
ExpressHeart: Web Portal to Visualize Transcriptome Profiles of Non-Cardiomyocyte Cells

 , , and
, , and
Abstract
:1. Introduction
2. Results
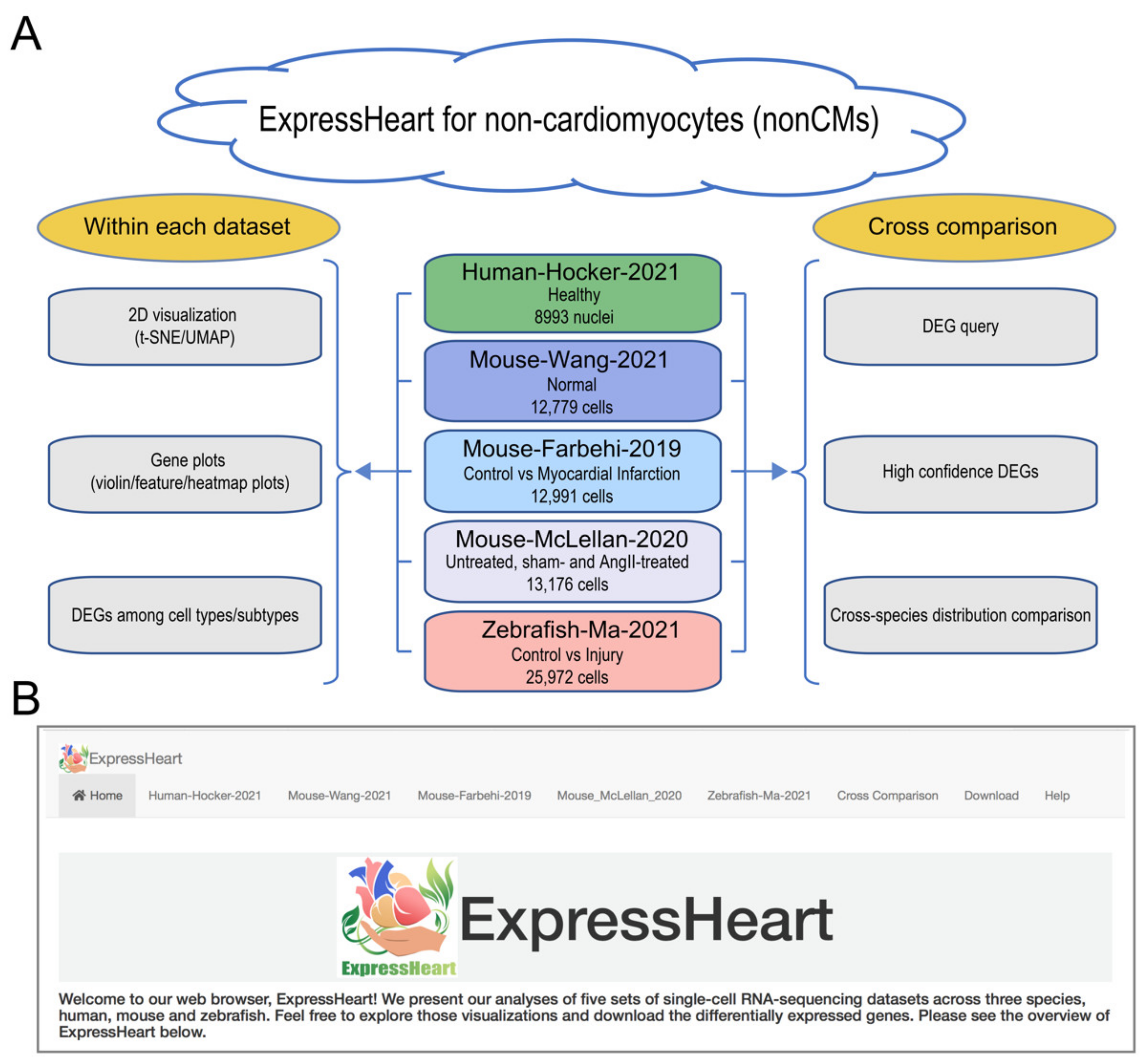
2.1. Overview of ExpressHeart
2.2. Datasets in ExpressHeart
- The Human-Hocker-2021 dataset comprises 8993 nonCM nuclei from the hearts of two healthy adult human donors. Nine cell types were identified, including fibroblasts, myofibroblasts, endothelial cells, pericytes, adipocytes, smooth muscle cells, nerve cells, and two groups of immune cells (macrophages and lymphocytes).
- The Mouse-Wang-2021 dataset consists of 12,779 cells from two adult mice, encompassing six major nonCMs cell types, namely fibroblasts, endothelial cells, pericytes, and three types of immune cells (macrophages, granulocytes, and lymphocytes). Of them, the three major cell types (fibroblasts, endothelial cells, and macrophages) are further clustered into three to five subtypes (Supplementary Table S2), with each subtype representing a distinct functional state.
- The Mouse-Farhebi-2019 dataset comprises of 12,991 nonCM cells from control and injured mouse hearts (3 and 7 days post-MI surgery), where 5658, 3825, and 3508 cells are from healthy, 3 days post-MI and 7 days post-MI hearts, respectively. Unbiased clustering identified 24 cell populations, including the major cell types, fibroblasts, endothelial cells, mural cells, and immune cells (macrophages, monocytes, dendritic cells, B cells, T cells, and natural killer (NK) cells). Similarly, there are multiple subtypes identified for the major cell types. For example, four subtypes are identified in fibroblasts, three in endothelial cells, and eight in macrophages/monocytes (Supplementary Table S2).
- The Mouse-McLellan-2020 dataset comprises of 13,176 nonCM cells from four untreated, four sham- and eight AngII-treated adult mice, where AngII-treatment could stimulate pathological remodeling of the heart. A total of 14 cell types were identified, including fibroblasts, epicardial cells, endothelial cells, lymphatic endothelial cells, endocardial cells, smooth muscle cells, pericytes, Schwann cells, proliferating mesenchymal cells, macrophages, dendritic-like cells, granulocytes, B cells, and T/NK cells. In addition, nine, three, four, and two subtypes were identified for cell types, fibroblasts, endothelial cells, macrophages, and smooth muscle cells, respectively (Supplementary Table S2).
- The Zebrafish-Ma-2021 dataset consists of 25,972 cells from uninjured (control) and injured zebrafish hearts. The authors performed scRNA-seq on nonCMs isolated from the adult zebrafish hearts before and after injury to investigate cellular functions of nonCMs during heart regeneration. The study generated transcriptome profiles for 6550, 9373, 7018, and 3031 cells before the injury, and at 2, 7, and 14 days post-injury, respectively. Nine clusters were identified in the uninjured hearts, namely, fibroblasts, endothelial cells, thrombocytes, and four types of immune cells (macrophages, neutrophils, resident mesenchymal cells, and lymphocytes). Analysis was performed for each of the three major cell types, fibroblasts, endothelial cells, and macrophages, and further identified four, four, and five subtypes, respectively (Supplementary Table S2).
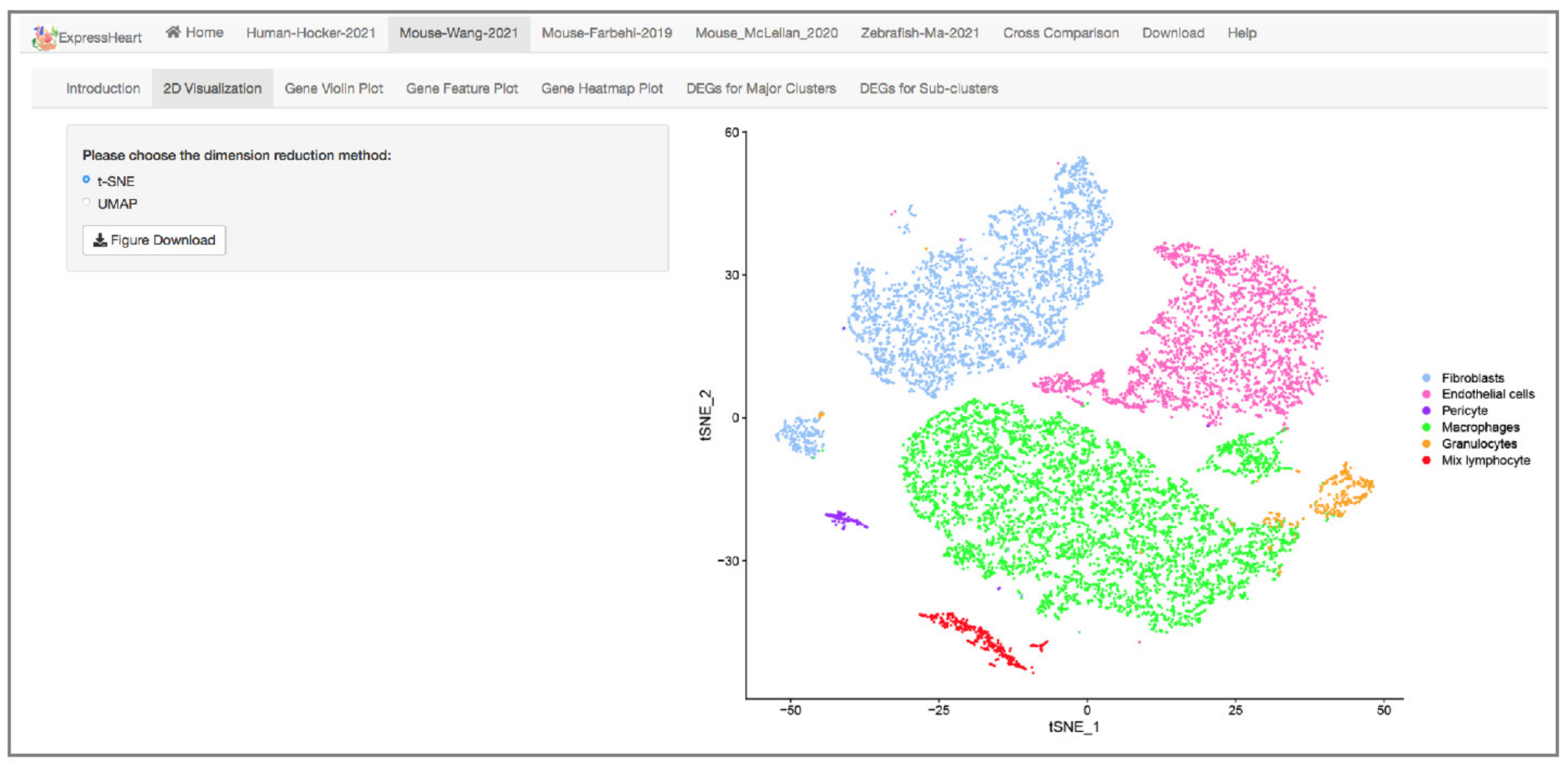
2.3. Visualization via UMAP and tSNE
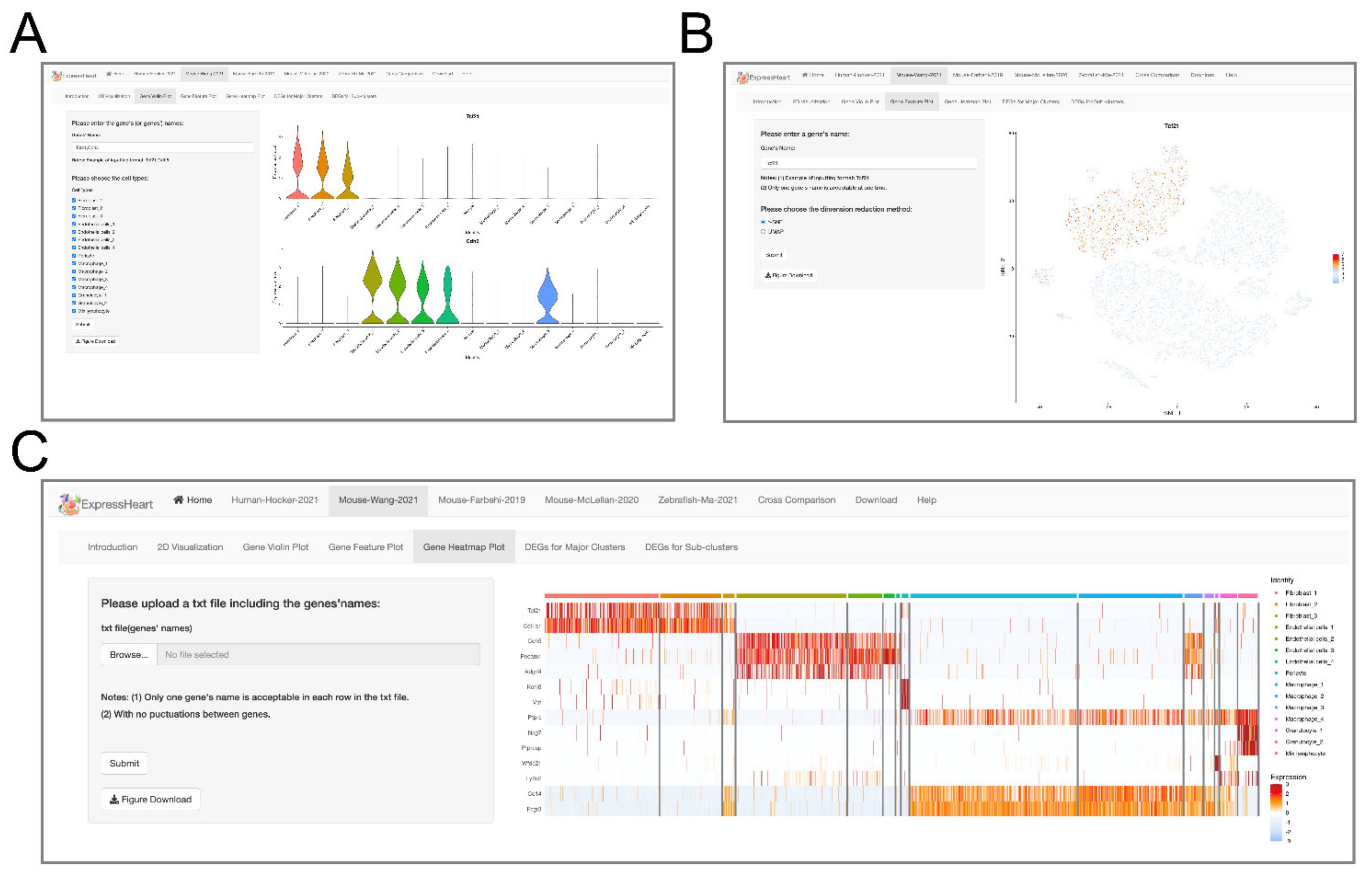
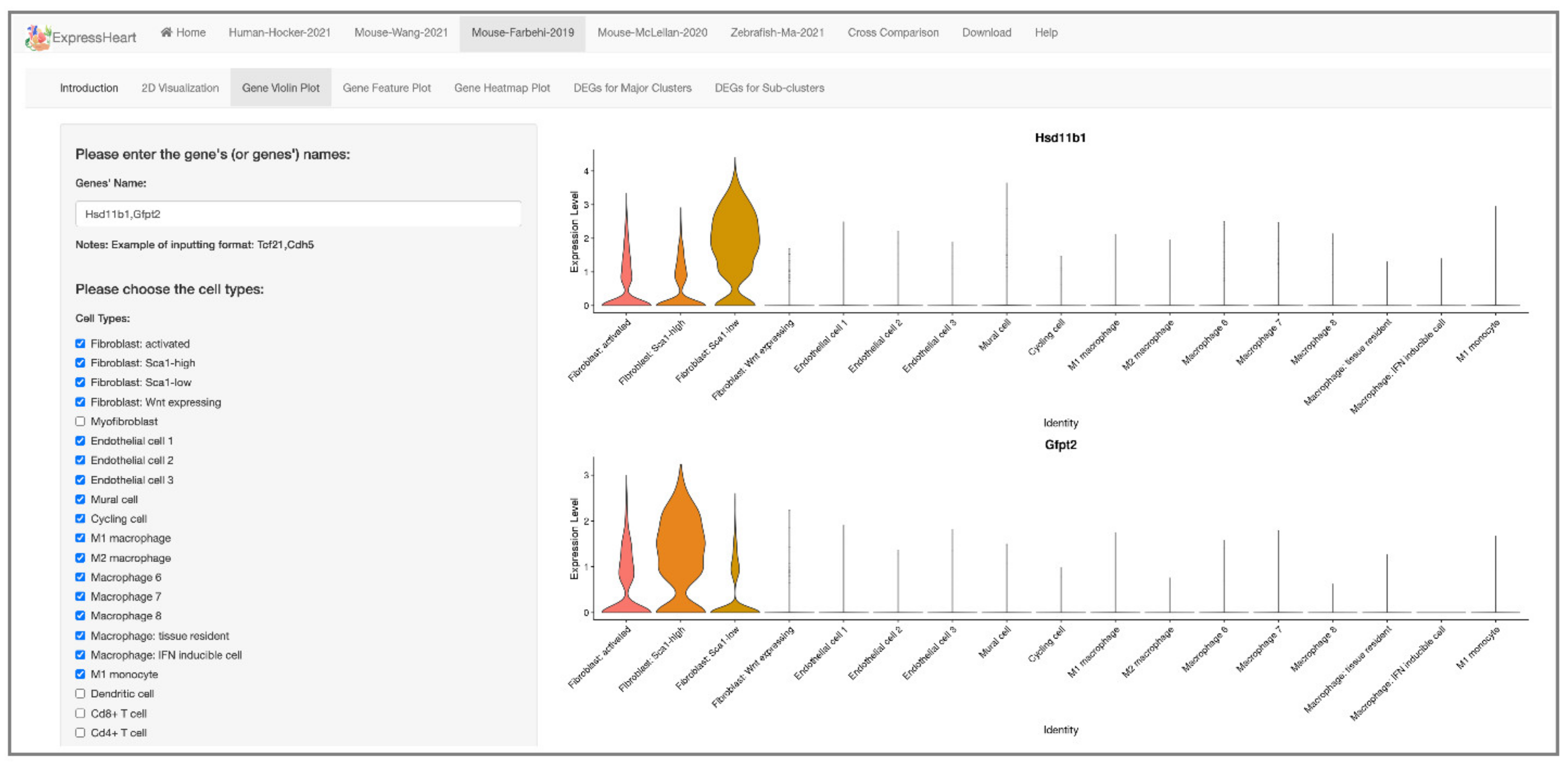
2.4. Visualization of Gene Expression Levels via Violin Plot, Feature Plot, and Heatmap
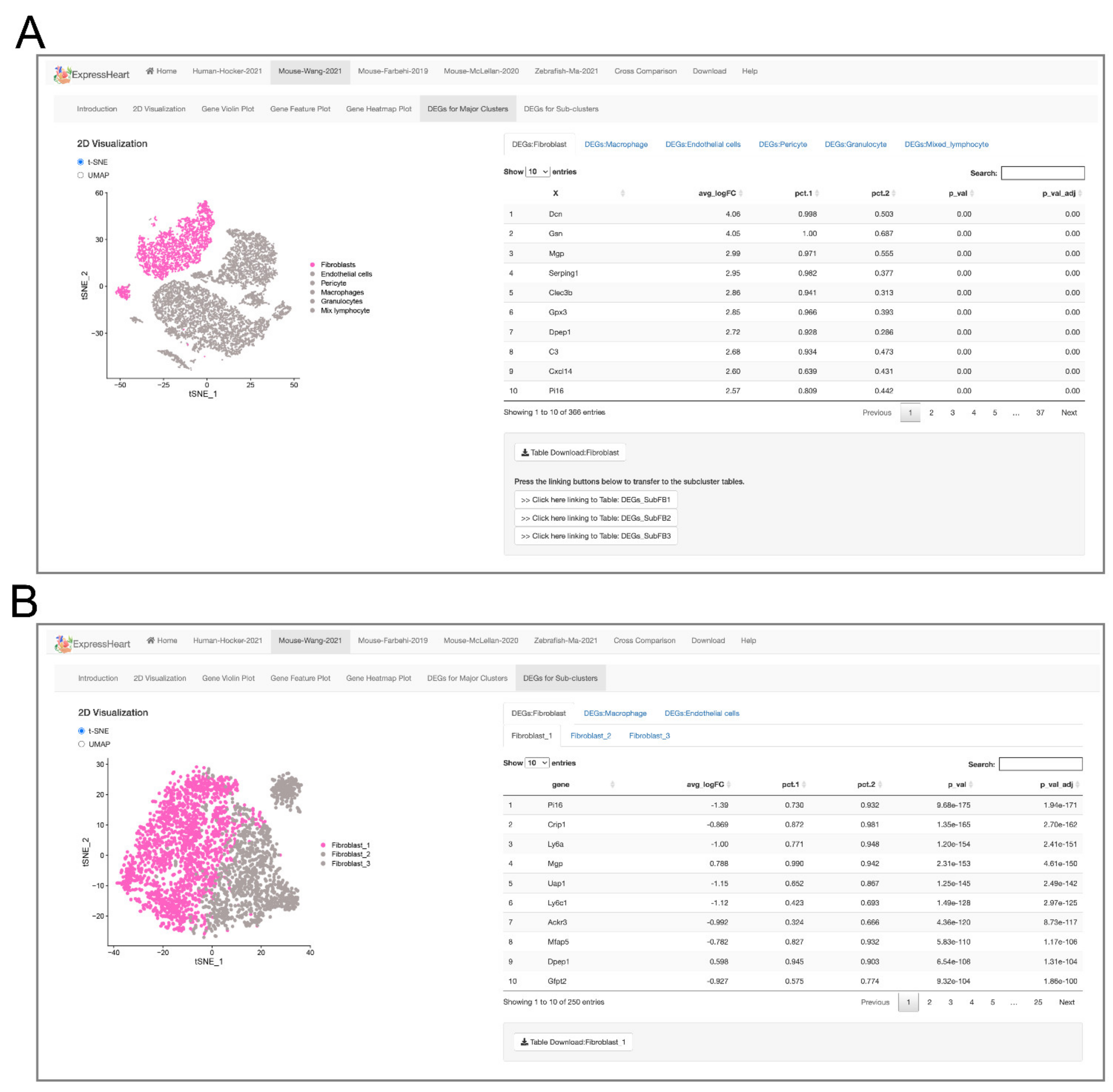
2.5. Browsing and Exporting DEGs
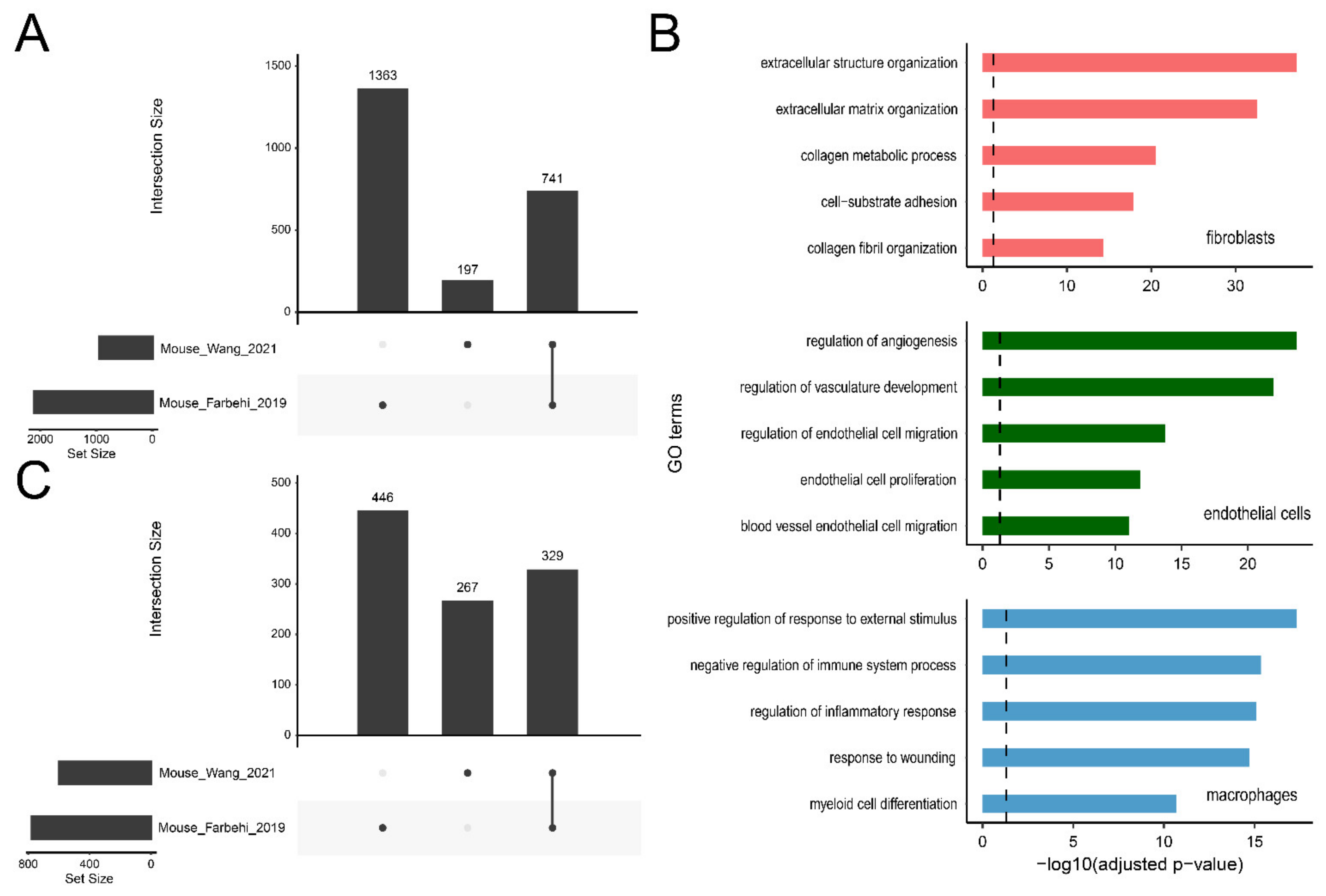
2.6. Applications of ExpressHeart-Cross-Dataset Comparison
2.6.1. DEG Query
2.6.2. High Confidence DEGs
2.6.3. Matching Cell Subtypes across Datasets
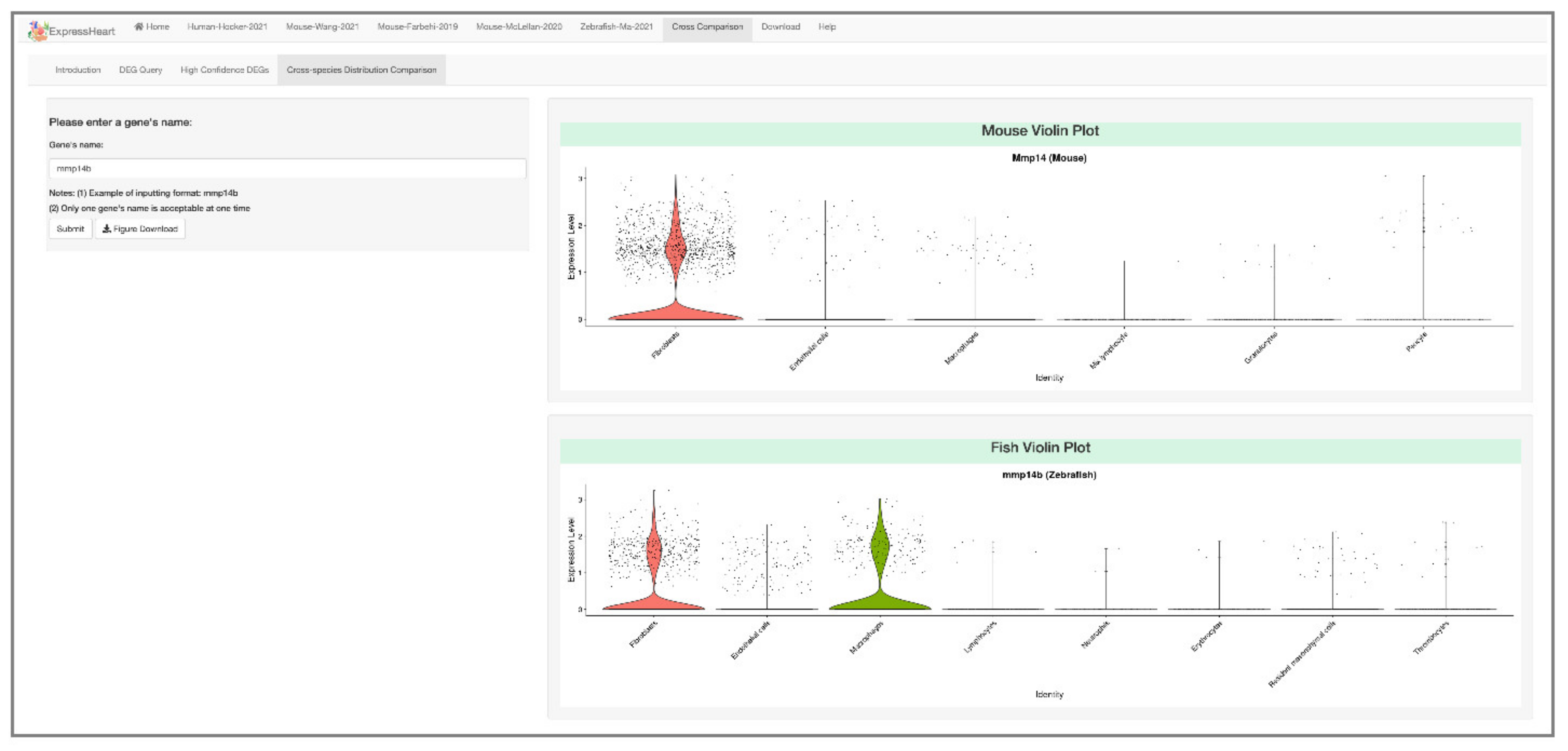
2.6.4. Cross-Species Distribution Comparison
3. Discussion
4. Materials and Methods
4.1. Processing of scRNA Sequencing Datasets
4.1.1. Human-Hocker-2021
4.1.2. Mouse-Wang-2021
4.1.3. Mouse-Farbehi-2019
4.1.4. Mouse-McLellan-2020
4.1.5. Zebrafish-Ma-2021
4.2. Data Visualization
4.3. Analysis of DEGs
4.4. High Confidence DEGs
4.5. Cross-Species Distribution Comparison
4.6. Web Server Implementation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bassat, E.; Mutlak, Y.E.; Genzelinakh, A.; Shadrin, I.Y.; Umansky, K.B.; Yifa, O.; Kain, D.; Rajchman, D.; Leach, J.; Bassat, D.R.; et al. The extracellular matrix protein agrin promotes heart regeneration in mice. Nature 2017, 547, 179–184. [Google Scholar] [CrossRef]
- Leach, J.P.; Heallen, T.; Zhang, M.; Rahmani, M.; Morikawa, Y.; Hill, M.C.; Segura, A.; Willerson, J.T.; Martin, J.F. Hippo pathway deficiency reverses systolic heart failure after infarction. Nature 2017, 550, 260–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morikawa, Y.; Heallen, T.; Leach, J.; Xiao, Y.; Martin, J.F. Dystrophin–glycoprotein complex sequesters Yap to inhibit cardiomyocyte proliferation. Nature 2017, 547, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Nakada, Y.; Canseco, D.C.; Thet, S.; Abdisalaam, S.; Asaithamby, A.; Santos, C.X.; Shah, A.M.; Zhang, H.; Faber, J.E.; Kinter, M.T.; et al. Hypoxia induces heart regeneration in adult mice. Nature 2017, 541, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Price, E.L.; Vieira, J.M.; Riley, P.R. Model organisms at the heart of regeneration. Dis. Model. Mech. 2019, 12, dmm040691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varga, I.; Kyselovič, J.; Galfiova, P.; Danisovic, L. The non-cardiomyocyte cells of the heart. Their possible roles in exercise-induced cardiac regeneration and remodeling. In Exercise for Cardiovascular Disease Prevention and Treatment. Advances in Experimental Medicine and Biology; Xiao, J., Ed.; Springer: Singapore, 2017; Volume 999, pp. 117–136. [Google Scholar]
- Feng, J.; Li, Y.; Nie, Y. Non-cardiomyocytes in heart regeneration. Curr. Drug Targets 2018, 19, 1077–1086. [Google Scholar] [CrossRef] [PubMed]
- Schafer, S.; Viswanathan, S.; Widjaja, A.A.; Lim, W.-W.; Moreno-Moral, A.; DeLaughter, D.M.; Ng, B.; Patone, G.; Chow, K.; Khin, E.; et al. IL-11 is a crucial determinant of cardiovascular fibrosis. Nature 2017, 552, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Skelly, D.A.; Squiers, G.T.; McLellan, M.A.; Bolisetty, M.T.; Robson, P.; Rosenthal, N.A.; Pinto, A.R. Single-cell transcriptional profiling reveals cellular diversity and intercommunication in the mouse heart. Cell Rep. 2018, 22, 600–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, Y.; Zheng, Y.; Liu, X.; Yan, L.; Fan, X.; Yong, J.; Hu, Y.; Dong, J.; Li, Q.; Wu, X.; et al. Single-cell transcriptome analysis maps the developmental track of the human heart. Cell Rep. 2019, 26, 1934–1950. [Google Scholar] [CrossRef] [Green Version]
- Farbehi, N.; Patrick, R.; Dorison, A.; Xaymardan, M.; Janbandhu, V.; Wystub-Lis, K.; Ho, J.W.K.; Nordon, R.E.; Harvey, R.P. Single-cell expression profiling reveals dynamic flux of cardiac stromal, vascular and immune cells in health and injury. eLife 2019, 8, 43882. [Google Scholar] [CrossRef]
- Forte, E.; Skelly, D.A.; Chen, M.; Daigle, S.; Morelli, K.A.; Hon, O.; Philip, V.M.; Costa, M.W.; Rosenthal, N.A.; Furtado, M.B. Dynamic interstitial cell response during myocardial infarction predicts resilience to rupture in genetically diverse mice. Cell Rep. 2020, 30, 3149–3163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Litviňuková, M.; Talavera-López, C.; Maatz, H.; Reichart, D.; Worth, C.L.; Lindberg, E.L.; Kanda, M.; Polanski, K.; Heinig, M.; Lee, M.; et al. Cells of the adult human heart. Nature 2020, 588, 466–472. [Google Scholar] [CrossRef] [PubMed]
- McLellan, M.A.; Skelly, D.A.; Dona, M.S.I.; Squiers, G.T.; Farrugia, G.E.; Gaynor, T.L.; Cohen, C.D.; Pandey, R.; Diep, H.; Vinh, A.; et al. High-resolution transcriptomic profiling of the heart during chronic stress reveals cellular drivers of cardiac fibrosis and hypertrophy. Circulation 2020, 142, 1448–1463. [Google Scholar] [CrossRef]
- Tucker, N.R.; Chaffin, M.; Fleming, S.J.; Hall, A.W.; Parsons, V.A.; Bedi, K.C., Jr.; Akkad, A.-D.; Herndon, C.N.; Arduini, A.; Papangeli, I.; et al. Transcriptional and cellular diversity of the human heart. Circulation 2020, 142, 466–482. [Google Scholar] [CrossRef]
- Wang, L.; Yu, P.; Zhou, B.; Song, J.; Li, Z.; Zhang, M.; Guo, G.; Wang, Y.; Chen, X.; Han, L.; et al. Single-cell reconstruction of the adult human heart during heart failure and recovery reveals the cellular landscape underlying cardiac function. Nat. Cell Biol. 2020, 22, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Hocker, J.D.; Poirion, O.B.; Zhu, F.; Buchanan, J.; Zhang, K.; Chiou, J.; Wang, T.-M.; Zhang, Q.; Hou, X.; Li, Y.E.; et al. Cardiac cell type–specific gene regulatory programs and disease risk association. Sci. Adv. 2021, 7, eabf1444. [Google Scholar] [CrossRef]
- Wang, L.; Yang, Y.; Ma, H.; Xie, Y.; Xu, J.; Near, D.; Wang, H.; Garbutt, T.; Li, Y.; Liu, J.; et al. Single-cell dual-omics reveals the transcriptomic and epigenomic diversity of cardiac non-myocytes. Cardiovasc. Res. 2021, cvab134. [Google Scholar] [CrossRef]
- Nicin, L.; Abplanalp, W.T.; Schänzer, A.; Sprengel, A.; John, D.; Mellentin, H.; Tombor, L.; Keuper, M.; Ullrich, E.; Klingel, K.; et al. Single nuclei sequencing reveals novel insights into the regulation of cellular signatures in children with dilated cardiomyopathy. Circulation 2021, 143, 1704–1719. [Google Scholar] [CrossRef]
- Wang, Z.; Cui, M.; Shah, A.M.; Tan, W.; Liu, N.; Bassel-Duby, R.; Olson, E.N. Cell-type-specific gene regulatory networks underlying murine neonatal heart regeneration at single-cell resolution. Cell Rep. 2020, 33, 108472. [Google Scholar] [CrossRef]
- Ma, H.; Liu, Z.; Yang, Y.; Feng, D.; Dong, Y.; Garbutt, T.A.; Hu, Z.; Wang, L.; Luan, C.; Cooper, C.D.; et al. Functional coordination of non-myocytes plays a key role in adult zebrafish heart regeneration. EMBO Rep. 2021, accepted. [Google Scholar]
- McInnes, L.; Healy, J.; Melville, J. Umap: Uniform manifold approximation and projection for dimension reduction. arXiv 2018, arXiv:1802.03426. [Google Scholar]
- Van der Maaten, L.; Hinton, G. Visualizing data using t-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- Karemaker, I.D.; Vermeulen, M. Single-cell DNA methylation profiling: Technologies and biological applications. Trends Biotechnol. 2018, 36, 952–965. [Google Scholar] [CrossRef]
- Pott, S.; Lieb, J.D. Single-cell ATAC-seq: Strength in numbers. Genome Biol. 2015, 16, 1–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satpathy, A.T.; Granja, J.M.; Yost, K.E.; Qi, Y.; Meschi, F.; McDermott, G.P.; Olsen, B.N.; Mumbach, M.R.; Pierce, S.E.; Corces, M.R.; et al. Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion. Nat. Biotechnol. 2019, 37, 925–936. [Google Scholar] [CrossRef]
- Ramani, V.; Deng, X.; Qiu, R.; Gunderson, K.L.; Steemers, F.J.; Disteche, C.M.; Noble, W.S.; Duan, Z.; Shendure, J. Massively multiplex single-cell Hi-C. Nat. Methods 2017, 14, 263–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck III, W.M.; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive integration of single-cell data. Cell 2019, 177, 1888–1902. [Google Scholar] [CrossRef] [PubMed]
- Korsunsky, I.; Millard, N.; Fan, J.; Slowikowski, K.; Zhang, F.; Wei, K.; Baglaenko, Y.; Brenner, M.; Loh, P.; Raychaudhuri, S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 2019, 16, 1289–1296. [Google Scholar] [CrossRef]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef]
- Welch, J.D.; Kozareva, V.; Ferreira, A.; Vanderburg, C.; Martin, C.; Macosko, E.Z. Single-cell multi-omic integration compares and contrasts features of brain cell identity. Cell 2019, 177, 1873–1887. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Beeley, C. Web Application Development with R Using Shiny, 2nd ed.; Packt Publishing Ltd.: Birmingham, UK, 2016. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | Species | Technologies | Condition | Cells/Nuclei | Genes | Ref |
|---|---|---|---|---|---|---|
| Human-Hocker-2021 | Human | snRNA-seq | Healthy | 8993 | 27,109 | [17] |
| Mouse-Wang-2021 | Mouse | scRNA-seq | Wildtype | 12,779 | 27,998 | [18] |
| Mouse-Farbehi-2019 | Mouse | scRNA-seq | Control vs. MI | 12,991 | 15,674 | [11] |
| Mouse-McLellan-2020 | Mouse | scRNA-seq | Untreated, sham- and Ang II-treated | 13,176 | 17,170 | [14] |
| Zebrafish-Ma-2021 | Zebrafish | scRNA-seq | Control vs. Injury | 25,972 | 35,117 | [21] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, G.; Luan, C.; Dong, Y.; Xie, Y.; Zentz, S.C.; Zelt, R.; Roach, J.; Liu, J.; Qian, L.; Li, Y.; et al. ExpressHeart: Web Portal to Visualize Transcriptome Profiles of Non-Cardiomyocyte Cells. Int. J. Mol. Sci. 2021, 22, 8943. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168943
Li G, Luan C, Dong Y, Xie Y, Zentz SC, Zelt R, Roach J, Liu J, Qian L, Li Y, et al. ExpressHeart: Web Portal to Visualize Transcriptome Profiles of Non-Cardiomyocyte Cells. International Journal of Molecular Sciences. 2021; 22(16):8943. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168943
Chicago/Turabian StyleLi, Gang, Changfei Luan, Yanhan Dong, Yifang Xie, Scott C. Zentz, Rob Zelt, Jeff Roach, Jiandong Liu, Li Qian, Yun Li, and et al. 2021. "ExpressHeart: Web Portal to Visualize Transcriptome Profiles of Non-Cardiomyocyte Cells" International Journal of Molecular Sciences 22, no. 16: 8943. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22168943