
Antagonizing S1P3 Receptor with Cell-Penetrating Pepducins in Skeletal Muscle Fibrosis

 ,
,  , ,
, ,  , , ,
, , ,  , and
, and
Abstract
:1. Introduction
2. Results
2.1. Design
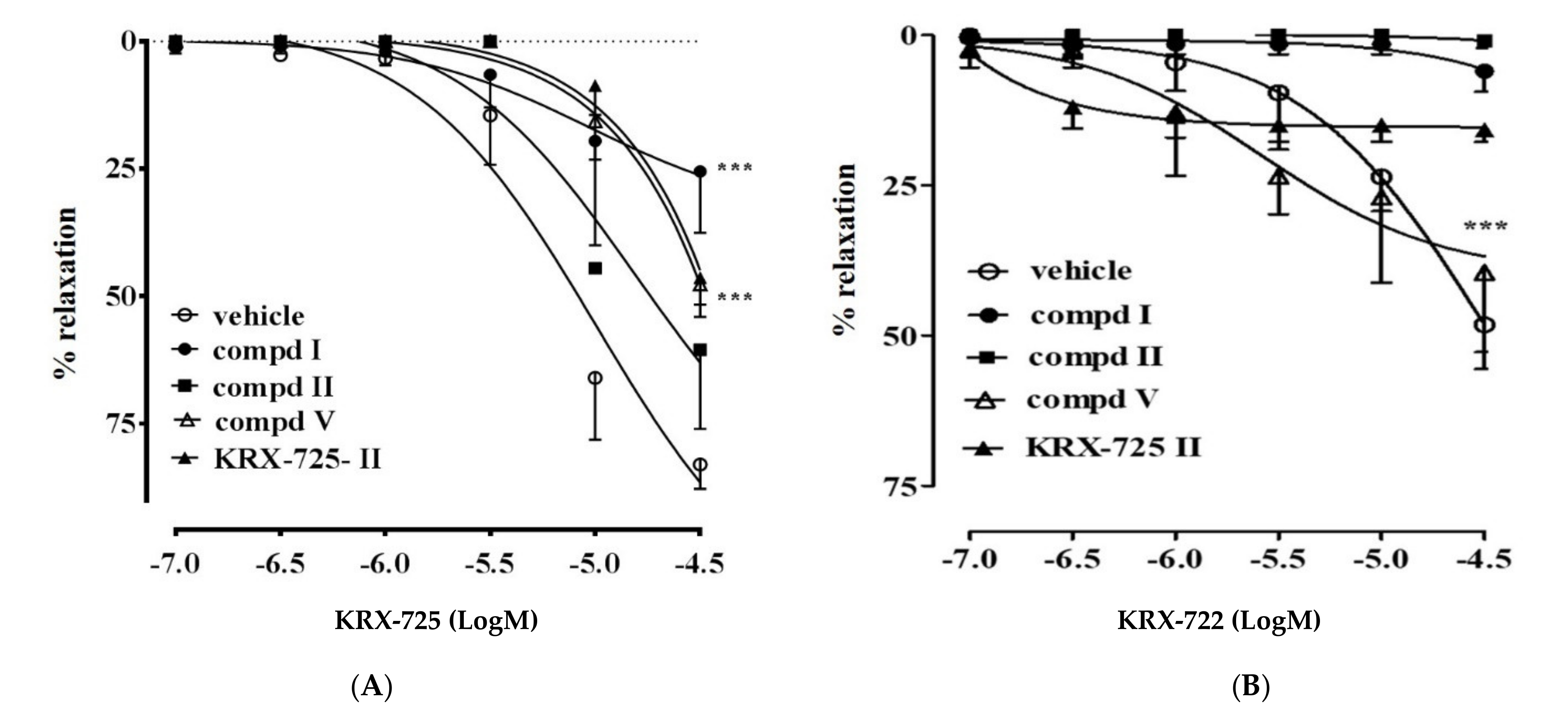
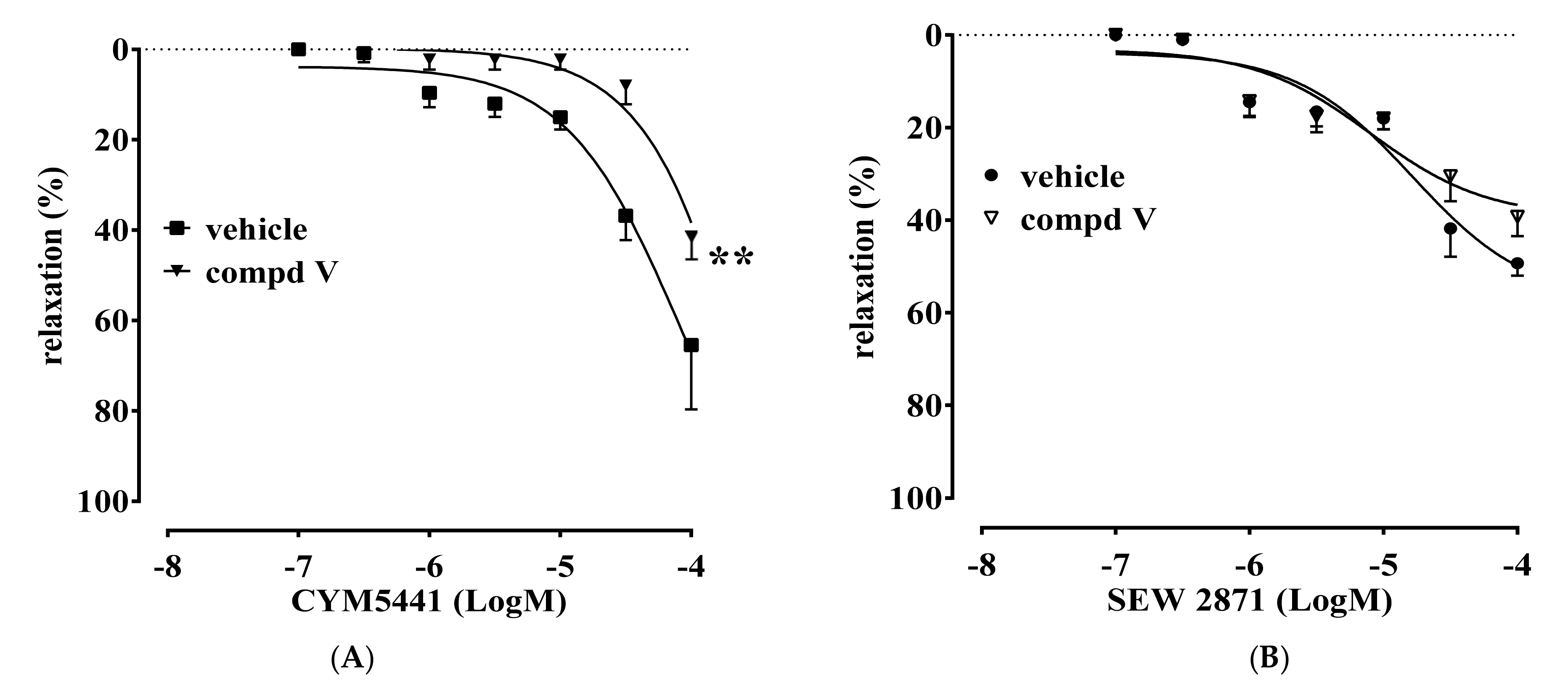
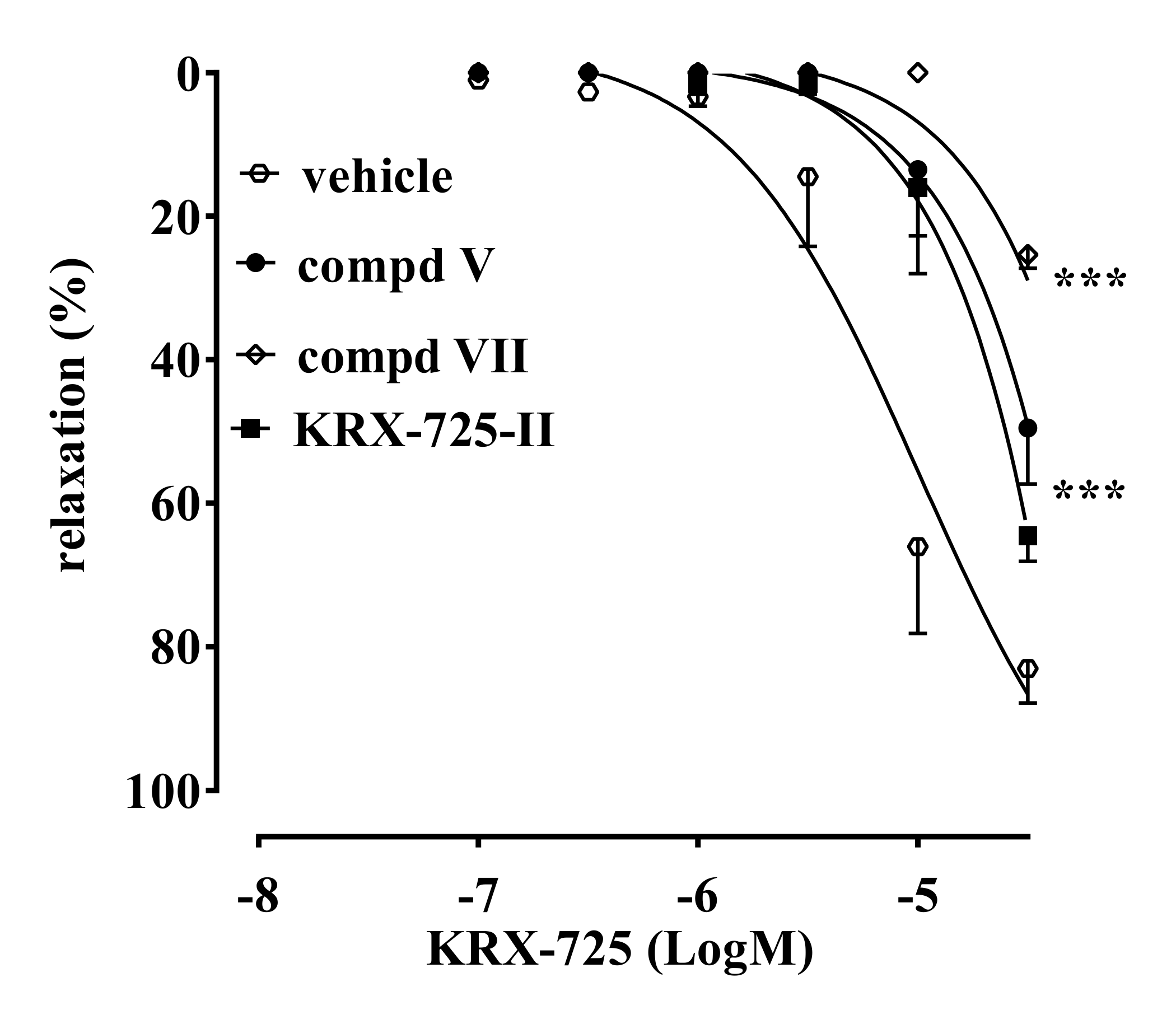
2.2. Vascular Activity
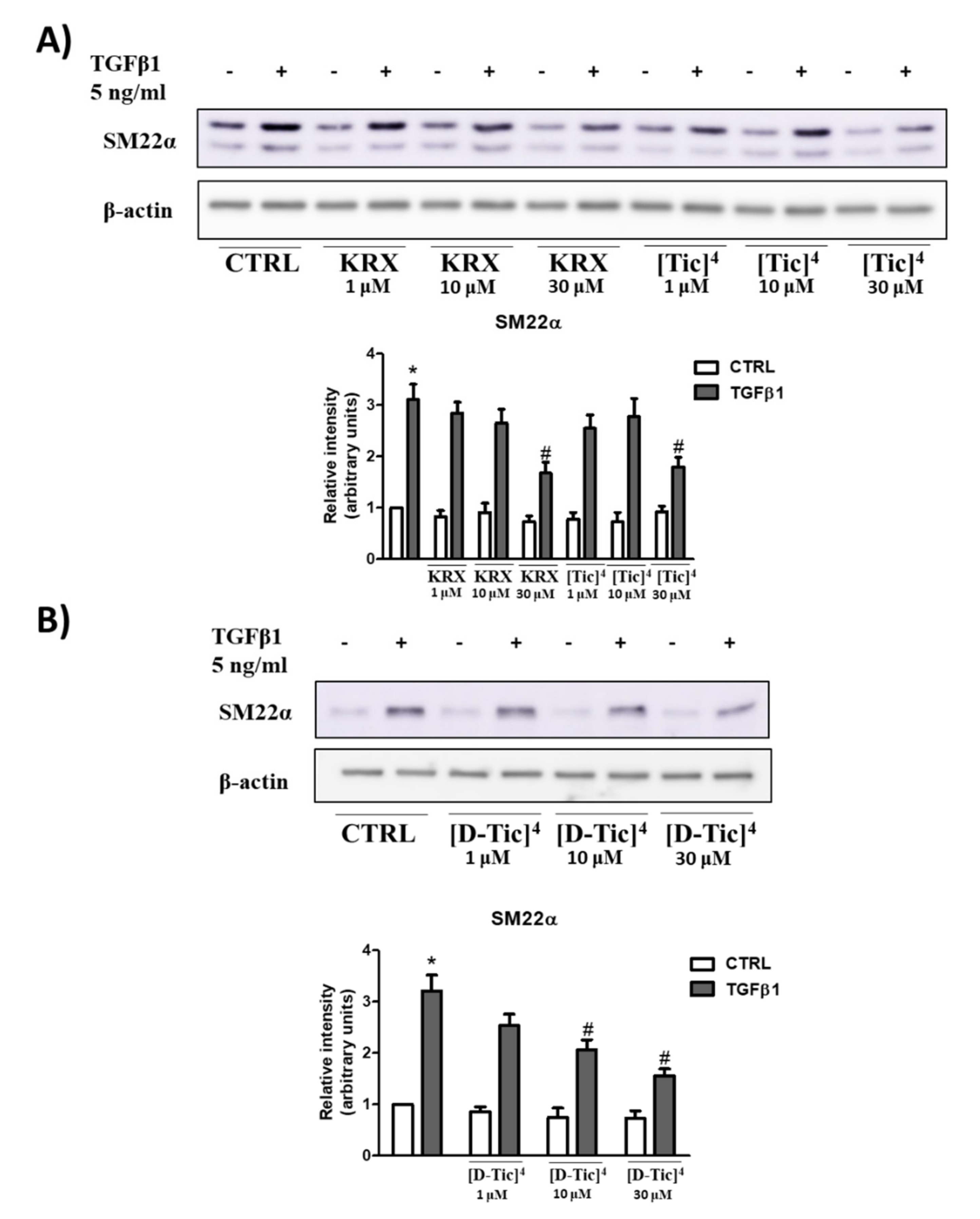
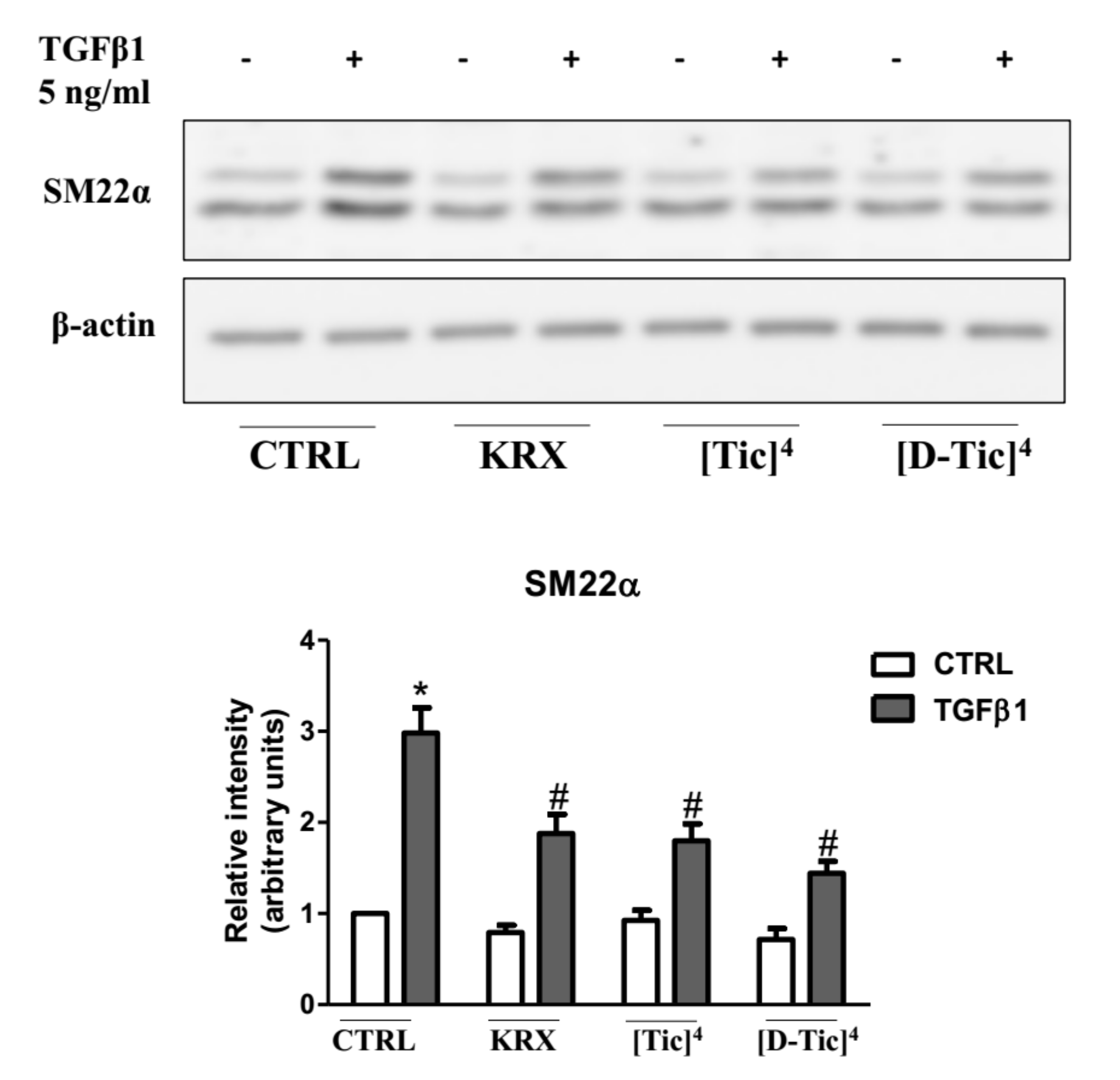
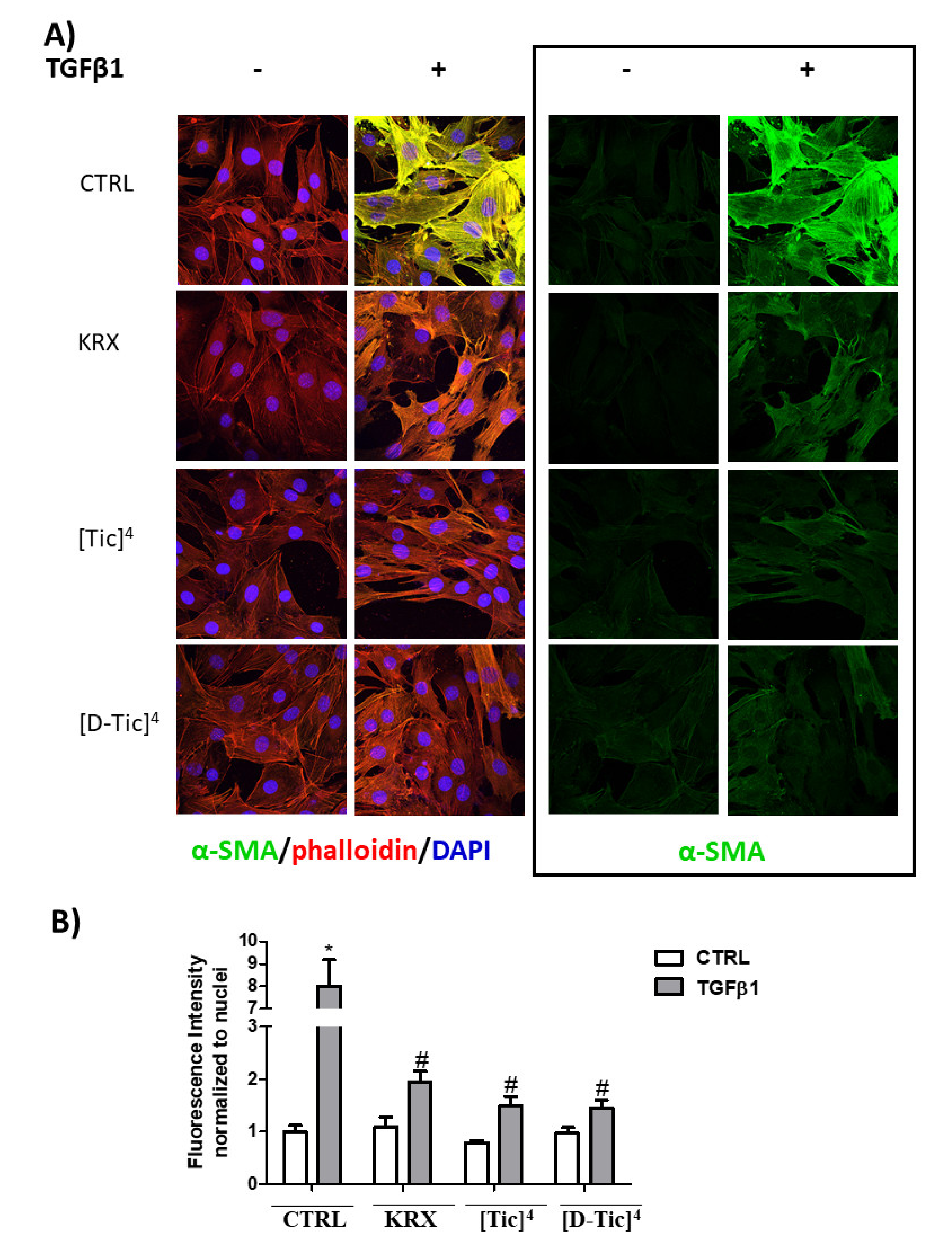
2.3. Anti-Fibrotic Activity
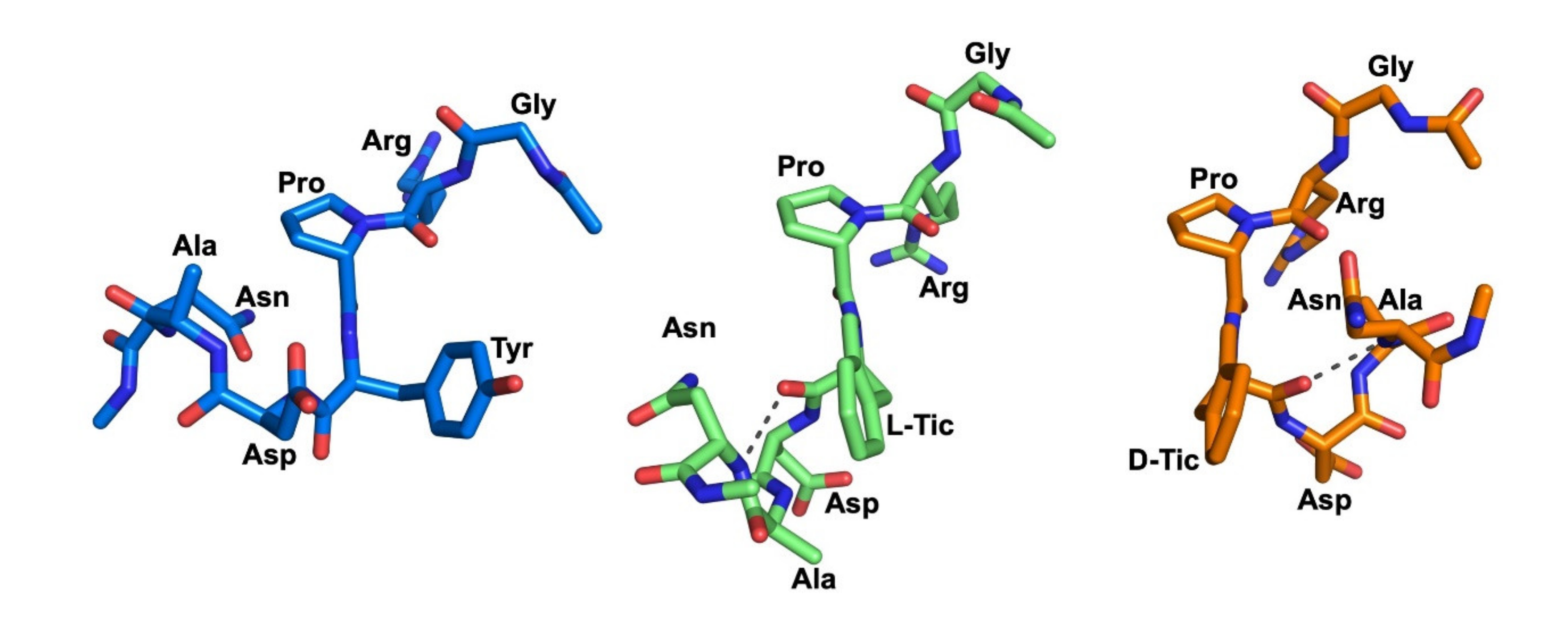
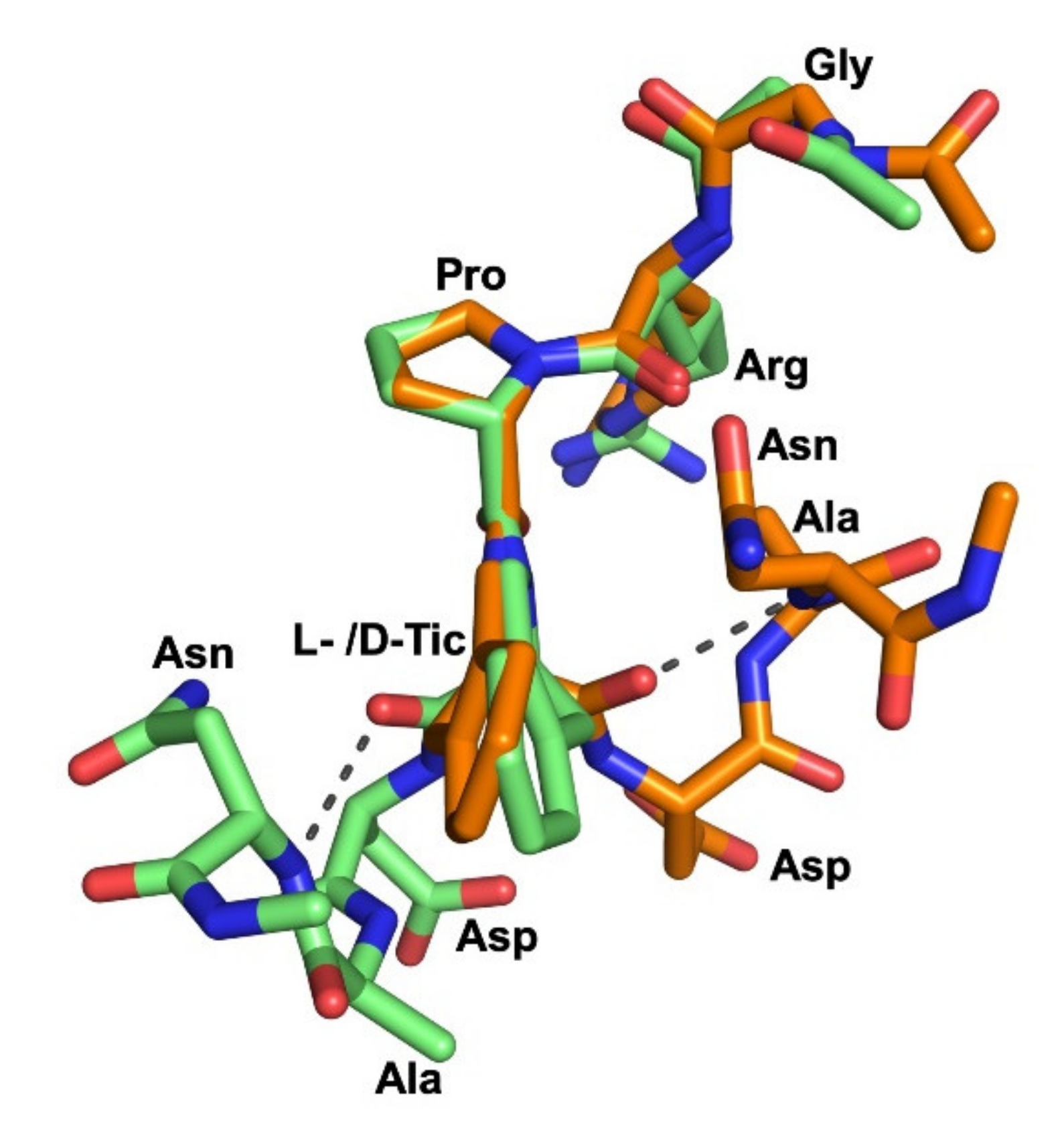
2.4. Molecular Dynamics Simulations
3. Discussion
4. Materials and Methods
4.1. Chemistry
4.1.1. Chemicals
4.1.2. Peptide Synthesis
4.2. Pharmacology
4.2.1. Aorta Rings Assay
4.2.2. Materials
4.2.3. Cell Culture and Agonist Treatment
4.2.4. Western Blot Analysis
4.2.5. Cell Immunofluorescence
4.2.6. Statistical Analysis
4.3. Computational Chemistry
4.3.1. Molecular Dynamics Simulation
4.3.2. Analysis of Molecular Dynamics Trajectory
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kolter, T.; Sandhoff, K. Sphingolipid metabolism diseases. Biochim. Biophys. Acta 2006, 1758, 2057–2079. [Google Scholar] [CrossRef] [Green Version]
- Blaho, V.A.; Hla, T. An update on the biology of sphingosine 1-phosphate receptors. J. Lipid Res. 2014, 55, 1596–1608. [Google Scholar] [CrossRef] [Green Version]
- Aarthi, J.J.; Darendeliler, M.A.; Pushparaj, P.N. Dissecting the role of the S1P/S1PR axis in health and disease. J. Dent. Res. 2011, 90, 841–854. [Google Scholar] [CrossRef]
- Donati, C.; Cencetti, F.; Bernacchioni, C.; Vannuzzi, V.; Bruni, P. Role of sphingosine 1-phosphate signalling in tissue fibrosis. Cell. Signal. 2021, 78, 10986. [Google Scholar] [CrossRef]
- Yang, L.; Chang, N.; Liu, X.; Han, Z.; Zhu, T.; Li, C.; Yang, L.; Li, L. Bone marrow-derived mesenchymal stem cells differentiate to hepatic myofibroblasts by transforming growth factor-β1 via sphingosine kinase/sphingosine 1-phosphate (S1P)/S1P receptor axis. Am. J. Pathol. 2012, 181, 85–97. [Google Scholar] [CrossRef]
- Liu, X.; Hong, Q.; Wang, Z.; Yu, Y.; Zou, X.; Xu, L. Transforming growth factor-β-sphingosine kinase 1/S1P signaling upregulates microRNA-21 to promote fibrosis in renal tubular epithelial cells. Exp. Biol. Med. 2016, 241, 265–272. [Google Scholar] [CrossRef] [Green Version]
- Cencetti, F.; Bernacchioni, C.; Nincheri, P.; Donati, C.; Bruni, P. Transforming growth factor-beta1 induces transdifferentiation of myoblasts into myofibroblasts via up-regulation of sphingosine kinase-1/S1P3 axis. Mol. Biol. Cell 2010, 21, 1111–1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bondì, M.; Germinario, E.; Pirazzini, M.; Zanetti, G.; Cencetti, F.; Donati, C.; Gorza, L.; Betto, R.; Bruni, P.; Danieli-Betto, D. Ablation of S1P3 receptor protects mouse soleus from age-related drop in muscle mass, force, and regenerative capacity. Am. J. Physiol. Cell Physiol. 2017, 313, C54–C67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Callaghan, K.; Kuliopulos, A.; Covic, L. Turning receptors on and off with intracellular pepducins: New insights into G-protein-coupled receptor drug development. J. Biol. Chem. 2012, 287, 12787–12796. [Google Scholar] [CrossRef] [Green Version]
- Severino, B.; Incisivo, G.M.; Fiorino, F.; Bertolino, A.; Frecentese, F.; Barbato, F.; Manganelli, S.; Maggioni, G.; Capasso, D.; Caliendo, G.; et al. Identification of a pepducin acting as S1P3 receptor antagonist. J. Pept. Sci. 2013, 19, 717–724. [Google Scholar] [CrossRef]
- Licht, T.; Tsirulnikov, L.; Reuveni, H.; Yarnitzky, T.; Ben-Sasson, S.A. Induction of pro-angiogenic signaling by a synthetic peptide derived from the second intracellular loop of S1P3 (EDG3). Blood 2003, 102, 2099–2107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerrero, M.; Poddutoori, R.; Urbano, M.; Peng, X.; Spicer, T.P.; Chase, P.S. Discovery, design and synthesis of a selective S1P3 receptor allosteric agonist. Bioorg. Med. Chem. Lett. 2013, 23, 6346–6349. [Google Scholar] [CrossRef] [Green Version]
- Gibson, S.E.; Guillo, N.; Tozer, M.J. Towards control of χ-space: Conformationally constrained analogs of Phe, Tyr, Trp and His. Tetrahedron 1999, 55, 585–615. [Google Scholar] [CrossRef]
- Langel, Ü. Handbook of Cell-Penetrating Peptides, 2nd ed.; CRC Press, Taylor & Francis Group: Boca Raton, FL, USA, 2007. [Google Scholar]
- Zhang, P.; Covic, L.; Kuliopulos, A. Pepducins and Other Lipidated Peptides as Mechanistic Probes and Therapeutics. In Cell-Penetrating Peptides; Methods in Molecular Biology; Langel, Ü., Ed.; Springer: New York, NY, USA, 2015; Volume 1324, pp. 191–203. ISBN 978-1-4939-2805-7. [Google Scholar]
- Ghosh, A. Hydrogen Bond Analysis of the EGFR-ErbB3 Heterodimer Related to Non-Small Cell Lung Cancer and Drug Resistance. J. Theor. Biol. 2019, 464, 63–71. [Google Scholar] [CrossRef]
- Lewis, P.N.; Momany, F.A.; Scheraga, H.A. Chain Reversals in Proteins. Biochim. Biophys. Acta 1973, 303, 211–229. [Google Scholar] [CrossRef]
- Deng, Q.; Clemas, J.A.; Chrebet, G.; Fischer, P.; Hale, J.J.; Li, Z.; Mills, S.G.; Bergstrom, J.; Mandala, S.; Mosley, R.; et al. Identification of Leu276 of the S1P1 receptor and Phe263 of the S1P3 receptor in interaction with receptor specific agonists by molecular modeling, site-directed mutagenesis, and affinity studies. Mol. Pharmacol. 2007, 71, 724–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wootten, D.; Christopoulos, A.; Sexton, P.M. Emerging paradigms in GPCR allostery: Implications for drug discovery. Nat. Rev. Drug Discov. 2013, 12, 630–644. [Google Scholar] [CrossRef] [PubMed]
- Marsters, J.C.; McDowell, R.S.; Reynolds, M.E.; Oare, D.A.; Somers, T.C.; Stanley, M.S.; Rawson, T.E.; Struble, M.E.; Burdick, D.J.; Chan, K.S.; et al. Benzodiazepine Peptidomimetic Inhibitors of Farnesyltransferase. Bioorg. Med. Chem. 1994, 2, 949–957. [Google Scholar] [CrossRef]
- Clerc, F.F.; Guitton, J.D.; Fromage, N.; Lelièvre, Y.; Duchesne, M.; Tocqué, B.; James-Surcouf, E.; Commerçon, A.; Becquart, J. Constrained Analogs of KCVFM with Improved Inhibitory Properties against Farnesyl Transferase. Bioorg. Med. Chem. Lett. 1995, 5, 1779–1784. [Google Scholar] [CrossRef]
- Leftheris, K.; Kline, T.; Vite, G.D.; Cho, Y.H.; Bhide, R.S.; Patel, D.V.; Patel, M.M.; Schmidt, R.J.; Weller, H.N.; Andahazy, M.L.; et al. Development of Highly Potent Inhibitors of Ras Farnesyltransferase Possessing Cellular and in Vivo Activity. J. Med. Chem. 1996, 39, 224–236. [Google Scholar] [CrossRef]
- Lukaszuk, A.; Demaegdt, H.; Van den Eynde, I.; Vanderheyden, P.; Vauquelin, G.; Tourwé, D. Conformational Constraints in Angiotensin IV to Probe the Role of Tyr2, Pro5 and Phe6. J. Pept. Sci. 2011, 17, 545–553. [Google Scholar] [CrossRef]
- Schiller, P.W.; Lemieux, C.; Chung, N.N. Differential stereochemical requirements of mu vs. delta opioid receptors for ligand binding and signal transduction: Development of a class of potent and highly delta-selective peptide antagonists. Proc. Natl. Acad. Sci. USA 1992, 89, 11871–11875. [Google Scholar] [CrossRef] [Green Version]
- Van der Poorten, O.; Knuhtsen, A.; Sejer Pedersen, D.; Ballet, S.; Tourwé, D. Side Chain Cyclized Aromatic Amino Acids: Great Tools as Local Constraints in Peptide and Peptidomimetic Design. J. Med. Chem. 2016, 59, 10865–10890. [Google Scholar] [CrossRef]
- Atherton, E.; Sheppard, R.C. Solid-Phase Peptide Synthesis: A Practical Approach; IRL Press: Oxford, UK, 1989. [Google Scholar]
- Bernacchioni, C.; Ghini, V.; Squecco, R.; Idrizaj, E.; Garella, R.; Puliti, E.; Cencetti, F.; Bruni, P.; Donati, C. Role of Sphingosine 1-Phosphate Signalling Axis in Muscle Atrophy Induced by TNFα in C2C12 Myotubes. Int. J. Mol. Sci. 2021, 22, 1280. [Google Scholar] [CrossRef] [PubMed]
- Bernacchioni, C.; Cencetti, F.; Ouro, A.; Bruno, M.; Gomez-Muñoz, A.; Donati, C.; Bruni, P. Lysophosphatidic Acid Signaling Axis Mediates Ceramide 1-Phosphate-Induced Proliferation of C2C12 Myoblasts. Int. J. Mol. Sci. 2018, 19, 139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruno, G.; Cencetti, F.; Bernacchioni, C.; Donati, C.; Blankenbach, K.V.; Thomas, D.; Heringdorf, D.M.Z.; Bruni, P. Bradykinin mediates myogenic differentiation in murine myoblasts through the involvement of SK1/Spns2/S1P2 axis. Cell. Signal. 2018, 45, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Martyna, G.J.; Klein, M.L.; Tuckerman, M. Nosé–Hoover Chains: The Canonical Ensemble via Continuous Dynamics. J. Chem. Phys. 1992, 97, 2635–2643. [Google Scholar] [CrossRef]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant Pressure Molecular Dynamics Algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Tuckerman, M.; Berne, B.J.; Martyna, G.J. Reversible Multiple Time Scale Molecular Dynamics. J. Chem. Phys. 1992, 97, 1990–2001. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | tR (min) a | MW b | ||
|---|---|---|---|---|
| Calcd | Found | |||
| KRX-725 | Myristoyl-GMRPYDANKR-NH2 | 13.751 | 1416.7 | 1417.1 |
| KRX-725-II | Myristoyl-G-RPYDAN-NH2 | 14.642 | 1001.2 | 1001.2 |
| I | [D-Tyr]4-KRX-725-II | 13.967 | 1001.2 | 1001.2 |
| II | [Trp]4-KRX-725-II | 16.156 | 1024.2 | 1024.4 |
| III | [Phe]4-KRX-725-II | 16.567 | 985.2 | 985.7 |
| IV | [p-F-Phe]4-KRX-725-II | 16.509 | 1003.2 | 1003.6 |
| V | [Tic]4-KRX-725-II | 17.690 | 997.2 | 997.9 |
| VI | [(4-NHSO2CH3)Phe]4-KRX-725-II | 17.562 | 1078.3 | 1078.2 |
| VII | [D-Tic]4-KRX-725-II | 17.705 | 997.2 | 997.8 |
| VIII | [Pip]4-KRX-725-II | 14.886 | 949.15 | 949.8 |
| IX | [Pro]4-KRX-725-II | 15.625 | 935.12 | 935.7 |
| Cluster 1 | Cluster 2 | Cluster 3 | Cluster 4 | Cluster 5 | |
|---|---|---|---|---|---|
| KRX-725-II (18) | 17% | 12% | 10.5% | 9.5% | 9% |
| Compound V (5) | 75% | 14% | 5.5% | 4% | 1.5% |
| Compound VII (8) | 47.5% | 33.5% | 6% | 5.5% | 3.5% |
 | ||||||
| Asp | Ala | |||||
| Φ | Ψ | Φ | Ψ | H-Bond Distance | L- or D-Tic/Asn Cα Distance | |
| Compound V | −73.9° | −4.9° | −99.3° | 22.8° | 3.2 Å | 5.8 Å |
| Compound VII | −59.4° | −11.6° | −76.9° | −28.1° | 2.8 Å | 5.7 Å |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Corvino, A.; Cerqua, I.; Lo Bianco, A.; Caliendo, G.; Fiorino, F.; Frecentese, F.; Magli, E.; Morelli, E.; Perissutti, E.; Santagada, V.; et al. Antagonizing S1P3 Receptor with Cell-Penetrating Pepducins in Skeletal Muscle Fibrosis. Int. J. Mol. Sci. 2021, 22, 8861. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168861
Corvino A, Cerqua I, Lo Bianco A, Caliendo G, Fiorino F, Frecentese F, Magli E, Morelli E, Perissutti E, Santagada V, et al. Antagonizing S1P3 Receptor with Cell-Penetrating Pepducins in Skeletal Muscle Fibrosis. International Journal of Molecular Sciences. 2021; 22(16):8861. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168861
Chicago/Turabian StyleCorvino, Angela, Ida Cerqua, Alessandra Lo Bianco, Giuseppe Caliendo, Ferdinando Fiorino, Francesco Frecentese, Elisa Magli, Elena Morelli, Elisa Perissutti, Vincenzo Santagada, and et al. 2021. "Antagonizing S1P3 Receptor with Cell-Penetrating Pepducins in Skeletal Muscle Fibrosis" International Journal of Molecular Sciences 22, no. 16: 8861. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168861