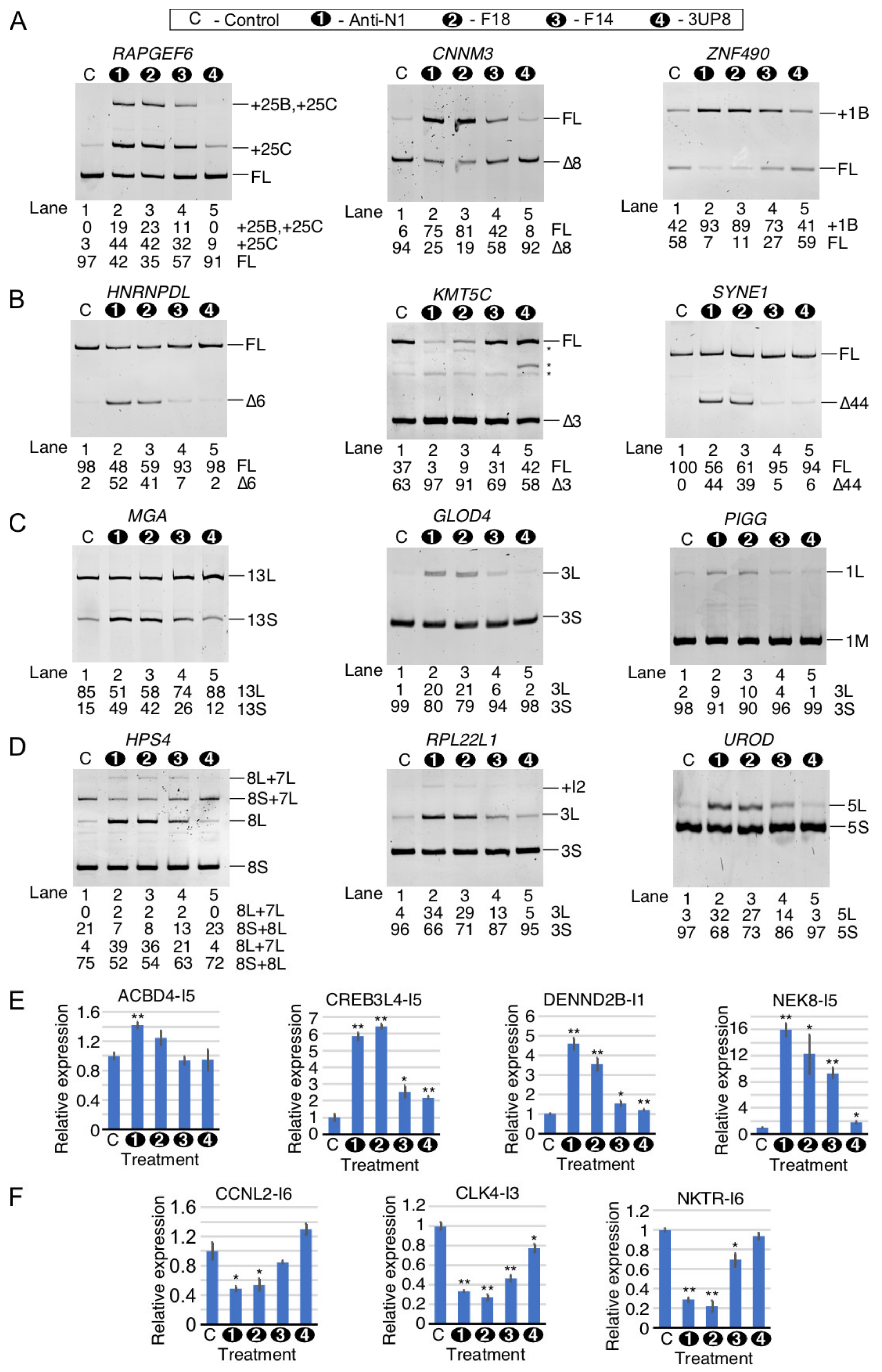
2.9. Comparison of Perturbed Splicing Events by ISS-N1-Targeting Splice-Switching Oligonucleotides
3. Discussion
4. Materials and Methods
4.1. Cell Culture and ASOs
4.2. ASO Transfections
4.3. Reverse Transcription and PCR (RT-PCR) and Quantitative PCR (qPCR)
4.4. Library Generation and RNA-Seq
4.5. RNA-Seq Mapping and Bioinformatic Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Crooke, S.T.; Witztum, J.L.; Bennett, C.F.; Baker, B.F. RNA-Targeted Therapeutics. Cell Metab. 2018, 27, 714–739. [Google Scholar] [CrossRef] [Green Version]
- Scharner, J.; Aznarez, I. Clinical Applications of Single-Stranded Oligonucleotides: Current Landscape of Approved and In-Development Therapeutics. Mol. Ther. 2021, 29, 540–554. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.F.; Krainer, A.R.; Cleveland, D.W. Antisense Oligonucleotide Therapies for Neurodegenerative Diseases. Annu. Rev. Neurosci. 2019, 42, 385–406. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.N.; Seo, J.; Singh, N.N. RNA in spinal muscular atrophy: Therapeutic implications of targeting. Expert Opin. Ther. Targets 2020, 24, 731–743. [Google Scholar] [CrossRef] [PubMed]
- Ottesen, E.W. ISS-N1 makes the First FDA-approved Drug for Spinal Muscular Atrophy. Transl. Neurosci. 2017, 8, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.N.; Howell, M.D.; Androphy, E.J.; Singh, R.N. How the discovery of ISS-N1 led to the first medical therapy for spinal muscular atrophy. Gene Ther. 2017, 24, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Scharner, J.; Ma, W.K.; Zhang, Q.; Lin, K.T.; Rigo, F.; Bennett, C.F.; Krainer, A.R. Hybridization-mediated off-target effects of splice-switching antisense oligonucleotides. Nucleic Acids Res. 2020, 48, 802–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, N.N.; Hoffman, S.; Reddi, P.P.; Singh, R.N. Spinal muscular atrophy: Broad disease spectrum and sex-specific phenotypes. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 166063. [Google Scholar] [CrossRef]
- Wirth, B.; Karakaya, M.; Kye, M.J.; Mendoza-Ferreira, N. Twenty-Five Years of Spinal Muscular Atrophy Research: From Phenotype to Genotype to Therapy, and What Comes Next. Annu. Rev. Genom. Hum. Genet. 2020, 21, 231–261. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Lotti, F.; Dittmar, K.; Younis, I.; Wan, L.; Kasim, M.; Dreyfuss, G. SMN deficiency causes tissue-specific perturbations in the repertoire of snRNAs and widespread defects in splicing. Cell 2008, 133, 585–600. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, S.; Bhatia, K.; Kannan, A.; Gangwani, L. Molecular Mechanisms of Neurodegeneration in Spinal Muscular Atrophy. J. Exp. Neurosci. 2016, 10, 39–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.N.; Howell, M.D.; Ottesen, E.W.; Singh, N.N. Diverse role of survival motor neuron protein. Biochim. Biophys. Acta Gene Regul. Mech. 2017, 1860, 299–315. [Google Scholar] [CrossRef] [Green Version]
- Kannan, A.; Bhatia, K.; Branzei, D.; Gangwani, L. Combined deficiency of Senataxin and DNA-PKcs causes DNA damage accumulation and neurodegeneration in spinal muscular atrophy. Nucleic Acids Res. 2018, 46, 8326–8346. [Google Scholar] [CrossRef] [Green Version]
- Ji, C.; Bader, J.; Ramanathan, P.; Hennlein, L.; Meissner, F.; Jablonka, S.; Mann, M.; Fischer, U.; Sendtner, M.; Briese, M. Interaction of 7SK with the Smn complex modulates snRNP production. Nat. Commun. 2021, 12, 1278. [Google Scholar] [CrossRef] [PubMed]
- Sansa, A.; de la Fuente, S.; Comella, J.X.; Garcera, A.; Soler, R.M. Intracellular pathways involved in cell survival are deregulated in mouse and human spinal muscular atrophy motoneurons. Neurobiol. Dis. 2021, 155, 105366. [Google Scholar] [CrossRef]
- Hensel, N.; Cieri, F.; Santonicola, P.; Tapken, I.; Schüning, T.; Taiana, M.; Pagliari, E.; Joseph, A.; Fischer, S.; Heidrich, N.; et al. Impairment of the neurotrophic signaling hub B-Raf contributes to motoneuron degeneration in spinal muscular atrophy. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef] [PubMed]
- Lorson, C.L.; Androphy, E.J. The domain encoded by exon 2 of the survival motor neuron protein mediates nucleic acid binding. Hum. Mol. Genet. 1998, 7, 1269–1275. [Google Scholar] [CrossRef] [Green Version]
- Bertrandy, S.; Burlet, P.; Clermont, O.; Huber, C.; Fondrat, C.; Thierry-Mieg, D.; Munnich, A.; Lefebvre, S. The RNA-binding properties of SMN: Deletion analysis of the zebrafish orthologue defines domains conserved in evolution. Hum. Mol. Genet. 1999, 8, 775–782. [Google Scholar] [CrossRef] [Green Version]
- Ottesen, E.W.; Singh, N.N.; Luo, D.; Singh, R.N. High-affinity RNA targets of the Survival Motor Neuron protein reveal diverse preferences for sequence and structural motifs. Nucleic Acids Res. 2018, 46, 10983–11001. [Google Scholar] [CrossRef] [PubMed]
- Lauria, F.; Bernabò, P.; Tebaldi, T.; Groen, E.J.N.; Perenthaler, E.; Maniscalco, F.; Rossi, A.; Donzel, D.; Clamer, M.; Marchioretto, M.; et al. SMN-primed ribosomes modulate the translation of transcripts related to spinal muscular atrophy. Nat. Cell Biol. 2020, 22, 1239–1251. [Google Scholar] [CrossRef] [PubMed]
- Lorson, C.L.; Hahnen, E.; Androphy, E.J.; Wirth, B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc. Natl. Acad. Sci. USA 1999, 96, 6307–6311. [Google Scholar] [CrossRef] [Green Version]
- Monani, U.R.; Lorson, C.L.; Parsons, D.W.; Prior, T.W.; Androphy, E.J.; Burghes, A.H.; McPherson, J.D. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum. Mol. Genet. 1999, 8, 1177–1183. [Google Scholar] [CrossRef] [PubMed]
- Ottesen, E.W.; Seo, J.; Singh, N.N.; Singh, R.N. A Multilayered Control of the Human. Front. Microbiol. 2017, 8, 2252. [Google Scholar] [CrossRef] [Green Version]
- Singh, N.N.; Seo, J.; Rahn, S.J.; Singh, R.N. A multi-exon-skipping detection assay reveals surprising diversity of splice isoforms of spinal muscular atrophy genes. PLoS ONE 2012, 7, e49595. [Google Scholar] [CrossRef] [Green Version]
- Seo, J.; Singh, N.N.; Ottesen, E.W.; Sivanesan, S.; Shishimorova, M.; Singh, R.N. Oxidative Stress Triggers Body-Wide Skipping of Multiple Exons of the Spinal Muscular Atrophy Gene. PLoS ONE 2016, 11, e0154390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, J.; Singh, N.N.; Ottesen, E.W.; Lee, B.M.; Singh, R.N. A novel human-specific splice isoform alters the critical C-terminus of Survival Motor Neuron protein. Sci. Rep. 2016, 6, 30778. [Google Scholar] [CrossRef] [Green Version]
- Woo, C.J.; Maier, V.K.; Davey, R.; Brennan, J.; Li, G.; Brothers, J.; Schwartz, B.; Gordo, S.; Kasper, A.; Okamoto, T.R.; et al. Gene activation of SMN by selective disruption of lncRNA-mediated recruitment of PRC2 for the treatment of spinal muscular atrophy. Proc. Natl. Acad. Sci. USA 2017, 114, E1509–E1518. [Google Scholar] [CrossRef] [Green Version]
- D’Ydewalle, C.; Ramos, D.M.; Pyles, N.J.; Ng, S.Y.; Gorz, M.; Pilato, C.M.; Ling, K.; Kong, L.; Ward, A.J.; Rubin, L.L.; et al. The Antisense Transcript SMN-AS1 Regulates SMN Expression and Is a Novel Therapeutic Target for Spinal Muscular Atrophy. Neuron 2017, 93, 66–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ottesen, E.W.; Luo, D.; Seo, J.; Singh, N.N.; Singh, R.N. Human Survival Motor Neuron genes generate a vast repertoire of circular RNAs. Nucleic Acids Res. 2019, 47, 2884–2905. [Google Scholar] [CrossRef] [Green Version]
- Pagliarini, V.; Jolly, A.; Bielli, P.; Di Rosa, V.; De la Grange, P.; Sette, C. Sam68 binds Alu-rich introns in SMN and promotes pre-mRNA circularization. Nucleic Acids Res. 2020, 48, 633–645. [Google Scholar] [CrossRef]
- Ottesen, E.W.; Singh, R.N. Characteristics of circular RNAs generated by human Survival Motor Neuron genes. Cell Signal. 2020, 73, 109696. [Google Scholar] [CrossRef]
- Singh, N.N.; Ottesen, E.W.; Singh, R.N. A survey of transcripts generated by spinal muscular atrophy genes. Biochim. Biophys. Acta Gene Regul. Mech. 2020, 1863, 194562. [Google Scholar] [CrossRef]
- Cho, S.; Dreyfuss, G. A degron created by SMN2 exon 7 skipping is a principal contributor to spinal muscular atrophy severity. Genes Dev. 2010, 24, 438–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Zaidy, S.A.; Mendell, J.R. From Clinical Trials to Clinical Practice: Practical Considerations for Gene Replacement Therapy in SMA Type 1. Pediatr. Neurol. 2019, 100, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.N.; Ottesen, E.W.; Singh, N.N. The First Orally Deliverable Small Molecule for the Treatment of Spinal Muscular Atrophy. Neurosci. Insights 2020, 15. [Google Scholar] [CrossRef]
- Singh, N.N.; Lee, B.M.; DiDonato, C.J.; Singh, R.N. Mechanistic principles of antisense targets for the treatment of spinal muscular atrophy. Future Med. Chem. 2015, 7, 1793–1808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, N.K.; Singh, N.N.; Androphy, E.J.; Singh, R.N. Splicing of a critical exon of human Survival Motor Neuron is regulated by a unique silencer element located in the last intron. Mol. Cell Biol. 2006, 26, 1333–1346. [Google Scholar] [CrossRef] [Green Version]
- Singh, N.N.; Hollinger, K.; Bhattacharya, D.; Singh, R.N. An antisense microwalk reveals critical role of an intronic position linked to a unique long-distance interaction in pre-mRNA splicing. RNA 2010, 16, 1167–1181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, N.N.; Lawler, M.N.; Ottesen, E.W.; Upreti, D.; Kaczynski, J.R.; Singh, R.N. An intronic structure enabled by a long-distance interaction serves as a novel target for splicing correction in spinal muscular atrophy. Nucleic Acids Res. 2013, 41, 8144–8165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, N.N.; Singh, R.N. How RNA structure dictates the usage of a critical exon of spinal muscular atrophy gene. Biochim. Biophys. Acta Gene Regul. Mech. 2019, 1862, 194403. [Google Scholar] [CrossRef]
- Sivanesan, S.; Howell, M.D.; Didonato, C.J.; Singh, R.N. Antisense oligonucleotide mediated therapy of spinal muscular atrophy. Transl. Neurosci. 2013, 4. [Google Scholar] [CrossRef] [Green Version]
- Singh, N.N.; Shishimorova, M.; Cao, L.C.; Gangwani, L.; Singh, R.N. A short antisense oligonucleotide masking a unique intronic motif prevents skipping of a critical exon in spinal muscular atrophy. RNA Biol. 2009, 6, 341–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, Y.; Vickers, T.A.; Okunola, H.L.; Bennett, C.F.; Krainer, A.R. Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am. J. Hum. Genet. 2008, 82, 834–848. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Janghra, N.; Mitrpant, C.; Dickinson, R.L.; Anthony, K.; Price, L.; Eperon, I.C.; Wilton, S.D.; Morgan, J.; Muntoni, F. A novel morpholino oligomer targeting ISS-N1 improves rescue of severe spinal muscular atrophy transgenic mice. Hum. Gene 2013, 24, 331–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitrpant, C.; Porensky, P.; Zhou, H.; Price, L.; Muntoni, F.; Fletcher, S.; Wilton, S.D.; Burghes, A.H. Improved antisense oligonucleotide design to suppress aberrant SMN2 gene transcript processing: Towards a treatment for spinal muscular atrophy. PLoS ONE 2013, 8, e62114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, Y.; Sahashi, K.; Rigo, F.; Hung, G.; Horev, G.; Bennett, C.F.; Krainer, A.R. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature 2011, 478, 123–126. [Google Scholar] [CrossRef]
- Porensky, P.N.; Mitrpant, C.; McGovern, V.L.; Bevan, A.K.; Foust, K.D.; Kaspar, B.K.; Wilton, S.D.; Burghes, A.H. A single administration of morpholino antisense oligomer rescues spinal muscular atrophy in mouse. Hum. Mol. Genet. 2012, 21, 1625–1638. [Google Scholar] [CrossRef] [PubMed]
- Arun, G.; Aggarwal, D.; Spector, D.L. Long Non-Coding RNA: Functional Implications. Noncoding RNA 2020, 6, 22. [Google Scholar] [CrossRef] [PubMed]
- Hai, T.; Wolford, C.C.; Chang, Y.S. ATF3, a hub of the cellular adaptive-response network, in the pathogenesis of diseases: Is modulation of inflammation a unifying component? Gene Expr. 2010, 15, 1–11. [Google Scholar] [CrossRef]
- Tsujino, H.; Kondo, E.; Fukuoka, T.; Dai, Y.; Tokunaga, A.; Miki, K.; Yonenobu, K.; Ochi, T.; Noguchi, K. Activating transcription factor 3 (ATF3) induction by axotomy in sensory and motoneurons: A novel neuronal marker of nerve injury. Mol. Cell Neurosci. 2000, 15, 170–182. [Google Scholar] [CrossRef]
- Jochum, W.; Passegué, E.; Wagner, E.F. AP-1 in mouse development and tumorigenesis. Oncogene 2001, 20, 2401–2412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robison, A.J.; Nestler, E.J. Transcriptional and epigenetic mechanisms of addiction. Nat. Rev. Neurosci. 2011, 12, 623–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowland, B.D.; Peeper, D.S. KLF4, p21 and context-dependent opposing forces in cancer. Nat. Rev. Cancer 2006, 6, 11–23. [Google Scholar] [CrossRef]
- Yamanaka, S. Strategies and new developments in the generation of patient-specific pluripotent stem cells. Cell Stem Cell 2007, 1, 39–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, D.; Dwyer, J.; Li, H.; Duan, W.; Liu, J.P. Ets2 maintains hTERT gene expression and breast cancer cell proliferation by interacting with c-Myc. J. Biol. Chem. 2008, 283, 23567–23580. [Google Scholar] [CrossRef] [Green Version]
- Safe, S.; Jin, U.H.; Morpurgo, B.; Abudayyeh, A.; Singh, M.; Tjalkens, R.B. Nuclear receptor 4A (NR4A) family-orphans no more. J. Steroid. Biochem. Mol. Biol. 2016, 157, 48–60. [Google Scholar] [CrossRef] [Green Version]
- Le, W.D.; Xu, P.; Jankovic, J.; Jiang, H.; Appel, S.H.; Smith, R.G.; Vassilatis, D.K. Mutations in NR4A2 associated with familial Parkinson disease. Nat. Genet. 2003, 33, 85–89. [Google Scholar] [CrossRef]
- Bluhm, A.; Viceconte, N.; Li, F.; Rane, G.; Ritz, S.; Wang, S.; Levin, M.; Shi, Y.; Kappei, D.; Butter, F. ZBTB10 binds the telomeric variant repeat TTGGGG and interacts with TRF2. Nucleic Acids Res. 2019, 47, 1896–1907. [Google Scholar] [CrossRef] [Green Version]
- Mertens-Talcott, S.U.; Chintharlapalli, S.; Li, X.; Safe, S. The oncogenic microRNA-27a targets genes that regulate specificity protein transcription factors and the G2-M checkpoint in MDA-MB-231 breast cancer cells. Cancer Res. 2007, 67, 11001–11011. [Google Scholar] [CrossRef] [Green Version]
- LeRoy, G.; Rickards, B.; Flint, S.J. The double bromodomain proteins Brd2 and Brd3 couple histone acetylation to transcription. Mol. Cell 2008, 30, 51–60. [Google Scholar] [CrossRef] [Green Version]
- Morey, L.; Pascual, G.; Cozzuto, L.; Roma, G.; Wutz, A.; Benitah, S.A.; Di Croce, L. Nonoverlapping functions of the Polycomb group Cbx family of proteins in embryonic stem cells. Cell Stem Cell 2012, 10, 47–62. [Google Scholar] [CrossRef] [Green Version]
- Hiraishi, N.; Ishida, Y.I.; Sudo, H.; Nagahama, M. WDR74 participates in an early cleavage of the pre-rRNA processing pathway in cooperation with the nucleolar AAA-ATPase NVL2. Biochem. Biophys. Res. Commun. 2018, 495, 116–123. [Google Scholar] [CrossRef]
- Yuniati, L.; Scheijen, B.; van der Meer, L.T.; van Leeuwen, F.N. Tumor suppressors BTG1 and BTG2: Beyond growth control. J. Cell Physiol. 2019, 234, 5379–5389. [Google Scholar] [CrossRef] [Green Version]
- Yasui, K.; Okamoto, H.; Arii, S.; Inazawa, J. Association of over-expressed TFDP1 with progression of hepatocellular carcinomas. J. Hum. Genet. 2003, 48, 609–613. [Google Scholar] [CrossRef] [Green Version]
- Fang, F.; Xia, N.; Angulo, B.; Carey, J.; Cady, Z.; Durruthy-Durruthy, J.; Bennett, T.; Sebastiano, V.; Reijo Pera, R.A. A distinct isoform of ZNF207 controls self-renewal and pluripotency of human embryonic stem cells. Nat. Commun. 2018, 9, 4384. [Google Scholar] [CrossRef] [Green Version]
- Kadoch, C.; Hargreaves, D.C.; Hodges, C.; Elias, L.; Ho, L.; Ranish, J.; Crabtree, G.R. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat. Genet. 2013, 45, 592–601. [Google Scholar] [CrossRef]
- Kadoch, C.; Crabtree, G.R. Reversible disruption of mSWI/SNF (BAF) complexes by the SS18-SSX oncogenic fusion in synovial sarcoma. Cell 2013, 153, 71–85. [Google Scholar] [CrossRef] [Green Version]
- Couture, J.P.; Nolet, G.; Beaulieu, E.; Blouin, R.; Gévry, N. The p400/Brd8 chromatin remodeling complex promotes adipogenesis by incorporating histone variant H2A.Z at PPARγ target genes. Endocrinology 2012, 153, 5796–5808. [Google Scholar] [CrossRef] [Green Version]
- Murachelli, A.G.; Ebert, J.; Basquin, C.; Le Hir, H.; Conti, E. The structure of the ASAP core complex reveals the existence of a Pinin-containing PSAP complex. Nat. Struct. Mol. Biol. 2012, 19, 378–386. [Google Scholar] [CrossRef]
- Shi, J.; Sugrue, S.P. Dissection of protein linkage between keratins and pinin, a protein with dual location at desmosome-intermediate filament complex and in the nucleus. J. Biol. Chem. 2000, 275, 14910–14915. [Google Scholar] [CrossRef] [Green Version]
- Brockschmidt, A.; Trost, D.; Peterziel, H.; Zimmermann, K.; Ehrler, M.; Grassmann, H.; Pfenning, P.N.; Waha, A.; Wohlleber, D.; Brockschmidt, F.F.; et al. KIAA1797/FOCAD encodes a novel focal adhesion protein with tumour suppressor function in gliomas. Brain 2012, 135, 1027–1041. [Google Scholar] [CrossRef] [Green Version]
- Jarvis, R.M.; Hughes, S.M.; Ledgerwood, E.C. Peroxiredoxin 1 functions as a signal peroxidase to receive, transduce, and transmit peroxide signals in mammalian cells. Free Radic. Biol. Med. 2012, 53, 1522–1530. [Google Scholar] [CrossRef]
- Kowal, J.; Arras, G.; Colombo, M.; Jouve, M.; Morath, J.P.; Primdal-Bengtson, B.; Dingli, F.; Loew, D.; Tkach, M.; Théry, C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. USA 2016, 113, E968–E977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyado, K.; Yamada, G.; Yamada, S.; Hasuwa, H.; Nakamura, Y.; Ryu, F.; Suzuki, K.; Kosai, K.; Inoue, K.; Ogura, A.; et al. Requirement of CD9 on the egg plasma membrane for fertilization. Science 2000, 287, 321–324. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.M. The anaphase promoting complex/cyclosome: A machine designed to destroy. Nat. Rev. Mol. Cell Biol. 2006, 7, 644–656. [Google Scholar] [CrossRef]
- Chung, C.Y.; Shin, H.R.; Berdan, C.A.; Ford, B.; Ward, C.C.; Olzmann, J.A.; Zoncu, R.; Nomura, D.K. Covalent targeting of the vacuolar H+-ATPase activates autophagy via mTORC1 inhibition. Nat. Chem. Biol. 2019, 15, 776–785. [Google Scholar] [CrossRef] [PubMed]
- Maeta, K.; Edamatsu, H.; Nishihara, K.; Ikutomo, J.; Bilasy, S.E.; Kataoka, T. Crucial Role of Rapgef2 and Rapgef6, a Family of Guanine Nucleotide Exchange Factors for Rap1 Small GTPase, in Formation of Apical Surface Adherens Junctions and Neural Progenitor Development in the Mouse Cerebral Cortex. eNeuro 2016, 3. [Google Scholar] [CrossRef] [Green Version]
- Hardy, S.; Uetani, N.; Wong, N.; Kostantin, E.; Labbé, D.P.; Bégin, L.R.; Mes-Masson, A.; Miranda-Saavedra, D.; Tremblay, M.L. The protein tyrosine phosphatase PRL-2 interacts with the magnesium transporter CNNM3 to promote oncogenesis. Oncogene 2015, 34, 986–995. [Google Scholar] [CrossRef]
- Li, R.Z.; Hou, J.; Wei, Y.; Luo, X.; Ye, Y.; Zhang, Y. hnRNPDL extensively regulates transcription and alternative splicing. Gene 2019, 687, 125–134. [Google Scholar] [CrossRef]
- Batlle, C.; Yang, P.; Coughlin, M.; Messing, J.; Pesarrodona, M.; Szulc, E.; Salvatella, X.; Kim, H.J.; Taylor, J.P.; Ventura, S. hnRNPDL Phase Separation Is Regulated by Alternative Splicing and Disease-Causing Mutations Accelerate Its Aggregation. Cell Rep. 2020, 30, 1117–1128.e1115. [Google Scholar] [CrossRef] [Green Version]
- Wickramasekara, R.N.; Stessman, H.A.F. Histone 4 Lysine 20 Methylation: A Case for Neurodevelopmental Disease. Biology 2019, 8, 11. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Xu, R.; Zhu, B.; Yang, X.; Ding, X.; Duan, S.; Xu, T.; Zhuang, Y.; Han, M. Syne-1 and Syne-2 play crucial roles in myonuclear anchorage and motor neuron innervation. Development 2007, 134, 901–908. [Google Scholar] [CrossRef] [Green Version]
- Hurlin, P.J.; Steingrìmsson, E.; Copeland, N.G.; Jenkins, N.A.; Eisenman, R.N. Mga, a dual-specificity transcription factor that interacts with Max and contains a T-domain DNA-binding motif. EMBO J. 1999, 18, 7019–7028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allaman, I.; Bélanger, M.; Magistretti, P.J. Methylglyoxal, the dark side of glycolysis. Front. Neurosci. 2015, 9, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makrythanasis, P.; Kato, M.; Zaki, M.S.; Saitsu, H.; Nakamura, K.; Santoni, F.A.; Miyatake, S.; Nakashima, M.; Issa, M.Y.; Guipponi, M.; et al. Pathogenic Variants in PIGG Cause Intellectual Disability with Seizures and Hypotonia. Am. J. Hum. Genet. 2016, 98, 615–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, T.; Li, W.; Zhang, Q.; Karim, A.; Novak, E.K.; Sviderskaya, E.V.; Hill, S.P.; Bennett, D.C.; Levin, A.V.; Nieuwenhuis, H.K.; et al. Hermansky-Pudlak syndrome is caused by mutations in HPS4, the human homolog of the mouse light-ear gene. Nat. Genet. 2002, 30, 321–324. [Google Scholar] [CrossRef]
- O’Leary, M.N.; Schreiber, K.H.; Zhang, Y.; Duc, A.C.; Rao, S.; Hale, J.S.; Academia, E.C.; Shah, S.R.; Morton, J.F.; Holstein, C.A.; et al. The ribosomal protein Rpl22 controls ribosome composition by directly repressing expression of its own paralog, Rpl22l1. PLoS Genet. 2013, 9, e1003708. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Duc, A.C.; Rao, S.; Sun, X.L.; Bilbee, A.N.; Rhodes, M.; Li, Q.; Kappes, D.J.; Rhodes, J.; Wiest, D.L. Control of hematopoietic stem cell emergence by antagonistic functions of ribosomal protein paralogs. Dev. Cell 2013, 24, 411–425. [Google Scholar] [CrossRef] [Green Version]
- Puy, H.; Gouya, L.; Deybach, J.C. Porphyrias. Lancet 2010, 375, 924–937. [Google Scholar] [CrossRef]
- Costello, J.L.; Castro, I.G.; Schrader, T.A.; Islinger, M.; Schrader, M. Peroxisomal ACBD4 interacts with VAPB and promotes ER-peroxisome associations. Cell Cycle 2017, 16, 1039–1045. [Google Scholar] [CrossRef]
- Adham, I.M.; Eck, T.J.; Mierau, K.; Müller, N.; Sallam, M.A.; Paprotta, I.; Schubert, S.; Hoyer-Fender, S.; Engel, W. Reduction of spermatogenesis but not fertility in Creb3l4-deficient mice. Mol. Cell Biol. 2005, 25, 7657–7664. [Google Scholar] [CrossRef] [Green Version]
- Ioannou, M.S.; Bell, E.S.; Girard, M.; Chaineau, M.; Hamlin, J.N.; Daubaras, M.; Monast, A.; Park, M.; Hodgson, L.; McPherson, P.S. DENND2B activates Rab13 at the leading edge of migrating cells and promotes metastatic behavior. J. Cell Biol. 2015, 208, 629–648. [Google Scholar] [CrossRef]
- Fry, A.M.; O’Regan, L.; Sabir, S.R.; Bayliss, R. Cell cycle regulation by the NEK family of protein kinases. J. Cell Sci. 2012, 125, 4423–4433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loyer, P.; Trembley, J.H. Roles of CDK/Cyclin complexes in transcription and pre-mRNA splicing: Cyclins L and CDK11 at the cross-roads of cell cycle and regulation of gene expression. Semin. Cell Dev. Biol. 2020, 107, 36–45. [Google Scholar] [CrossRef]
- Fedorov, O.; Huber, K.; Eisenreich, A.; Filippakopoulos, P.; King, O.; Bullock, A.N.; Szklarczyk, D.; Jensen, L.J.; Fabbro, D.; Trappe, J.; et al. Specific CLK inhibitors from a novel chemotype for regulation of alternative splicing. Chem. Biol. 2011, 18, 67–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chambers, C.A.; Gallinger, S.; Anderson, S.K.; Giardina, S.; Ortaldo, J.R.; Hozumi, N.; Roder, J. Expression of the NK-TR gene is required for NK-like activity in human T cells. J. Immunol. 1994, 152, 2669–2674. [Google Scholar]
- Singh, N.N.; Lee, B.M.; Singh, R.N. Splicing regulation in spinal muscular atrophy by an RNA structure formed by long-distance interactions. Ann. N. Y. Acad. Sci. 2015, 1341, 176–187. [Google Scholar] [CrossRef] [Green Version]
- Singh, N.N.; Luo, D.; Singh, R.N. Pre-mRNA Splicing Modulation by Antisense Oligonucleotides. Methods Mol. Biol. 2018, 1828, 415–437. [Google Scholar] [CrossRef]
- Seo, J.; Ottesen, E.W.; Singh, R.N. Antisense methods to modulate pre-mRNA splicing. Methods Mol. Biol. 2014, 1126, 271–283. [Google Scholar] [CrossRef]
- Osman, E.Y.; Yen, P.F.; Lorson, C.L. Bifunctional RNAs targeting the intronic splicing silencer N1 increase SMN levels and reduce disease severity in an animal model of spinal muscular atrophy. Mol. Ther. 2012, 20, 119–126. [Google Scholar] [CrossRef] [Green Version]
- Hammond, S.M.; Hazell, G.; Shabanpoor, F.; Saleh, A.F.; Bowerman, M.; Sleigh, J.N.; Meijboom, K.E.; Zhou, H.; Muntoni, F.; Talbot, K.; et al. Systemic peptide-mediated oligonucleotide therapy improves long-term survival in spinal muscular atrophy. Proc. Natl. Acad. Sci. USA 2016, 113, 10962–10967. [Google Scholar] [CrossRef] [Green Version]
- Shabanpoor, F.; Hammond, S.M.; Abendroth, F.; Hazell, G.; Wood, M.J.A.; Gait, M.J. Identification of a Peptide for Systemic Brain Delivery of a Morpholino Oligonucleotide in Mouse Models of Spinal Muscular Atrophy. Nucleic Acid Ther. 2017, 27, 130–143. [Google Scholar] [CrossRef] [Green Version]
- Robin, V.; Griffith, G.; Carter, J.L.; Leumann, C.J.; Garcia, L.; Goyenvalle, A. Efficient SMN Rescue following Subcutaneous Tricyclo-DNA Antisense Oligonucleotide Treatment. Mol. Ther. Nucleic Acids 2017, 7, 81–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Touznik, A.; Maruyama, R.; Hosoki, K.; Echigoya, Y.; Yokota, T. LNA/DNA mixmer-based antisense oligonucleotides correct alternative splicing of the SMN2 gene and restore SMN protein expression in type 1 SMA fibroblasts. Sci. Rep. 2017, 7, 3672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramirez, A.; Crisafulli, S.G.; Rizzuti, M.; Bresolin, N.; Comi, G.P.; Corti, S.; Nizzardo, M. Investigation of New Morpholino Oligomers to Increase Survival Motor Neuron Protein Levels in Spinal Muscular Atrophy. Int. J. Mol. Sci 2018, 19, 167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, L.; Rigo, F.; Bennett, C.F.; Krainer, A.R.; Hua, Y. Comparison of the efficacy of MOE and PMO modifications of systemic antisense oligonucleotides in a severe SMA mouse model. Nucleic Acids Res. 2020, 48, 2853–2865. [Google Scholar] [CrossRef] [PubMed]
- Bilanges, B.; Stokoe, D. Direct comparison of the specificity of gene silencing using antisense oligonucleotides and RNAi. Biochem. J. 2005, 388, 573–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartys, N.; Kierzek, R.; Lisowiec-Wachnicka, J. The regulation properties of RNA secondary structure in alternative splicing. Biochim. Biophys. Acta Gene Regul. Mech. 2019, 1862, 194401. [Google Scholar] [CrossRef]
- Andrews, R.J.; Moss, W.N. Computational approaches for the discovery of splicing regulatory RNA structures. Biochim. Biophys. Acta Gene Regul. Mech. 2019, 1862, 194380. [Google Scholar] [CrossRef]
- Baralle, F.E.; Singh, R.N.; Stamm, S. RNA structure and splicing regulation. Biochim. Biophys. Acta Gene Regul. Mech. 2019, 1862, 194448. [Google Scholar] [CrossRef]
- Xu, B.; Meng, Y.; Jin, Y. RNA structures in alternative splicing and back-splicing. Wiley Interdiscip. Rev. RNA 2021, 12, e1626. [Google Scholar] [CrossRef] [PubMed]
- Howell, M.D.; Ottesen, E.W.; Singh, N.N.; Anderson, R.L.; Singh, R.N. Gender-Specific Amelioration of SMA Phenotype upon Disruption of a Deep Intronic Structure by an Oligonucleotide. Mol. Ther. 2017, 25, 1328–1341. [Google Scholar] [CrossRef] [Green Version]
- Marcucci, R.; Baralle, F.E.; Romano, M. Complex splicing control of the human Thrombopoietin gene by intronic G runs. Nucleic Acids Res. 2007, 35, 132–142. [Google Scholar] [CrossRef] [Green Version]
- Berget, S.M. Exon recognition in vertebrate splicing. J. Biol. Chem. 1995, 270, 2411–2414. [Google Scholar] [CrossRef] [Green Version]
- Fox-Walsh, K.L.; Dou, Y.; Lam, B.J.; Hung, S.P.; Baldi, P.F.; Hertel, K.J. The architecture of pre-mRNAs affects mechanisms of splice-site pairing. Proc. Natl. Acad. Sci. USA 2005, 102, 16176–16181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keren, H.; Lev-Maor, G.; Ast, G. Alternative splicing and evolution: Diversification, exon definition and function. Nat. Rev. Genet. 2010, 11, 345–355. [Google Scholar] [CrossRef]
- Ke, S.; Chasin, L.A. Intronic motif pairs cooperate across exons to promote pre-mRNA splicing. Genome Biol. 2010, 11, R84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Contreras, R.; Fisette, J.F.; Nasim, F.U.; Madden, R.; Cordeau, M.; Chabot, B. Intronic binding sites for hnRNP A/B and hnRNP F/H proteins stimulate pre-mRNA splicing. PLoS Biol. 2006, 4, e21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, J.M.; Phizicky, D.V.; Neugebauer, K.M. Nuclear mechanisms of gene expression control: Pre-mRNA splicing as a life or death decision. Curr. Opin. Genet. Dev. 2021, 67, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Bentley, D.L. Coupling mRNA processing with transcription in time and space. Nat. Rev. Genet. 2014, 15, 163–175. [Google Scholar] [CrossRef] [Green Version]
- García-Moreno, J.F.; Romão, L. Perspective in Alternative Splicing Coupled to Nonsense-Mediated mRNA Decay. Int. J. Mol. Sci. 2020, 21, 9424. [Google Scholar] [CrossRef]
- Li, X.; Liu, S.; Jiang, J.; Zhang, L.; Espinosa, S.; Hill, R.C.; Hansen, K.C.; Zhou, Z.H.; Zhao, R. CryoEM structure of Saccharomyces cerevisiae U1 snRNP offers insight into alternative splicing. Nat. Commun. 2017, 8, 1035. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Aibara, S.; Vos, S.M.; Agafonov, D.E.; Lührmann, R.; Cramer, P. Structure of a transcribing RNA polymerase II-U1 snRNP complex. Science 2021, 371, 305–309. [Google Scholar] [CrossRef]
- Di, C.; So, B.R.; Cai, Z.; Arai, C.; Duan, J.; Dreyfuss, G. U1 snRNP Telescripting Roles in Transcription and Its Mechanism. Cold Spring Harb. Symp. Quant. Biol. 2019, 84, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Liang, X.H.; Crooke, S.T. Phosphorothioate oligonucleotides can displace NEAT1 RNA and form nuclear paraspeckle-like structures. Nucleic Acids Res. 2014, 42, 8648–8662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crooke, S.T.; Vickers, T.A.; Liang, X.H. Phosphorothioate modified oligonucleotide-protein interactions. Nucleic Acids Res. 2020, 48, 5235–5253. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.H.; De Hoyos, C.L.; Shen, W.; Zhang, L.; Fazio, M.; Crooke, S.T. Solid-Phase Separation of Toxic Phosphorothioate Antisense Oligonucleotide-Protein Nucleolar Aggregates Is Cytoprotective. Nucleic Acid Ther. 2021, 31, 126–144. [Google Scholar] [CrossRef]
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in oligonucleotide drug delivery. Nat. Rev. Drug Discov. 2020, 19, 673–694. [Google Scholar] [CrossRef]
- Ratni, H.; Ebeling, M.; Baird, J.; Bendels, S.; Bylund, J.; Chen, K.S.; Denk, N.; Feng, Z.; Green, L.; Guerard, M.; et al. Discovery of Risdiplam, a Selective Survival of Motor Neuron-2 ( SMN2) Gene Splicing Modifier for the Treatment of Spinal Muscular Atrophy (SMA). J. Med. Chem. 2018, 61, 6501–6517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, A.K.; Hurley, B.; Kerrigan, R.; Shu, L.; Chin, D.N.; Shen, Y.; O’Brien, G.; Sung, M.J.; Hou, Y.; Axford, J.; et al. Discovery of Small Molecule Splicing Modulators of Survival Motor Neuron-2 (SMN2) for the Treatment of Spinal Muscular Atrophy (SMA). J. Med. Chem. 2018, 61, 11021–11036. [Google Scholar] [CrossRef]
- Garcia-Lopez, A.; Tessaro, F.; Jonker, H.R.A.; Wacker, A.; Richter, C.; Comte, A.; Berntenis, N.; Schmucki, R.; Hatje, K.; Petermann, O.; et al. Targeting RNA structure in SMN2 reverses spinal muscular atrophy molecular phenotypes. Nat. Commun. 2018, 9, 2032. [Google Scholar] [CrossRef]
- Ando, S.; Suzuki, S.; Okubo, S.; Ohuchi, K.; Takahashi, K.; Nakamura, S.; Shimazawa, M.; Fuji, K.; Hara, H. Discovery of a CNS penetrant small molecule SMN2 splicing modulator with improved tolerability for spinal muscular atrophy. Sci. Rep. 2020, 10, 17472. [Google Scholar] [CrossRef]
- Singh, R.N. More is needed to complement the available therapies of spinal muscular atrophy. Future Med. Chem. 2019, 11, 2873–2876. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrow, J.; Frankish, A.; Gonzalez, J.M.; Tapanari, E.; Diekhans, M.; Kokocinski, F.; Aken, B.L.; Barrell, D.; Zadissa, A.; Searle, S.; et al. GENCODE: The reference human genome annotation for The ENCODE Project. Genome Res. 2012, 22, 1760–1774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. FeatureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, S.; Park, J.W.; Lu, Z.X.; Lin, L.; Henry, M.D.; Wu, Y.N.; Zhou, Q.; Xing, Y. rMATS: Robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. USA 2014, 111, E5593–E5601. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Keenan, A.B.; Torre, D.; Lachmann, A.; Leong, A.K.; Wojciechowicz, M.L.; Utti, V.; Jagodnik, K.M.; Kropiwnicki, E.; Wang, Z.; Ma’ayan, A. ChEA3: Transcription factor enrichment analysis by orthogonal omics integration. Nucleic Acids Res. 2019, 47, W212–W224. [Google Scholar] [CrossRef] [Green Version]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Acronym | Full Name |
|---|---|
| 2′OMe | 2′-O-methyl ribose sugar modification |
| 3′ss | 3′ splice site |
| 3UP8 | 8mer ASO that partially targets ISS-N1 |
| 5′ss | 5′ splice site |
| A3S | Alternative 3′ splice site usage |
| A5S | Alternative 5′ splice site usage |
| Anti-N1 | 20mer ASO targeting ISS-N1 |
| ASO | antisense oligonucleotide |
| EIN | Increased exon inclusion |
| ESK | Increased exon skipping |
| F14 | 14mer ASO that targets ISS-N1 |
| F18 | 18mer ASO that targets ISS-N1 |
| HiA | High concentration (100 nM) of Anti-N1 |
| IRM | Improved intron removal |
| IRT | Increased intron retention |
| ISO | ISS-N1 targeting splice switching ASO |
| ISS-N1 | Intronic splicing silencer N1 |
| L2FC | log(2) fold change |
| LoA | Low concentration (5 nM) of Anti-N1 |
| MOE | 2′-O-methoxyethyl ribose sugar modification |
| MXE | Mutually exclusive exons and/or mixed events |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ottesen, E.W.; Luo, D.; Singh, N.N.; Singh, R.N. High Concentration of an ISS-N1-Targeting Antisense Oligonucleotide Causes Massive Perturbation of the Transcriptome. Int. J. Mol. Sci. 2021, 22, 8378. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168378
Ottesen EW, Luo D, Singh NN, Singh RN. High Concentration of an ISS-N1-Targeting Antisense Oligonucleotide Causes Massive Perturbation of the Transcriptome. International Journal of Molecular Sciences. 2021; 22(16):8378. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168378
Chicago/Turabian StyleOttesen, Eric William, Diou Luo, Natalia Nikolaevna Singh, and Ravindra Narayan Singh. 2021. "High Concentration of an ISS-N1-Targeting Antisense Oligonucleotide Causes Massive Perturbation of the Transcriptome" International Journal of Molecular Sciences 22, no. 16: 8378. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168378