
Co-Aggregation of S100A9 with DOPA and Cyclen-Based Compounds Manifested in Amyloid Fibril Thickening without Altering Rates of Self-Assembly

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Results
2.1. Computation Analysis of the Physico-Chemical Properties of Molecular Compounds and Their Prospective Drug Likeness
2.2. Kinetic Analysis of S100A9 Amyloid Formation in the Absence and Presence of DOPA and Cyclen-Based Compounds Monitored by ThT Fluorescence
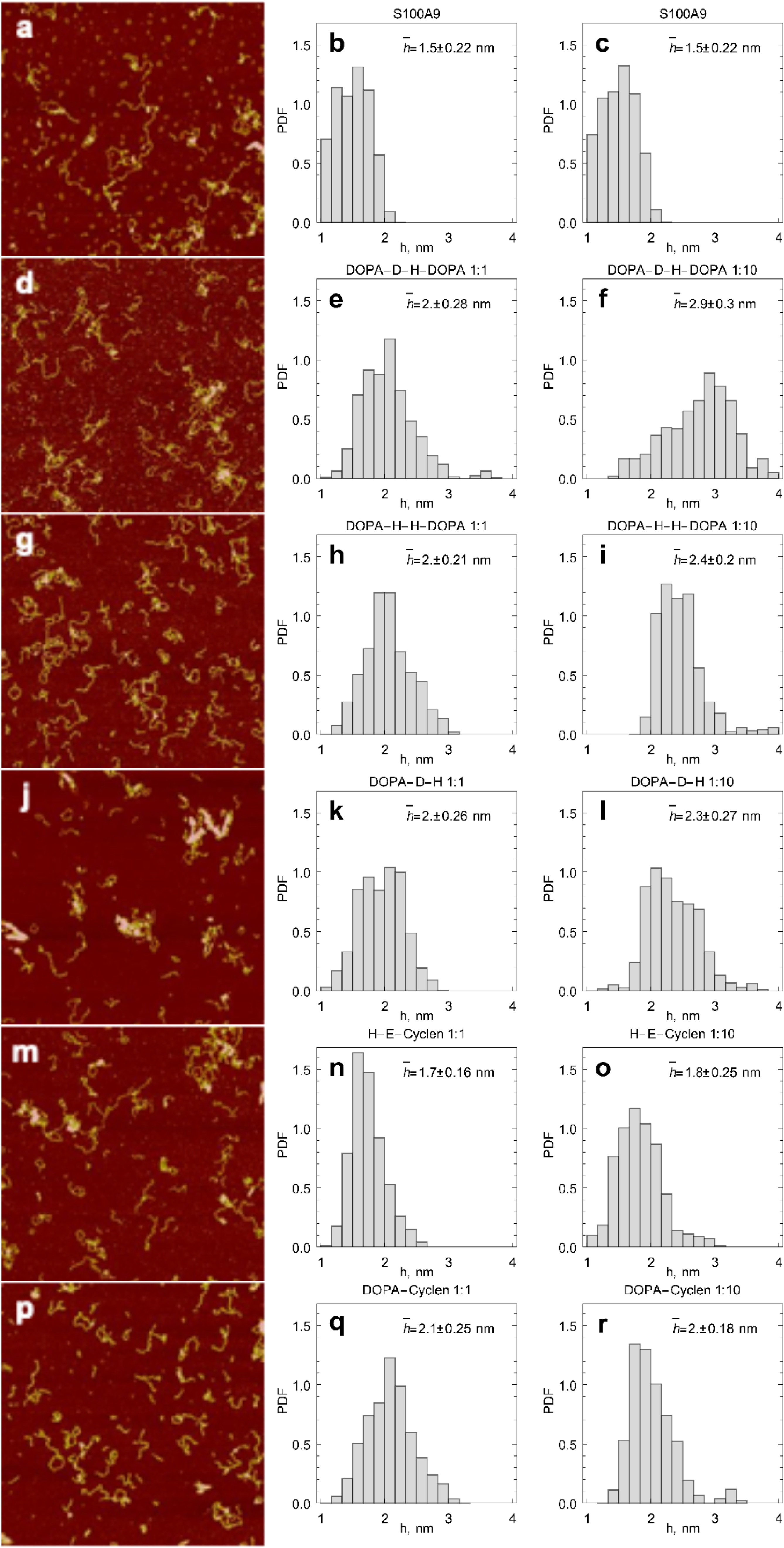
2.3. AFM Imaging of S100A9 Amyloids Aggregated Alone and Together with Corresponding Compounds
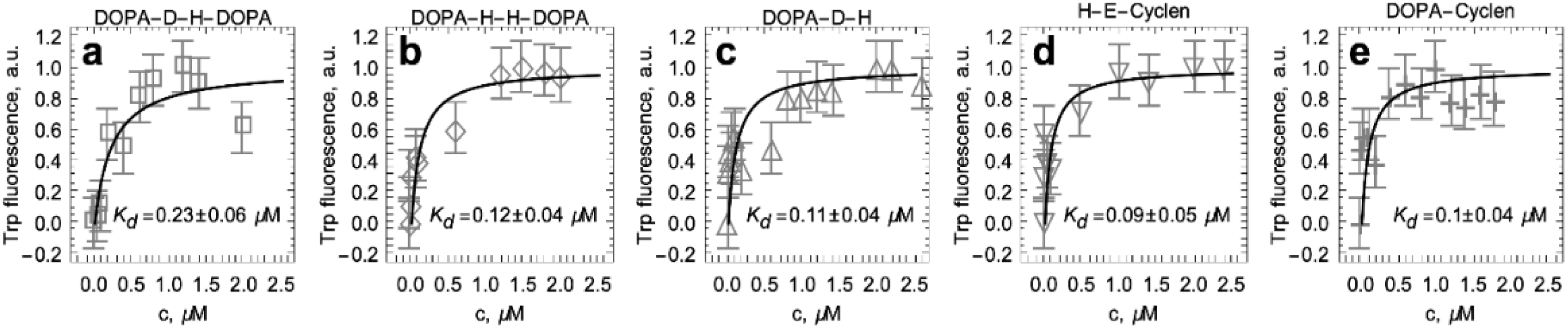
2.4. Titration of S100A9 with DOPA and Cyclen-Based Compounds Followed by Intrinsic Fluorescence
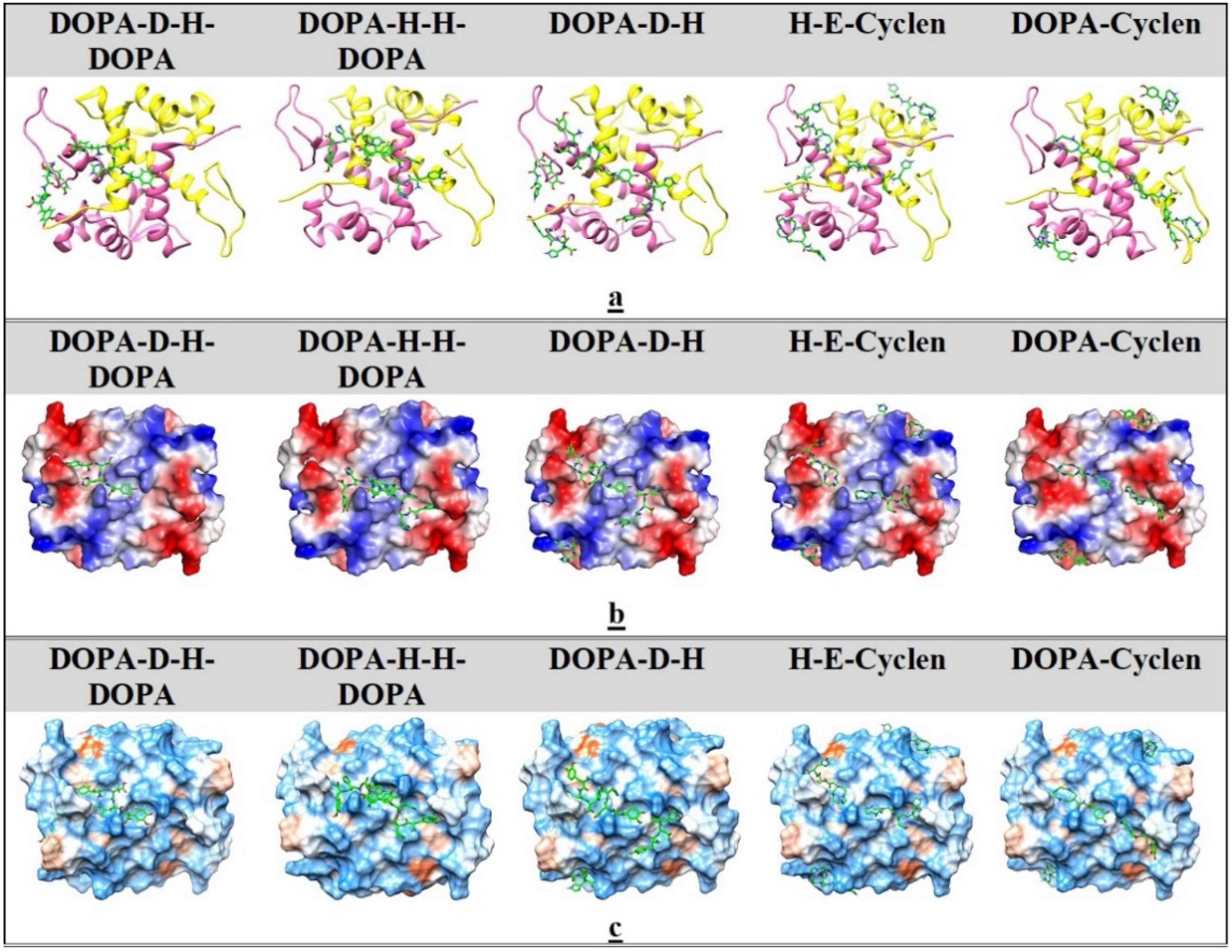
2.5. Ligand Docking
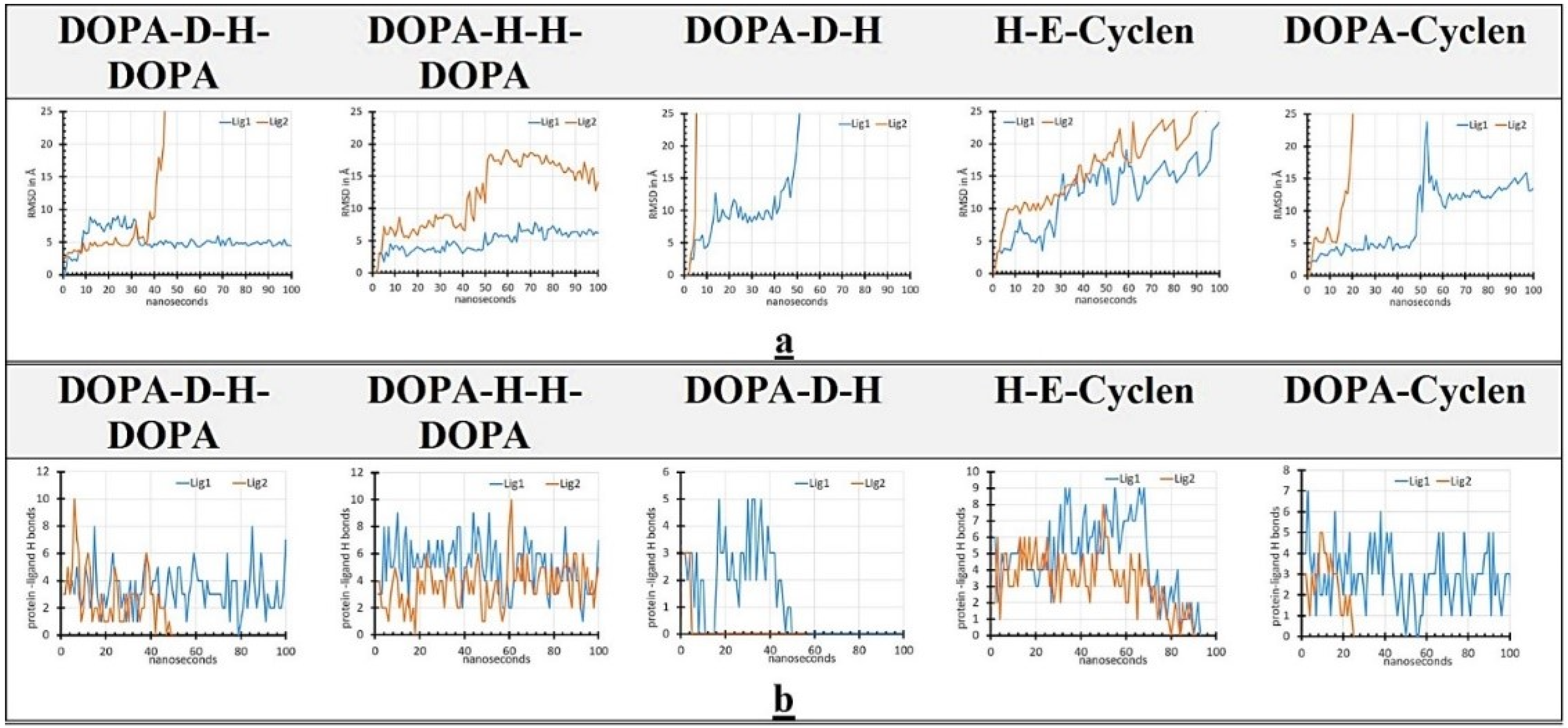
2.6. MD Simulation
3. Discussion
4. Materials and Methods
4.1. Amyloid Fibril Formation
4.2. ThT Fluorescence Assay
4.3. Kinetic Curve Fitting
4.4. AFM Imaging
4.5. Titration of S100A9 with DOPA and Cyclen-Based Compounds Monitored by Intrinsic Fluorescence
4.6. Ligand Docking Studies
4.7. All-Atom MD Studies
4.8. DOPA and Cyclen Ligand Synthesis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AFM | Atomic force microscopy |
| Aβ | Amyloid β |
| Cyclen | 1,4,7,10-tetraazacyclododecane |
| DOPA | L-3,4-dihydroxyphenylalanine |
| MD | Molecular dynamic |
| ThT | Thioflavin T |
| RMSD | Root mean square deviation |
References
- Fitzpatrick, A.W.P.; Debelouchina, G.T.; Bayro, M.J.; Clare, D.K.; Caporini, M.A.; Bajaj, V.S.; Jaroniec, C.P.; Wang, L.; Ladizhansky, V.; Müller, S.A.; et al. Atomic structure and hierarchical assembly of a cross-β amyloid fibril. Proc. Natl. Acad. Sci. USA 2013, 110, 5468–5473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaham-Niv, S.; Adler-Abramovich, L.; Schnaider, L.; Gazit, E. Extension of the generic amyloid hypothesis to nonproteinaceous metabolite assemblies. Sci. Adv. 2015, 1, e1500137. [Google Scholar] [CrossRef] [Green Version]
- Sunde, M.; Serpell, L.C.; Bartlam, M.; Fraser, P.E.; Pepys, M.B.; Blake, C.C.F. Common core structure of amyloid fibrils by synchrotron X-ray diffraction. J. Mol. Biol. 1997, 273, 729–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benson, M.D.; Buxbaum, J.N.; Eisenberg, D.S.; Merlini, G.; Saraiva, M.J.M.; Sekijima, Y.; Sipe, J.D.; Westermark, P. Amyloid nomenclature 2018: Recommendation of the International Society of Amyloidosis (ISA) nomenclature committee. Amyloid 2018, 25, 215–219. [Google Scholar] [CrossRef] [Green Version]
- Hardy, J.; Selkoe, D.J. The Amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gazit, E. A possible role for pi-stacking in the self-assembly of amyloid fibrils. FASEB J. 2002, 16, 77–83. [Google Scholar] [CrossRef]
- Pujols, J.; Peña-Díaz, S.; Pallarès, I.; Ventura, S. Chemical chaperones as novel drugs for Parkinson’s disease. Trends Mol. Med. 2020, 26, 408–421. [Google Scholar] [CrossRef]
- Ferreon, A.C.M.; Moosa, M.M.; Gambin, Y.; Deniz, A.A. Counteracting chemical chaperone effects on the single-molecule α-synuclein structural landscape. Proc. Natl. Acad. Sci. USA 2012, 109, 17826–17831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svenningsson, P.; Odin, P.; Dizdar, N.; Johansson, A.; Grigoriou, S.; Tsitsi, P.; Wictorin, K.; Bergquist, F.; Nyholm, D.; Rinne, J.; et al. A Phase 2a trial investigating the safety and tolerability of the novel cortical enhancer IRL752 in Parkinson’s disease dementia. Mov. Disord. 2020, 35, 1046–1054. [Google Scholar] [CrossRef]
- Sidorova, Y.A.; Volcho, K.P.; Salakhutdinov, N.F. Neuroregeneration in Parkinson’s disease: From proteins to small molecules. Curr. Neuropharmacol. 2018, 17, 268–287. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Klechikov, A.G.; Gharibyan, A.L.; Wärmländer, S.K.T.S.; Jarvet, J.; Zhao, L.; Jia, X.; Narayana, V.K.; Shankar, S.K.; Olofsson, A.; et al. The role of pro-inflammatory S100A9 in Alzheimer’s disease amyloid-neuroinflammatory cascade. Acta Neuropathol. 2014, 127, 507–522. [Google Scholar] [CrossRef] [Green Version]
- Horvath, I.; Iashchishyn, I.A.; Wang, C.; Moskalenko, R.A.; Wärmländer, S.K.T.S.; Wallin, C.; Gräslund, A.; Kovacs, G.G.; Morozova-Roche, L.A. Co-aggregation of pro-inflammatory S100A9 with α-synuclein in Parkinson’s disease: Ex vivo and in vitro studies. J. Neuroimmun. 2018, 15, 172. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Iashchishyn, I.A.; Nyström, S.; Klementieva, O.; Kara, J.; Horvath, I.; Moskalenko, R.A.; Rofougaran, R.; Gouras, G.; Kovacs, G.G.; et al. S100A9-driven amyloid-neuroinflammatory cascade in traumatic brain injury and Alzheimer’s disease. Sci. Rep. 2018, 8, 12836. [Google Scholar] [CrossRef]
- Vogl, T.; Gharibyan, A.L.; Morozova-Roche, L.A. Pro-inflammatory S100A8 and S100A9 proteins: Self-assembly into multifunctional native and amyloid complexes. Int. J. Mol. Sci. 2012, 13, 2893–2917. [Google Scholar] [CrossRef] [Green Version]
- Iashchishyn, I.A.; Sulskis, D.; Nguyen Ngoc, M.; Smirnovas, V.; Morozova-Roche, L.A. Finke-Watzky two-step nucleation-autocatalysis model of S100A9 amyloid formation: Protein misfolding as “nucleation” event. ACS Chem. Neurosci. 2017, 8, 2152–2158. [Google Scholar] [CrossRef]
- Iashchishyn, I.A.; Gruden, M.; Moskalenko, R.A.; Davydova, T.V.; Wang, C.; Sewell, R.D.E.; Morozova-Roche, L.A. Intranasally administered S100A9 amyloids induced cellular stress, amyloid seeding and behavioral impairment in aged mice. ACS Chem. Neurosci. 2018, 9, 1338–1348. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Iashchishyn, I.A.; Kara, J.; Foderà, V.; Vetri, V.; Sancataldo, G.; Marklund, M.; Morozova-Roche, L.A. Proinflammatory and amyloidogenic S100A9 induced by traumatic brain injury in mouse model. Neurosci. Lett. 2019, 699, 199–205. [Google Scholar] [CrossRef]
- Pansieri, J.; Iashchishyn, I.A.; Fakhouri, H.; Ostojić, L.; Malisauskas, M.; Musteikyte, G.; Smirnovas, V.; Schneider, M.M.; Scheidt, T.; Xu, C.K.; et al. Templating S100A9 amyloids on A-beta fibrillar surfaces revealed by charge detection mass spectrometry, microscopy, kinetic and microfluidic analyses. Chem. Sci. 2020, 11, 7031–7039. [Google Scholar] [CrossRef]
- Otzen, D.; Riek, R. Functional amyloids. Cold Spring Harb. Perspect. Biol. 2019, 11, a033860. [Google Scholar] [CrossRef] [PubMed]
- Ke, P.C.; Zhou, R.; Serpell, L.C.; Riek, R.; Knowles, T.P.J.; Lashuel, H.A.; Gazit, E.; Hamley, I.W.; Davis, T.P.; Fändrich, M.; et al. Half a century of amyloids: Past, present and future. Chem. Soc. Rev. 2020, 49, 5473–5509. [Google Scholar] [CrossRef] [PubMed]
- Maji, S.K.; Perrin, M.H.; Sawaya, M.R.; Jessberger, S.; Vadodaria, K.; Rissman, R.A.; Singru, P.S.; Nilsson, K.P.R.; Simon, R.; Schubert, D.; et al. Functional amyloids as natural storage of peptide hormones in pituitary secretory granules. Science 2009, 325, 328–332. [Google Scholar] [CrossRef] [Green Version]
- Wickner, R.B.; Edskes, H.K.; Shewmaker, F.P.; Kryndushkin, D.; Nemecek, J.; McGlinchey, R.; Bateman, D. The relationship of prions and translation. Wiley Interdiscip. Rev. RNA 2010, 1, 81–89. [Google Scholar] [CrossRef] [Green Version]
- Javed, I.; Zhang, Z.; Adamcik, J.; Andrikopoulos, N.; Li, Y.; Otzen, D.E.; Lin, S.; Mezzenga, R.; Davis, T.P.; Ding, F.; et al. Accelerated amyloid beta pathogenesis by bacterial amyloid FapC. Adv. Sci. 2020, 7, 2001299. [Google Scholar] [CrossRef] [PubMed]
- Christensen, L.F.B.; Alijanvand, S.H.; Burdukiewicz, M.; Herbst, F.-A.; Kjeldal, H.; Dueholm, M.S.; Otzen, D.E. Identification of amyloidogenic proteins in the microbiomes of a rat Parkinson’s disease model and wild-type rats. Protein Sci. 2021, 6, 417. [Google Scholar] [CrossRef]
- Rodgers, K.J. Non-protein amino acids and neurodegeneration: The enemy within. Exp. Neurol. 2014, 253, 192–196. [Google Scholar] [CrossRef]
- Hornykiewicz, O. Dopamine (3-hydroxytyramine) and brain function. Pharmacol. Rev. 1966, 18, 925–964. [Google Scholar] [PubMed]
- Drozak, J.; Bryła, J. Dopamine: Not just a neurotransmitter. Postepy Hig. Med. Dosw. 2005, 59, 405–420. [Google Scholar]
- Conway, K.A.; Rochet, J.C.; Bieganski, R.M.; Lansbury, J. Kinetic stabilization of the alpha-synuclein protofibril by a dopamine-alpha-synuclein adduct. Science 2001, 294, 1346–1349. [Google Scholar] [CrossRef]
- Li, J.; Zhu, M.; Manning-Bog, A.B.; Di Monte, D.A.; Fink, A.L. Factors affecting the fibrillation of α-synuclein, a natively unfolded protein. FASEB J. 2004, 18, 962–964. [Google Scholar] [CrossRef]
- Bisaglia, M.; Tosatto, L.; Munari, F.; Tessari, I.; de Laureto, P.P.; Mammi, S.; Bubacco, L. Dopamine quinones interact with alpha-synuclein to form unstructured adducts. Biochem. Biophys. Res. Commun. 2010, 394, 424–428. [Google Scholar] [CrossRef]
- Herrera, F.E.; Chesi, A.; Paleologou, K.E.; Schmid, A.; Munoz, A.; Vendruscolo, M.; Gustincich, S.; Lashuel, H.A.; Carloni, P. Inhibition of alpha-synuclein fibrillization by dopamine is mediated by interactions with five C-terminal residues and with E83 in the NAC region. PLoS ONE 2008, 3, e3394. [Google Scholar] [CrossRef]
- Yedlapudi, D.; Joshi, G.S.; Luo, D.; Todi, S.V.; Dutta, A.K. Inhibition of alpha-synuclein aggregation by multifunctional dopamine agonists assessed by a novel in vitro assay and an in vivo Drosophila synucleinopathy model. Sci. Rep. 2016, 6, 38510. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.; Huber, A.; Rand, D.; Gosselet, F.; Cooper, I.; Gazit, E.; Segal, D. Naphthoquinone-dopamine hybrids inhibit α-synuclein aggregation, disrupt preformed fibrils, and attenuate aggregate-induced toxicity. Chem. Eur. J. 2020, 26, 16486–16496. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.; Viswanathan, G.K.; Huber, A.; Arad, E.; Engel, H.; Jelinek, R.; Gazit, E.; Segal, D. Inhibition of tau amyloid formation and disruption of its preformed fibrils by Naphthoquinone–Dopamine hybrid. FEBS J. 2021, 1, 21. [Google Scholar] [CrossRef]
- Ramesh, M.; Makam, P.; Voshavar, C.; Khare, H.; Rajasekhar, K.; Ramakumar, S.; Govindaraju, T. L-Dopa and dopamine conjugated naphtalenediimides modulate amyloid β toxicity. Org. Biomol. Chem. 2018, 16, 7682–7692. [Google Scholar] [CrossRef]
- Saha, A.; Bolisetty, S.; Handschin, S.; Mezzenga, R. Self-assembly and fibrillization of a Fmoc-functionalized polyphenolic amino acid. Soft Matter 2013, 9, 10239–10242. [Google Scholar] [CrossRef]
- Ceylan, H.; Urel, M.; Erkal, T.S.; Tekinay, A.B.; Dana, A.; Guler, M.O. Mussel inspired dynamic cross-linking of self-healing peptide nanofiber network. Adv. Funct. Mater. 2013, 23, 2081–2090. [Google Scholar] [CrossRef]
- Fichman, G.; Adler-Abramovich, L.; Manohar, S.; Mironi-Harpaz, I.; Guterman, T.; Seliktar, D.; Messersmith, P.B.; Gazit, E. Seamless metallic coating and surface adhesion of self-assembled bio-inspired nanostructures based on di-(3,4-dihydroxy-L-phenylalanine) peptide motif. ACS Nano 2014, 8, 7220–7228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fichman, G.; Guterman, T.; Adler-Abramovich, L.; Gazit, E. The Use of the calcitonin minimal recognition module for the design of DOPA-containing fibrillar assemblies. Nanomaterials 2014, 4, 726–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mewis, R.E.; Archibald, S.J. Biomedical applications of macrocyclic ligand complexes. Coord. Chem. Rev. 2010, 254, 1686–1712. [Google Scholar] [CrossRef]
- Lejault, P.; Duskova, K.; Bernhard, C.; Valverde, I.E.; Romieu, A.; Monchaud, D. The scope of application of macrocyclic polyamines beyond metal chelation. Eur. JOC 2019, 36, 6146–6157. [Google Scholar] [CrossRef]
- Watanabe, H.; Kawasaki, A.; Sano, K.; Ono, M.; Saji, H. Synthesis and evaluation of copper-64 labeled benzofuran derivatives targeting β-amyloid aggregates. Bioorg. Med. Chem. 2016, 24, 3618–3623. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Wang, X.; He, Y.; Zhang, C.; Wu, Z.; Liao, K.; Wang, J.; Guo, Z. Effects of cyclen and cyclam on Zn(II)- and Cu(II)-induced amyloid β-peptide aggregation and neurotoxicity. Inorg. Chem. 2009, 48, 5801–5809. [Google Scholar] [CrossRef]
- Jeong, K.; Chung, W.Y.; Kye, Y.S.; Kim, D. Cu (II) cyclen cleavage agent for human islet amyloid peptide. Bioorg. Med. Chem. 2010, 18, 2598–2601. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Yu, Y.P.; Cui, W.; Fang, C.L.; Wu, W.H.; Zhao, Y.F.; Li, Y.M. Cyclen-hybrid compounds captures copper to protect INS-1 cells from islet amyloid polypeptide cytotoxicity by inhibiting and lysing effects. Chem. Commun. 2010, 46, 8023–8025. [Google Scholar] [CrossRef]
- Wu, W.H.; Lei, P.; Liu, Q.; Hu, J.; Gunn, A.P.; Chen, M.S.; Rui, Y.F.; Su, X.Y.; Xie, Z.P.; Zhao, Y.F.; et al. Sequestration of copper from -amyloid promotes selective lysis by cyclen-hybrid cleavage agents. J. Biol. Chem. 2008, 283, 31657–31664. [Google Scholar] [CrossRef] [Green Version]
- Chem Axon. Marvin Was Used for Drawing, Displaying and Characterizing Chemical Structures, Substructures and Reactions, Marvin 19.20.0, ChemAxon. Available online: https://www.chemaxon.com (accessed on 27 June 2021).
- Patrick, G.L. An Introduction to Medicinal Chemistry, 4th ed.; OUP Oxford: Oxford, UK, 2009; p. 776. [Google Scholar]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Flory, P.J. Molecular size distribution in linear condensation polymers. Am. Chem. Soc. 1936, 58, 1877–1885. [Google Scholar] [CrossRef]
- Blatz, P.J.; Tobolsky, A.V. Note on the kinetics of systems manifesting simultaneous polymerization-depolymerization phenomena. J. Phys. Chem. 1945, 49, 77–80. [Google Scholar] [CrossRef]
- Leri, M.; Chaudhary, H.; Iashchishyn, I.A.; Pansieri, J.; Svedružić, Z.M.; Alcalde, S.G.; Musteikyte, G.; Smirnovas, V.; Stefani, M.; Bucciantini, M.; et al. Natural compound from olive oil inhibits S100A9 amyloid formation and cytotoxicity: Implications for preventing Alzheimer’s disease. ACS Chem. NeuroSci. 2021, 12, 1905–1918. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, H.; Iashchishyn, I.A.; Romanova, N.V.; Rambaran, M.A.; Musteikyte, G.; Smirnovas, V.; Holmboe, M.; Ohlin, C.A.; Svedružić, Z.M.; Morozova-Roche, L.A. Polyoxometalates as effective nano-inhibitors of amyloid aggregation of pro-inflammatory S100A9 protein involved in neurodegenerative diseases. ACS Appl. Mater. Interfaces 2021, 13, 26721–26734. [Google Scholar] [CrossRef]
- Pansieri, K.; Ostojić, L.; Iashchishyn, I.A.; Magzoub, M.; Wallin, C.; Wärmländer, S.K.T.S.; Gräslund, A.; Ngoc, M.N.; Smirnovas, V.; Svedružić, Z.; et al. Pro-inflammatory S100A9 protein aggregation promoted by NCAM1 peptide constructs. ACS Chem. Biol. 2019, 14, 1410–1417. [Google Scholar] [CrossRef]
- Chang, C.C.; Khan, I.; Tsai, K.L.; Li, H.; Yang, L.W.; Chou, R.H.; Yu, C. Blocking the interaction between S100A9 and RAGE V domain using CHAPS molecule: A novel route to drug development against cell proliferation. Biochim. Biophys. Acta 2016, 1864, 1558–1569. [Google Scholar] [CrossRef] [PubMed]
- Svedružić, Ž.M.; Vrbnjak, K.; Martinović, M.; Miletić, V. Structural analysis of the simultaneous activation and inhibition of γ-secretase activity in the development of drugs for Alzheimer’s disease. Pharmaceutics 2021, 13, 514. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis version. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar] [CrossRef] [Green Version]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Fändrich, M.; Nyström, S.; Nilsson, K.P.R.; Böckmann, A.; Le Vine, H., 3rd; Hammarström, P. Amyloid fibril polymorphism: A challenge for molecular imaging and therapy. J. Intern. Med. 2018, 283, 218–237. [Google Scholar] [CrossRef]
- Tycko, R. Amyloid polymorphism: Structural basis and neurobiological relevance. Neuron 2015, 86, 632–645. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, J.S.; Otzen, D.E. Amyloid-a state in many guises: Survival of the fittest fibril fold. Protein Sci. 2008, 17, 2–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scialò, C.; De Cecco, E.; Manganotti, P.; Legname, G. Prion and prion-like protein strains: Deciphering the molecular basis of heterogeneity in neurodegeneration. Viruses 2019, 11, 261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morales, R. Prion strains in mammals: Different conformations leading to disease. PLoS Pathog. 2017, 13, e1006323. [Google Scholar] [CrossRef] [Green Version]
- Vogl, T.; Leukert, N.; Barczyk, K.; Strupat, K.; Roth, J. Biophysical characterization of S100A8 and S100A9 in the absence and presence of bivalent cations. Biochim. Biophys. Acta 2006, 1763, 1298–1306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horvath, I.; Jia, X.; Johansson, P.; Wang, C.; Moskalenko, R.; Steinau, A.; Forsgren, L.; Wågberg, T.; Svensson, J.; Zetterberg, H.; et al. Pro-inflammatory S100A9 protein as a robust biomarker differentiating early stages of cognitive impairment in Alzheimer’s disease. ACS Chem. NeuroSci. 2016, 7, 34–39. [Google Scholar] [CrossRef]
- Baldassarre, M.; Baronio, C.M.; Morozova-Roche, L.A.; Barth, A. Amyloid β-peptides 1–40 and 1–42 form oligomers with mixed β-sheets. Chem. Sci. 2017, 8, 8247–8254. [Google Scholar] [CrossRef] [Green Version]
- Yanamandra, K.; Alexeyev, O.; Zamotin, V.; Srivastava, V.; Shchukarev, A.; Brorsson, A.-C.; Tartaglia, G.G.; Vogl, T.; Kayed, R.; Wingsle, G.; et al. Amyloid formation by the pro-inflammatory S100A8/A9 proteins in the ageing prostate. PLoS ONE 2009, 4, e5562. [Google Scholar] [CrossRef] [Green Version]
- LeVine, H. Thioflavine T interaction with amyloid β-sheet structures. Amyloid 1995, 2, 1–6. [Google Scholar] [CrossRef]
- Miletić, V.; Odorčić, I.; Nikolić, P.; Svedružić, Ž.M. In silico design of the first DNA-independent mechanism-based inhibitor of mammalian DNA methyltransferase Dnmt1. PLoS ONE 2017, 12, e0174410. [Google Scholar] [CrossRef]
- Kim, S.; Lee, J.; Jo, S.; Brooks, C.L., 3rd; Lee, H.S.; Im, W. CHARMM-GUI ligand reader and modeler for CHARMM force field generation of small molecules. J. Comput. Chem. 2017, 38, 1879–1886. [Google Scholar] [CrossRef]
- Arabuli, L.; Jezek, R.; Macek, T.; Lovecka, P.; Nikoleishvili, E.; Sulashvili, N.; Maisuradze, I. Solid-phase synthesis and in vitro cytotoxicity of new peptide functionalized cyclencarboxymethylen and L-DOPA hybrids. Med. Chem. 2017, 7, 398–405. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein and Compounds | Kinetic Rate Constants and Their Rations | |||
|---|---|---|---|---|
| 1:1 | 1:10 | |||
| k, μM−1s−1 | k/kS100A9 | k, μM−1s−1 | k/kS100A9 | |
| S100A9 | 0.066 | 1 | 0.066 | 1 |
| DOPA-D-H-DOPA | 0.06 | 0.91 | 0.066 | 1 |
| DOPA-H-H-DOPA | 0.077 | 1.17 | 0.134 | 2.03 |
| DOPA-D-H | 0.063 | 0.95 | 0.045 | 0.69 |
| H-E-cyclen | 0.069 | 1.05 | 0.048 | 0.72 |
| DOPA-cyclen | 0.062 | 0.93 | 0.133 | 2.02 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arabuli, L.; Iashchishyn, I.A.; Romanova, N.V.; Musteikyte, G.; Smirnovas, V.; Chaudhary, H.; Svedružić, Ž.M.; Morozova-Roche, L.A. Co-Aggregation of S100A9 with DOPA and Cyclen-Based Compounds Manifested in Amyloid Fibril Thickening without Altering Rates of Self-Assembly. Int. J. Mol. Sci. 2021, 22, 8556. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168556
Arabuli L, Iashchishyn IA, Romanova NV, Musteikyte G, Smirnovas V, Chaudhary H, Svedružić ŽM, Morozova-Roche LA. Co-Aggregation of S100A9 with DOPA and Cyclen-Based Compounds Manifested in Amyloid Fibril Thickening without Altering Rates of Self-Assembly. International Journal of Molecular Sciences. 2021; 22(16):8556. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168556
Chicago/Turabian StyleArabuli, Lili, Igor A. Iashchishyn, Nina V. Romanova, Greta Musteikyte, Vytautas Smirnovas, Himanshu Chaudhary, Željko M. Svedružić, and Ludmilla A. Morozova-Roche. 2021. "Co-Aggregation of S100A9 with DOPA and Cyclen-Based Compounds Manifested in Amyloid Fibril Thickening without Altering Rates of Self-Assembly" International Journal of Molecular Sciences 22, no. 16: 8556. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168556