
Nitric Oxide-Releasing Drug Glyceryl Trinitrate Targets JAK2/STAT3 Signaling, Migration and Invasion of Triple-Negative Breast Cancer Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
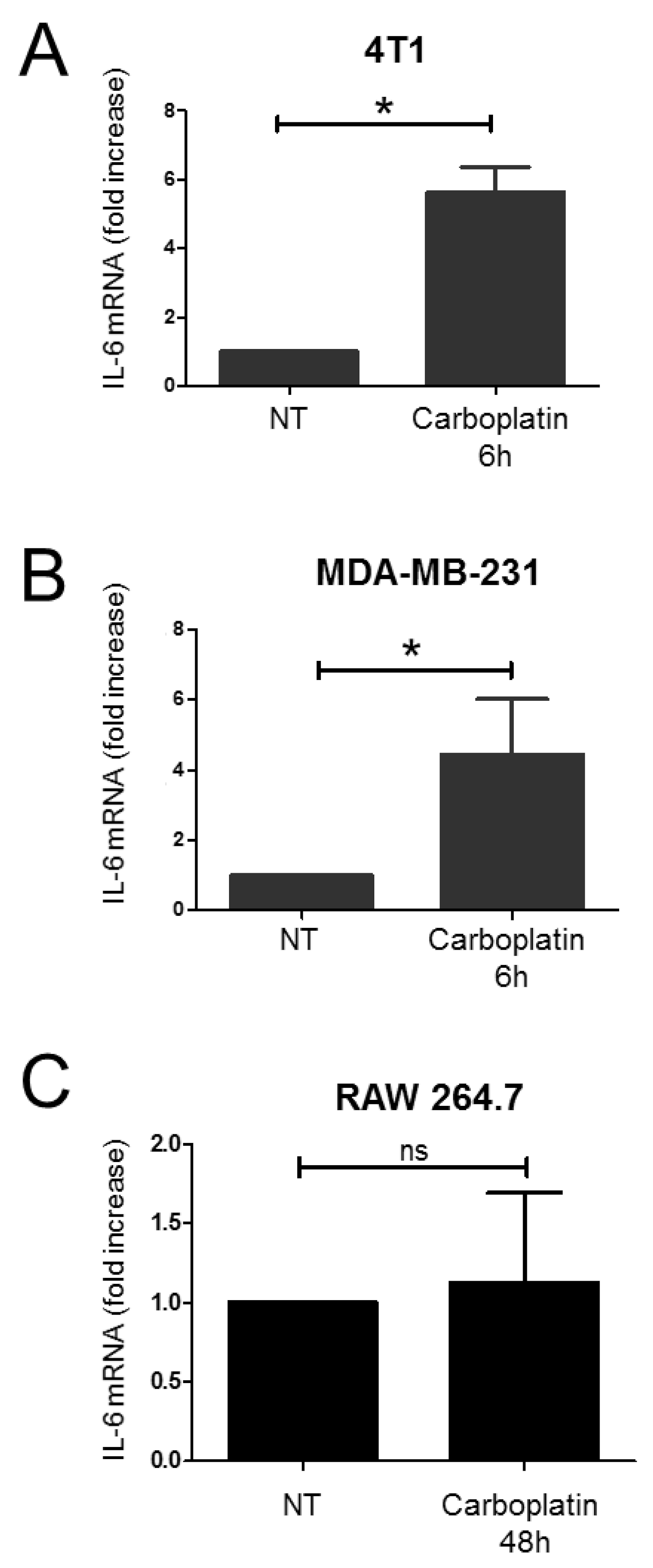
2.1. Effect of Carboplatin on the Production of IL-6 in TNBC Cells
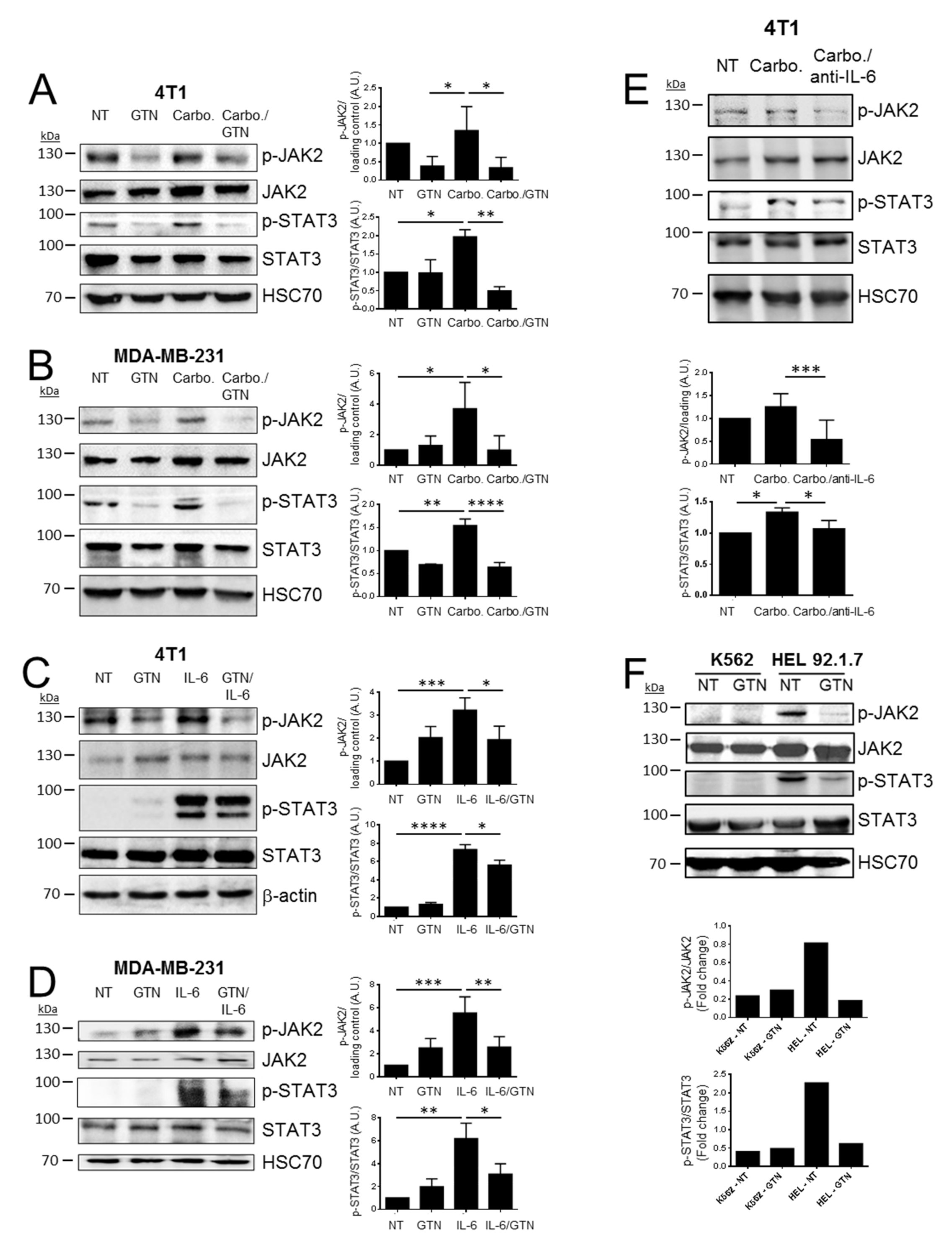
2.2. GTN Inhibits JAK2/STAT3 Signaling Pathway
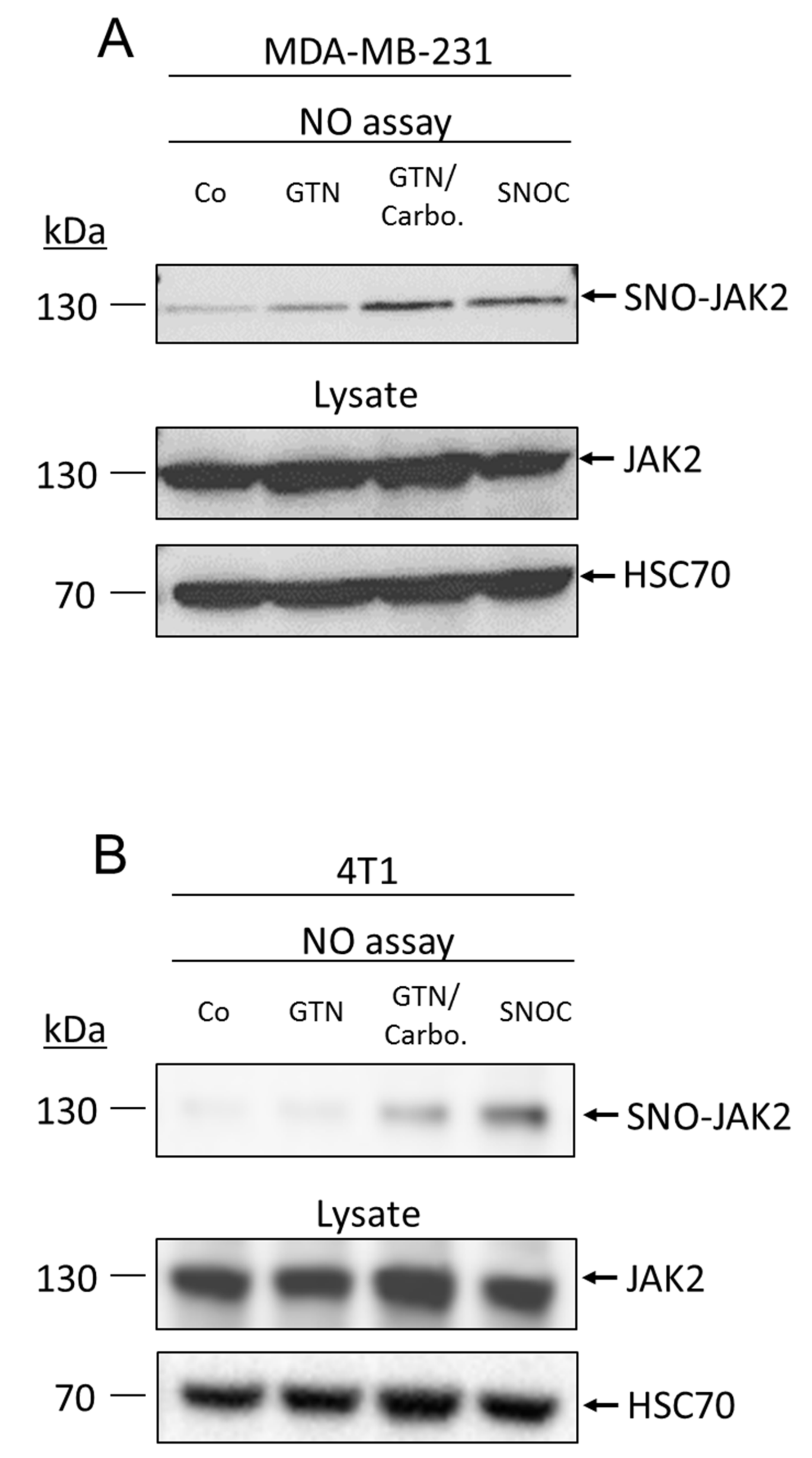
2.3. NO Promotes JAK2 S-Nitrosylation
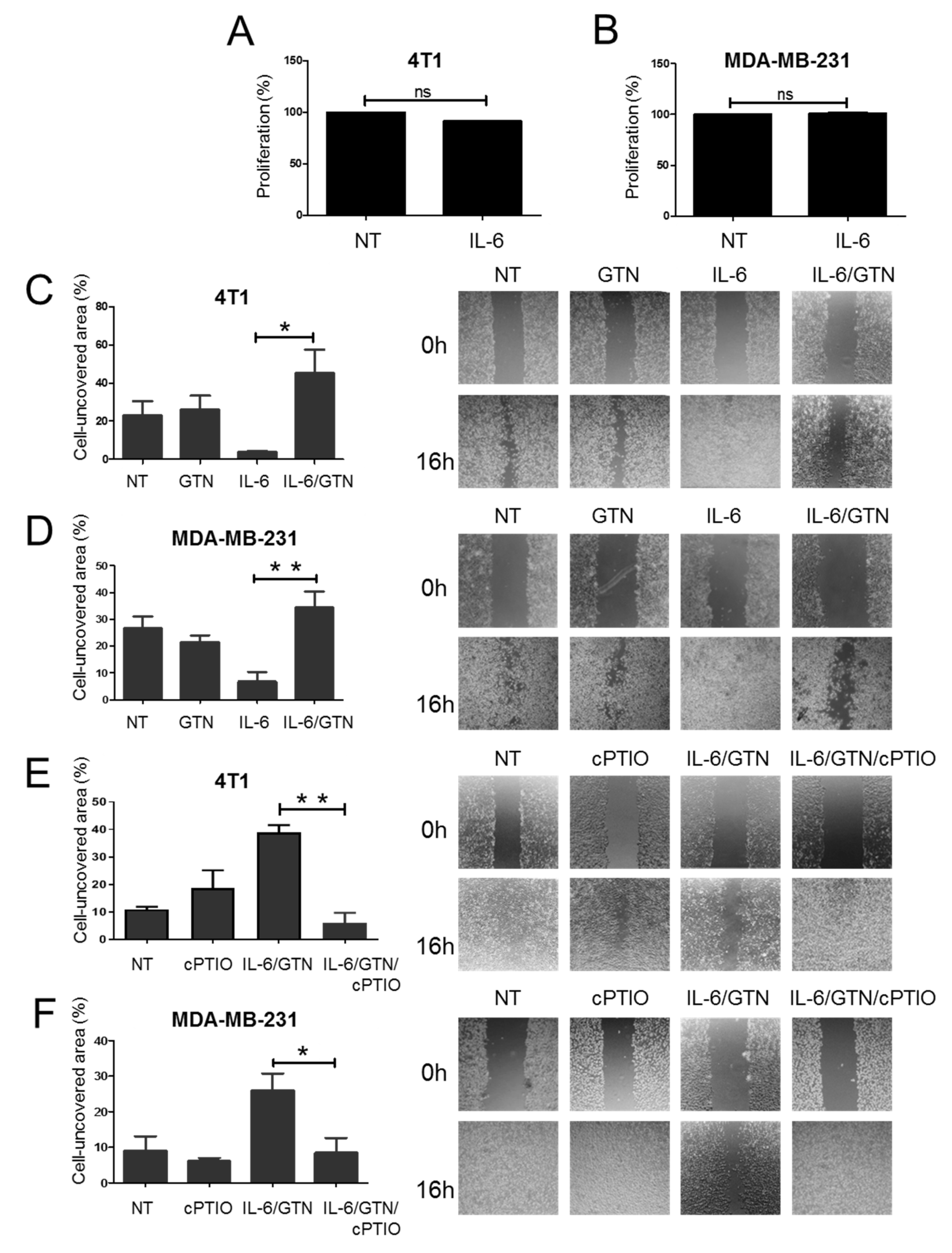
2.4. Effect of GTN on IL-6-Mediated Wound Healing in TNBC Cells
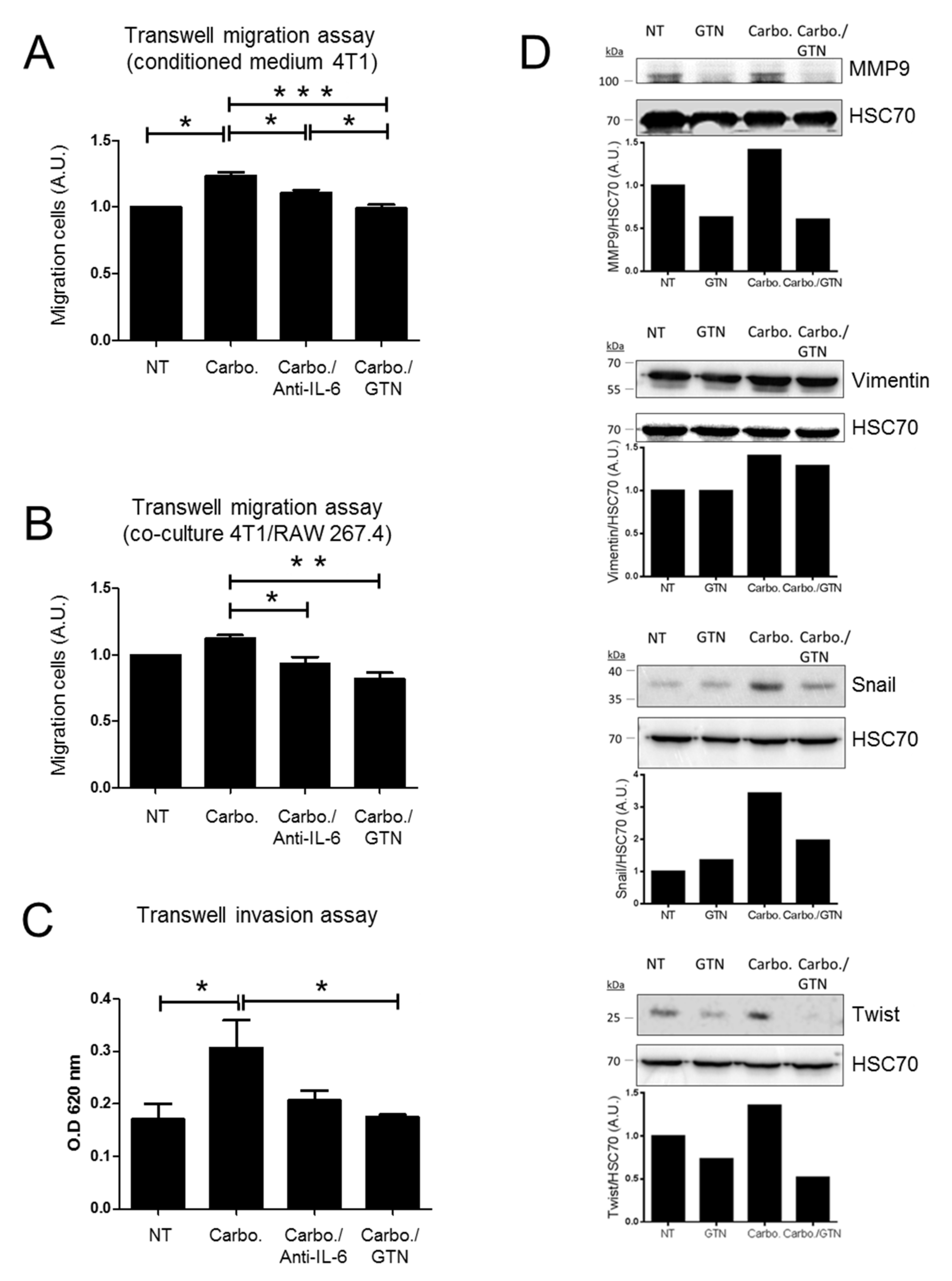
2.5. In Vitro Effects of GTN on Migration and Invasion of TNBC Cells
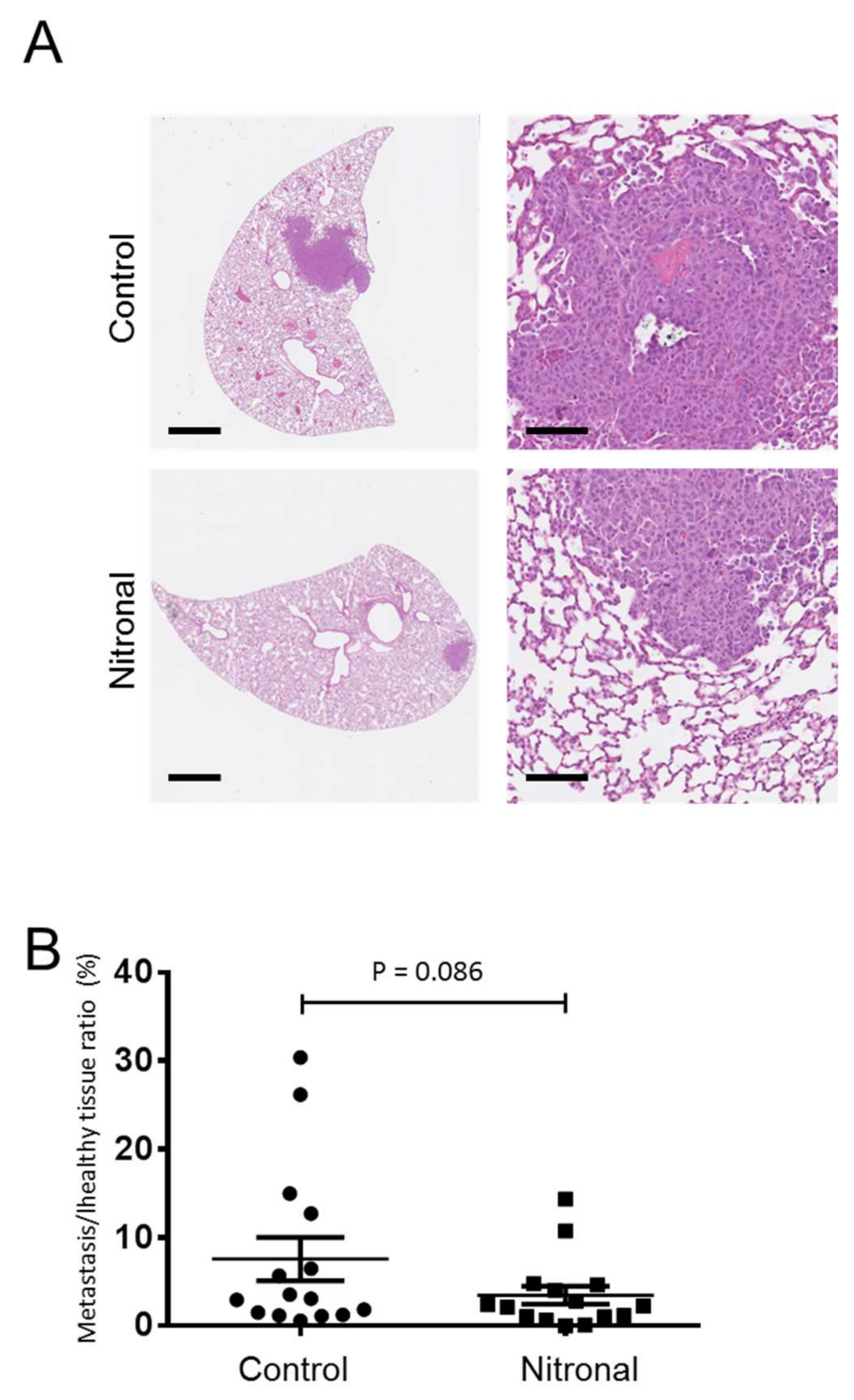
2.6. In Vivo Effect of GTN in a Syngeneic Model of Murine Metastatic TNBC
3. Discussion
4. Materials and Methods
4.1. Cancer Cell Lines
4.2. Drug, Reagents and Recombinant Proteins
4.3. Quantitative Real-Time Polymerase Chain Reaction (RT-qPCR)
4.4. Griess Assay
4.5. Proliferation Assay (MTS)
4.6. Apoptosis Quantification
4.7. Wound Healing Assay
4.8. Transwell Migration and Invasion Assays
4.9. Biotin Switch Assay (BSA)
4.10. Animal and Ethics Statement
4.11. In Vivo Syngeneic Mouse Model of Breast Metastasis Formation
4.12. Histology and Metastases Assessment
4.13. Western Blotting
4.14. Antibodies
4.15. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016, 13, 674–690. [Google Scholar] [CrossRef]
- Di Leo, A.; Curigliano, G.; Diéras, V.; Malorni, L.; Sotiriou, C.; Swanton, C.; Thompson, A.; Tutt, A.; Piccart, M. New approaches for improving outcomes in breast cancer in Europe. Breast 2015, 24, 321–330. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.H.; Qin, L.; Li, X. Role of STAT3 signaling pathway in breast cancer. Cell Commun. Signal. 2020, 18, 33. [Google Scholar] [CrossRef] [Green Version]
- Qiu, S.Q.; Waaijer, S.J.H.; Zwager, M.C.; de Vries, E.G.E.; van der Vegt, B.; Schröder, C.P. Tumor-associated macrophages in breast cancer: Innocent bystander or important player? Cancer Treat. Rev. 2018, 70, 178–189. [Google Scholar] [CrossRef] [Green Version]
- Martins, D.; Schmitt, F. Microenvironment in breast tumorigenesis: Friend or foe? Histol. Histopathol. 2018, 18021. [Google Scholar] [CrossRef]
- Barbazán, J.; Matic Vignjevic, D. Cancer associated fibroblasts: Is the force the path to the dark side? Curr. Opin. Cell Biol. 2018, 56, 71–79. [Google Scholar] [CrossRef]
- Yaacoub, K.; Pedeux, R.; Tarte, K.; Guillaudeux, T. Role of the tumor microenvironment in regulating apoptosis and cancer progression. Cancer Lett. 2016, 378, 150–159. [Google Scholar] [CrossRef]
- Heinrich, P.C.; Behrmann, I.; Haan, S.; Hermanns, H.M.; Müller-Newen, G.; Schaper, F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem. J. 2003, 374, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Grivennikov, S.; Karin, M. Autocrine IL-6 signaling: A key event in tumorigenesis? Cancer Cell 2008, 13, 7–9. [Google Scholar] [CrossRef] [Green Version]
- Landskron, G.; De la Fuente, M.; Thuwajit, P.; Thuwajit, C.; Hermoso, M.A. Chronic inflammation and cytokines in the tumor microenvironment. J. Immunol. Res. 2014, 2014, 149185. [Google Scholar] [CrossRef] [Green Version]
- Hou, L.; Xie, S.; Li, G.; Xiong, B.; Gao, Y.; Zhao, X.; Hu, J.; Deng, S.; Jiang, J. IL-6 Triggers the Migration and Invasion of Oestrogen Receptor-Negative Breast Cancer Cells via Regulation of Hippo Pathways. Basic Clin. Pharmacol. Toxicol. 2018, 123, 549–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, Y.A.; Sung, M.K.; Yeon, J.Y.; Ro, J.; Kim, J. Prognostic role of interleukin-6, interleukin-8, and leptin levels according to breast cancer subtype. Cancer Res. Treat. 2013, 45, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Dethlefsen, C.; Højfeldt, G.; Hojman, P. The role of intratumoral and systemic IL-6 in breast cancer. Breast Cancer Res. Treat. 2013, 138, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, J.; Tachibana, M.; Horiguchi, Y.; Oya, M.; Ohigashi, T.; Asakura, H.; Murai, M. Serum interleukin 6 as a prognostic factor in patients with prostate cancer. Clin. Cancer Res. 2000, 6, 2702–2706. [Google Scholar]
- Nakashima, J. Prognostic significance of interleukin-6 in patients with prostate cancer. Nihon Rinsho 2002, 60, 165–169. [Google Scholar]
- Gudbrandsdottir, G.; Aarstad, H.H.; Bostad, L.; Hjelle, K.M.; Aarstad, H.J.; Bruserud, Ø.; Tvedt, T.H.A.; Beisland, C. Serum levels of the IL-6 family of cytokines predict prognosis in renal cell carcinoma (RCC). Cancer Immunol. Immunother. 2021, 70, 19–30. [Google Scholar] [CrossRef]
- Lippitz, B.E.; Harris, R.A. Cytokine patterns in cancer patients: A review of the correlation between interleukin 6 and prognosis. Oncoimmunology 2016, 5, e1093722. [Google Scholar] [CrossRef] [Green Version]
- Bournazou, E.; Bromberg, J. Targeting the tumor microenvironment: JAK-STAT3 signaling. JAKSTAT 2013, 2, e23828. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Lee, H.; Herrmann, A.; Buettner, R.; Jove, R. Revisiting STAT3 signalling in cancer: New and unexpected biological functions. Nat. Rev. Cancer 2014, 14, 736–746. [Google Scholar] [CrossRef]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef]
- Rossi, J.F.; Lu, Z.Y.; Jourdan, M.; Klein, B. Interleukin-6 as a therapeutic target. Clin. Cancer Res. 2015, 21, 1248–1257. [Google Scholar] [CrossRef] [Green Version]
- Plenchette, S.; Romagny, S.; Laurens, V.; Bettaieb, A. NO and cancer: Itinerary of a double agent. Med. Sci. (Paris) 2016, 32, 625–633. [Google Scholar] [CrossRef] [Green Version]
- Yasuda, H.; Yamaya, M.; Nakayama, K.; Sasaki, T.; Ebihara, S.; Kanda, A.; Asada, M.; Inoue, D.; Suzuki, T.; Okazaki, T.; et al. Randomized phase II trial comparing nitroglycerin plus vinorelbine and cisplatin with vinorelbine and cisplatin alone in previously untreated stage IIIB/IV non-small-cell lung cancer. J. Clin. Oncol. 2006, 24, 688–694. [Google Scholar] [CrossRef]
- Siemens, D.R.; Heaton, J.P.; Adams, M.A.; Kawakami, J.; Graham, C.H. Phase II study of nitric oxide donor for men with increasing prostate-specific antigen level after surgery or radiotherapy for prostate cancer. Urology 2009, 74, 878–883. [Google Scholar] [CrossRef]
- Kiss, E.; Abdelwahab, E.H.M.M.; Steib, A.; Papp, E.; Torok, Z.; Jakab, L.; Smuk, G.; Sarosi, V.; Pongracz, J.E. Cisplatin treatment induced interleukin 6 and 8 production alters lung adenocarcinoma cell migration in an oncogenic mutation dependent manner. Respir Res 2020, 21, 120. [Google Scholar] [CrossRef]
- Davidson, A.; Veillard, A.S.; Tognela, A.; Chan, M.M.; Hughes, B.G.; Boyer, M.; Briscoe, K.; Begbie, S.; Abdi, E.; Crombie, C.; et al. A phase III randomized trial of adding topical nitroglycerin to first-line chemotherapy for advanced nonsmall-cell lung cancer: The Australasian lung cancer trials group NITRO trial. Ann. Oncol. 2015, 26, 2280–2286. [Google Scholar] [CrossRef]
- Hao, Y.; Baker, D.; Ten Dijke, P. TGF-β-Mediated Epithelial-Mesenchymal Transition and Cancer Metastasis. Int. J. Mol. Sci. 2019, 20, 2767. [Google Scholar] [CrossRef] [Green Version]
- Wahba, H.A.; El-Hadaad, H.A. Current approaches in treatment of triple-negative breast cancer. Cancer Biol. Med. 2015, 12, 106–116. [Google Scholar] [CrossRef]
- Korde, L.A.; Somerfield, M.R.; Carey, L.A.; Crews, J.R.; Denduluri, N.; Hwang, E.S.; Khan, S.A.; Loibl, S.; Morris, E.A.; Perez, A.; et al. Neoadjuvant Chemotherapy, Endocrine Therapy, and Targeted Therapy for Breast Cancer: ASCO Guideline. J. Clin. Oncol. 2021, JCO2003399. [Google Scholar] [CrossRef]
- Heo, T.H.; Wahler, J.; Suh, N. Potential therapeutic implications of IL-6/IL-6R/gp130-targeting agents in breast cancer. Oncotarget 2016, 7, 15460–15473. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.; Tanaka, T.; Narazaki, M.; Kishimoto, T. Targeting Interleukin-6 Signaling in Clinic. Immunity 2019, 50, 1007–1023. [Google Scholar] [CrossRef]
- Stover, D.G.; Gil Del Alcazar, C.R.; Brock, J.; Guo, H.; Overmoyer, B.; Balko, J.; Xu, Q.; Bardia, A.; Tolaney, S.M.; Gelman, R.; et al. Phase II study of ruxolitinib, a selective JAK1/2 inhibitor, in patients with metastatic triple-negative breast cancer. NPJ Breast Cancer 2018, 4, 10. [Google Scholar] [CrossRef] [PubMed]
- Plenchette, S.; Romagny, S.; Laurens, V.; Bettaieb, A. S-Nitrosylation in TNF superfamily signaling pathway: Implication in cancer. Redox Biol. 2015, 6, 507–515. [Google Scholar] [CrossRef] [Green Version]
- Elsasser, T.H.; Kahl, S.; Li, C.J.; Sartin, J.L.; Garrett, W.M.; Rodrigo, J. Caveolae nitration of Janus kinase-2 at the 1007Y-1008Y site: Coordinating inflammatory response and metabolic hormone readjustment within the somatotropic axis. Endocrinology 2007, 148, 3803–3813. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.K.; Patil, C.N.; Patlolla, S.; Gunter, B.W.; Booz, G.W.; Duhé, R.J. Identification of a redox-sensitive switch within the JAK2 catalytic domain. Free Radic. Biol. Med. 2012, 52, 1101–1110. [Google Scholar] [CrossRef] [Green Version]
- Singh, I.; Kim, J.; Singh, A.K.; Sharma, A.K.; Won, J.S. STAT3 Regulation By S-Nitrosylation: Implication In Cancer. Redox Biol. 2015, 5, 416–417. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Won, J.S.; Singh, A.K.; Sharma, A.K.; Singh, I. STAT3 regulation by S-nitrosylation: Implication for inflammatory disease. Antioxid. Redox Signal. 2014, 20, 2514–2527. [Google Scholar] [CrossRef]
- Cheteh, E.H.; Sarne, V.; Ceder, S.; Bianchi, J.; Augsten, M.; Rundqvist, H.; Egevad, L.; Östman, A.; Wiman, K.G. Interleukin-6 derived from cancer-associated fibroblasts attenuates the p53 response to doxorubicin in prostate cancer cells. Cell Death Discov. 2020, 6, 42. [Google Scholar] [CrossRef]
- Walsh, E.M.; Keane, M.M.; Wink, D.A.; Callagy, G.; Glynn, S.A. Review of Triple Negative Breast Cancer and the Impact of Inducible Nitric Oxide Synthase on Tumor Biology and Patient Outcomes. Crit Rev. Oncog. 2016, 21, 333–351. [Google Scholar] [CrossRef]
- Granados-Principal, S.; Liu, Y.; Guevara, M.L.; Blanco, E.; Choi, D.S.; Qian, W.; Patel, T.; Rodriguez, A.A.; Cusimano, J.; Weiss, H.L.; et al. Inhibition of iNOS as a novel effective targeted therapy against triple-negative breast cancer. Breast Cancer Res. 2015, 17, 25. [Google Scholar] [CrossRef]
- Dávila-González, D.; Choi, D.S.; Rosato, R.R.; Granados-Principal, S.M.; Kuhn, J.G.; Li, W.F.; Qian, W.; Chen, W.; Kozielski, A.J.; Wong, H.; et al. Pharmacological Inhibition of NOS Activates ASK1/JNK Pathway Augmenting Docetaxel-Mediated Apoptosis in Triple-Negative Breast Cancer. Clin. Cancer Res. 2018, 24, 1152–1162. [Google Scholar] [CrossRef] [Green Version]
- Jin, K.; Pandey, N.B.; Popel, A.S. Simultaneous blockade of IL-6 and CCL5 signaling for synergistic inhibition of triple-negative breast cancer growth and metastasis. Breast Cancer Res. 2018, 20, 54. [Google Scholar] [CrossRef] [Green Version]
- Hartman, Z.C.; Poage, G.M.; den Hollander, P.; Tsimelzon, A.; Hill, J.; Panupinthu, N.; Zhang, Y.; Mazumdar, A.; Hilsenbeck, S.G.; Mills, G.B.; et al. Growth of triple-negative breast cancer cells relies upon coordinate autocrine expression of the proinflammatory cytokines IL-6 and IL-8. Cancer Res. 2013, 73, 3470–3480. [Google Scholar] [CrossRef] [Green Version]
- Chavey, C.; Bibeau, F.; Gourgou-Bourgade, S.; Burlinchon, S.; Boissière, F.; Laune, D.; Roques, S.; Lazennec, G. Oestrogen receptor negative breast cancers exhibit high cytokine content. Breast Cancer Res. 2007, 9, R15. [Google Scholar] [CrossRef] [Green Version]
- Jayatilaka, H.; Tyle, P.; Chen, J.J.; Kwak, M.; Ju, J.; Kim, H.J.; Lee, J.S.H.; Wu, P.H.; Gilkes, D.M.; Fan, R.; et al. Synergistic IL-6 and IL-8 paracrine signalling pathway infers a strategy to inhibit tumour cell migration. Nat. Commun. 2017, 8, 15584. [Google Scholar] [CrossRef] [Green Version]
- Salgado, R.; Junius, S.; Benoy, I.; Van Dam, P.; Vermeulen, P.; Van Marck, E.; Huget, P.; Dirix, L.Y. Circulating interleukin-6 predicts survival in patients with metastatic breast cancer. Int. J. Cancer 2003, 103, 642–646. [Google Scholar] [CrossRef]
- Bachelot, T.; Ray-Coquard, I.; Menetrier-Caux, C.; Rastkha, M.; Duc, A.; Blay, J.Y. Prognostic value of serum levels of interleukin 6 and of serum and plasma levels of vascular endothelial growth factor in hormone-refractory metastatic breast cancer patients. Br. J. Cancer 2003, 88, 1721–1726. [Google Scholar] [CrossRef] [Green Version]
- Benoy, I.H.; Salgado, R.; Van Dam, P.; Geboers, K.; Van Marck, E.; Scharpé, S.; Vermeulen, P.B.; Dirix, L.Y. Increased serum interleukin-8 in patients with early and metastatic breast cancer correlates with early dissemination and survival. Clin. Cancer Res. 2004, 10, 7157–7162. [Google Scholar] [CrossRef] [Green Version]
- Dingemans, A.M.; Groen, H.J.; Herder, G.J.; Stigt, J.A.; Smit, E.F.; Bahce, I.; Burgers, J.A.; van den Borne, B.E.; Biesma, B.; Vincent, A.; et al. A randomized phase II study comparing paclitaxel-carboplatin-bevacizumab with or without nitroglycerin patches in patients with stage IV nonsquamous nonsmall-cell lung cancer: NVALT12 (NCT01171170)†. Ann. Oncol. 2015, 26, 2286–2293. [Google Scholar] [CrossRef]
- Yasuda, H.; Nakayama, K.; Watanabe, M.; Suzuki, S.; Fuji, H.; Okinaga, S.; Kanda, A.; Zayasu, K.; Sasaki, T.; Asada, M.; et al. Nitroglycerin treatment may enhance chemosensitivity to docetaxel and carboplatin in patients with lung adenocarcinoma. Clin. Cancer Res. 2006, 12, 6748–6757. [Google Scholar] [CrossRef] [Green Version]
- Arrieta, O.; Blake, M.; de la Mata-Moya, M.D.; Corona, F.; Turcott, J.; Orta, D.; Alexander-Alatorre, J.; Gallardo-Rincón, D. Phase II study. Concurrent chemotherapy and radiotherapy with nitroglycerin in locally advanced non-small cell lung cancer. Radiother. Oncol. 2014, 111, 311–315. [Google Scholar] [CrossRef]
- Jourd’heuil, D.; Gray, L.; Grisham, M.B. S-nitrosothiol formation in blood of lipopolysaccharide-treated rats. Biochem. Biophys. Res. Commun. 2000, 273, 22–26. [Google Scholar] [CrossRef]
- Bettaieb, A.; Paul, C.; Plenchette, S. Exploration of Fas S-Nitrosylation by the Biotin Switch Assay. Methods Mol. Biol. 2017, 1557, 199–206. [Google Scholar] [CrossRef]
- Burgy, O.; Bellaye, P.S.; Causse, S.; Beltramo, G.; Wettstein, G.; Boutanquoi, P.M.; Goirand, F.; Garrido, C.; Bonniaud, P. Pleural inhibition of the caspase-1/IL-1β pathway diminishes profibrotic lung toxicity of bleomycin. Respir. Res. 2016, 17, 162. [Google Scholar] [CrossRef] [Green Version]
- Bankhead, P.; Loughrey, M.B.; Fernández, J.A.; Dombrowski, Y.; McArt, D.G.; Dunne, P.D.; McQuaid, S.; Gray, R.T.; Murray, L.J.; Coleman, H.G.; et al. QuPath: Open source software for digital pathology image analysis. Sci. Rep. 2017, 7, 16878. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bouaouiche, S.; Ghione, S.; Sghaier, R.; Burgy, O.; Racoeur, C.; Derangère, V.; Bettaieb, A.; Plenchette, S. Nitric Oxide-Releasing Drug Glyceryl Trinitrate Targets JAK2/STAT3 Signaling, Migration and Invasion of Triple-Negative Breast Cancer Cells. Int. J. Mol. Sci. 2021, 22, 8449. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168449
Bouaouiche S, Ghione S, Sghaier R, Burgy O, Racoeur C, Derangère V, Bettaieb A, Plenchette S. Nitric Oxide-Releasing Drug Glyceryl Trinitrate Targets JAK2/STAT3 Signaling, Migration and Invasion of Triple-Negative Breast Cancer Cells. International Journal of Molecular Sciences. 2021; 22(16):8449. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168449
Chicago/Turabian StyleBouaouiche, Sarra, Silvia Ghione, Randa Sghaier, Olivier Burgy, Cindy Racoeur, Valentin Derangère, Ali Bettaieb, and Stéphanie Plenchette. 2021. "Nitric Oxide-Releasing Drug Glyceryl Trinitrate Targets JAK2/STAT3 Signaling, Migration and Invasion of Triple-Negative Breast Cancer Cells" International Journal of Molecular Sciences 22, no. 16: 8449. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168449