
Enhancer of Zeste Homolog 2 (EZH2) Contributes to Rod Photoreceptor Death Process in Several Forms of Retinal Degeneration and Its Activity Can Serve as a Biomarker for Therapy Efficacy

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
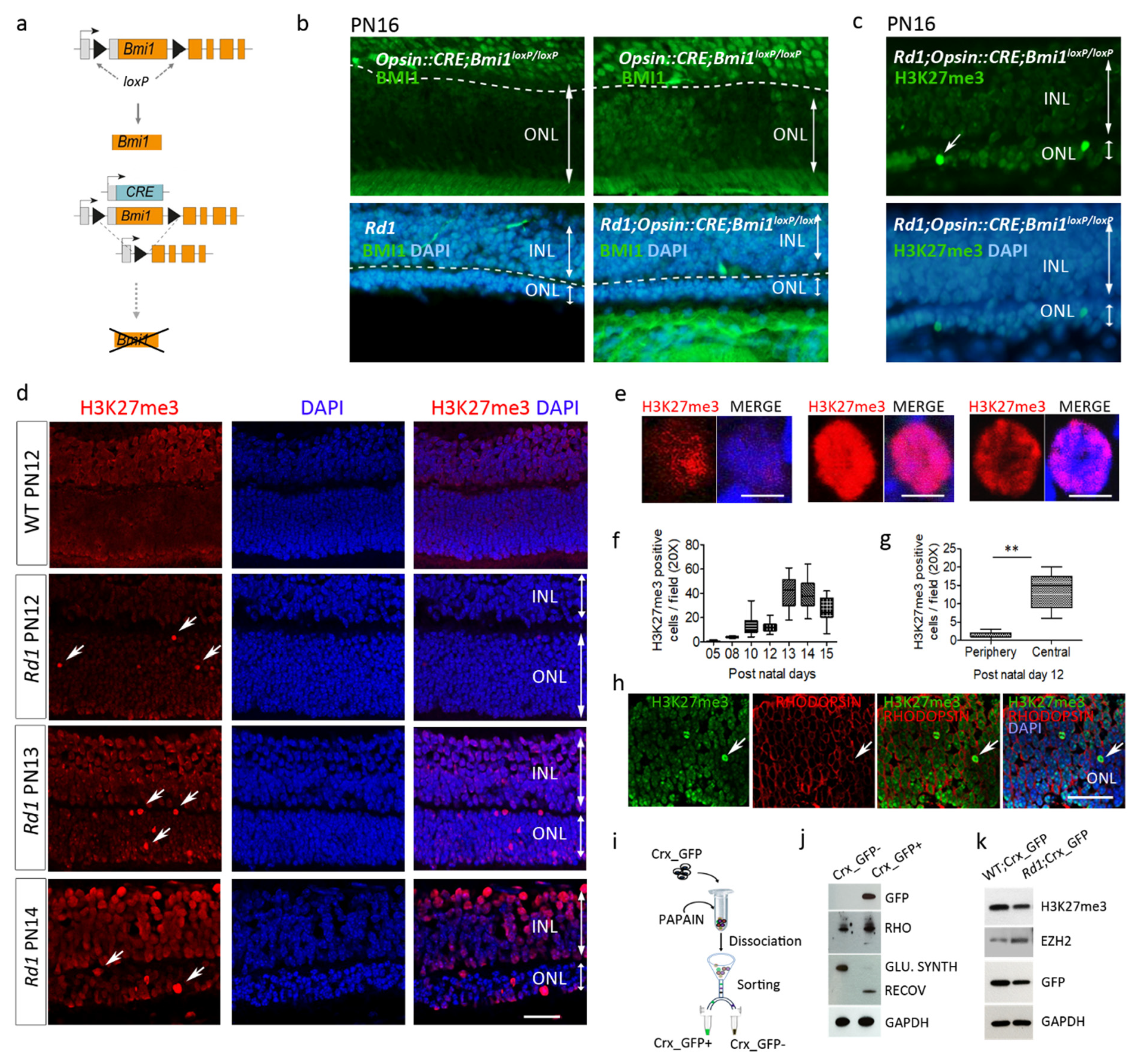
2.1. EZH2 Activity Precedes BMI1 Action to Mediate Photoreceptor Death in the Rd1 Retina
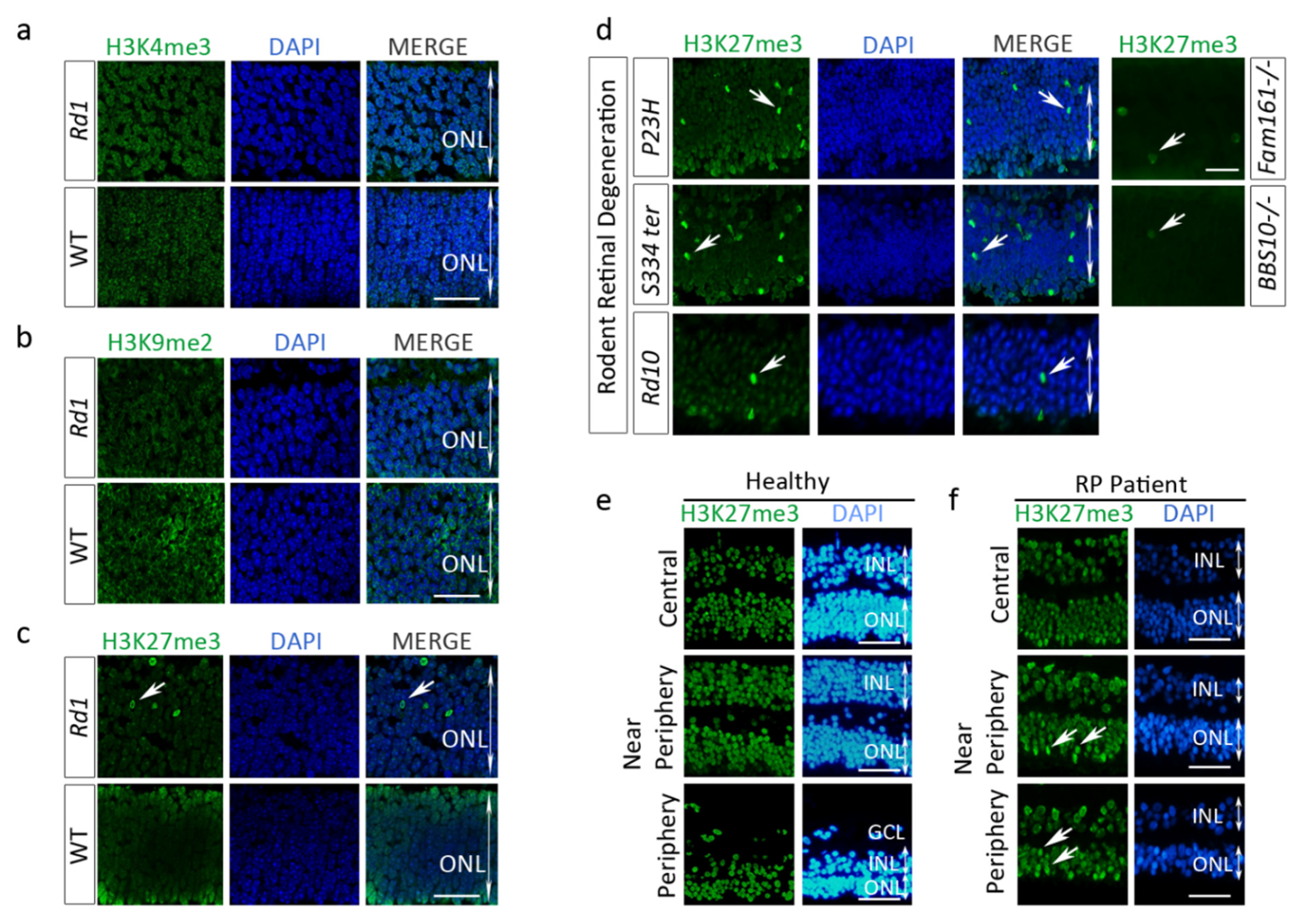
2.2. Enhanced H3K27 Trimethylation (H3K27me3) in Rd1 Photoreceptors and Other IRD Models
2.3. H3K27me3 Accumulates in a Human Retinitis Pigmentosa (RP) Patient Retina
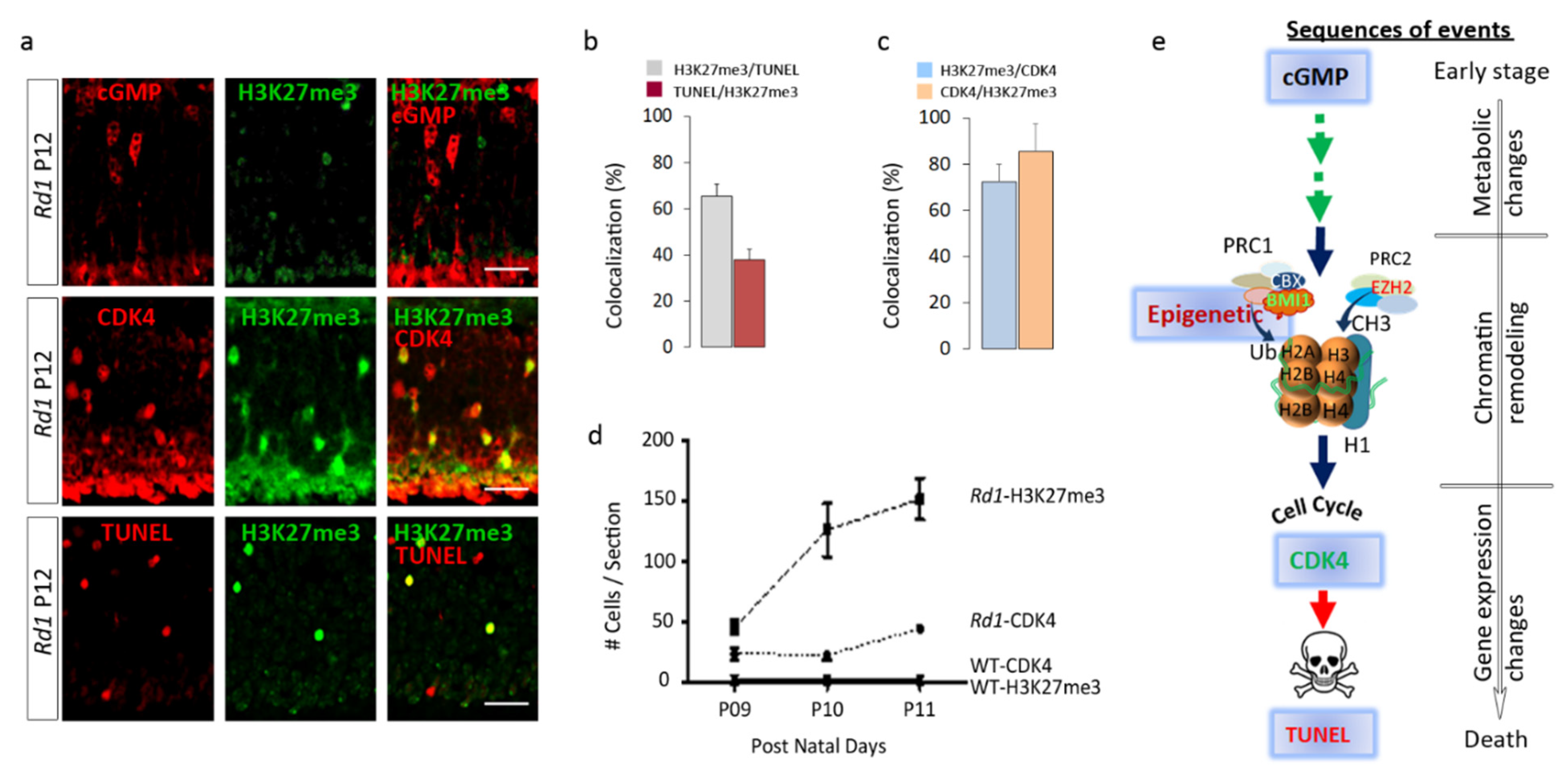
2.4. H3K27me3 Accumulation in the Rd1 Retina Precedes Late Events of Photoreceptor Death
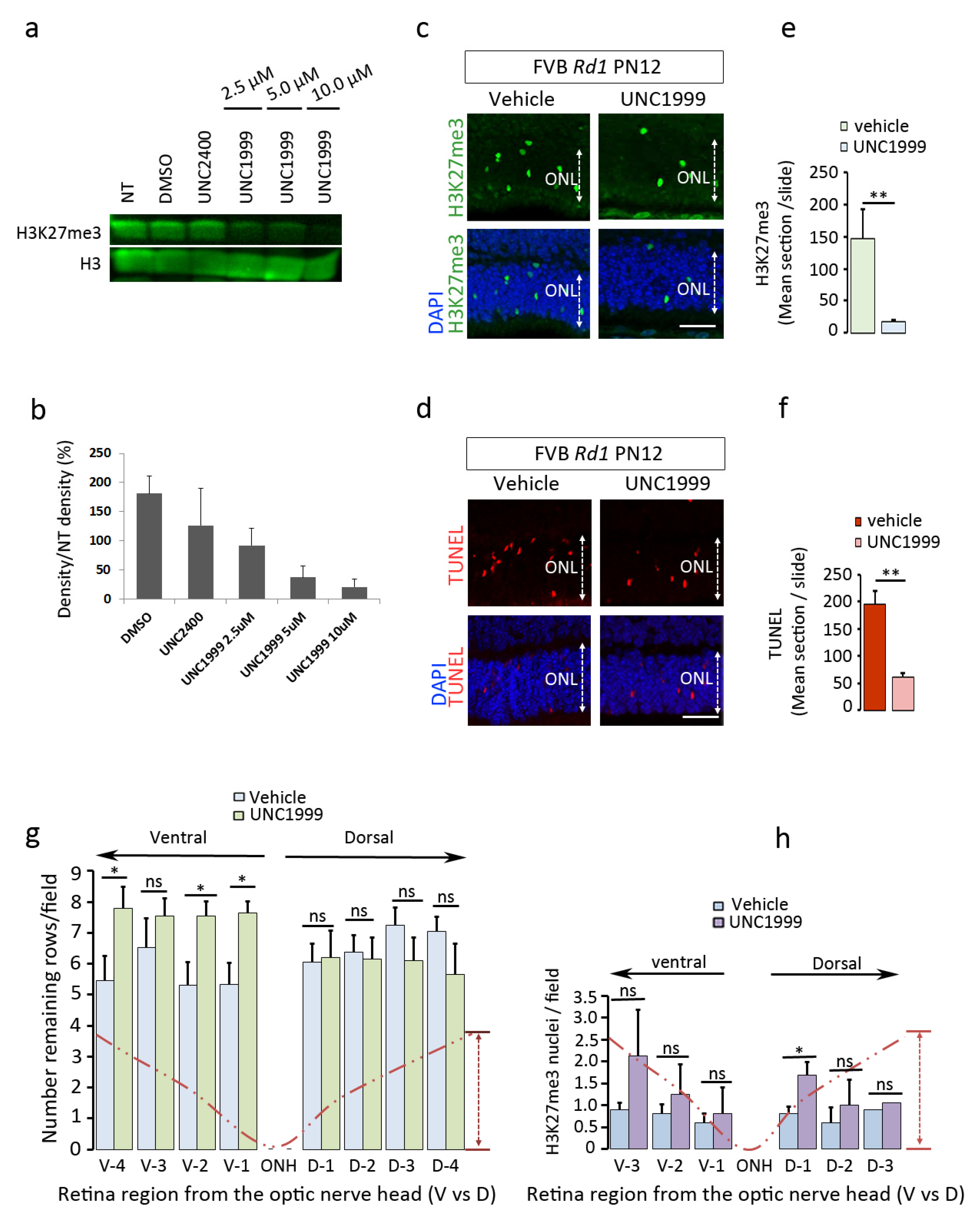
2.5. EZH2 Contributes to the Process of Cell Death
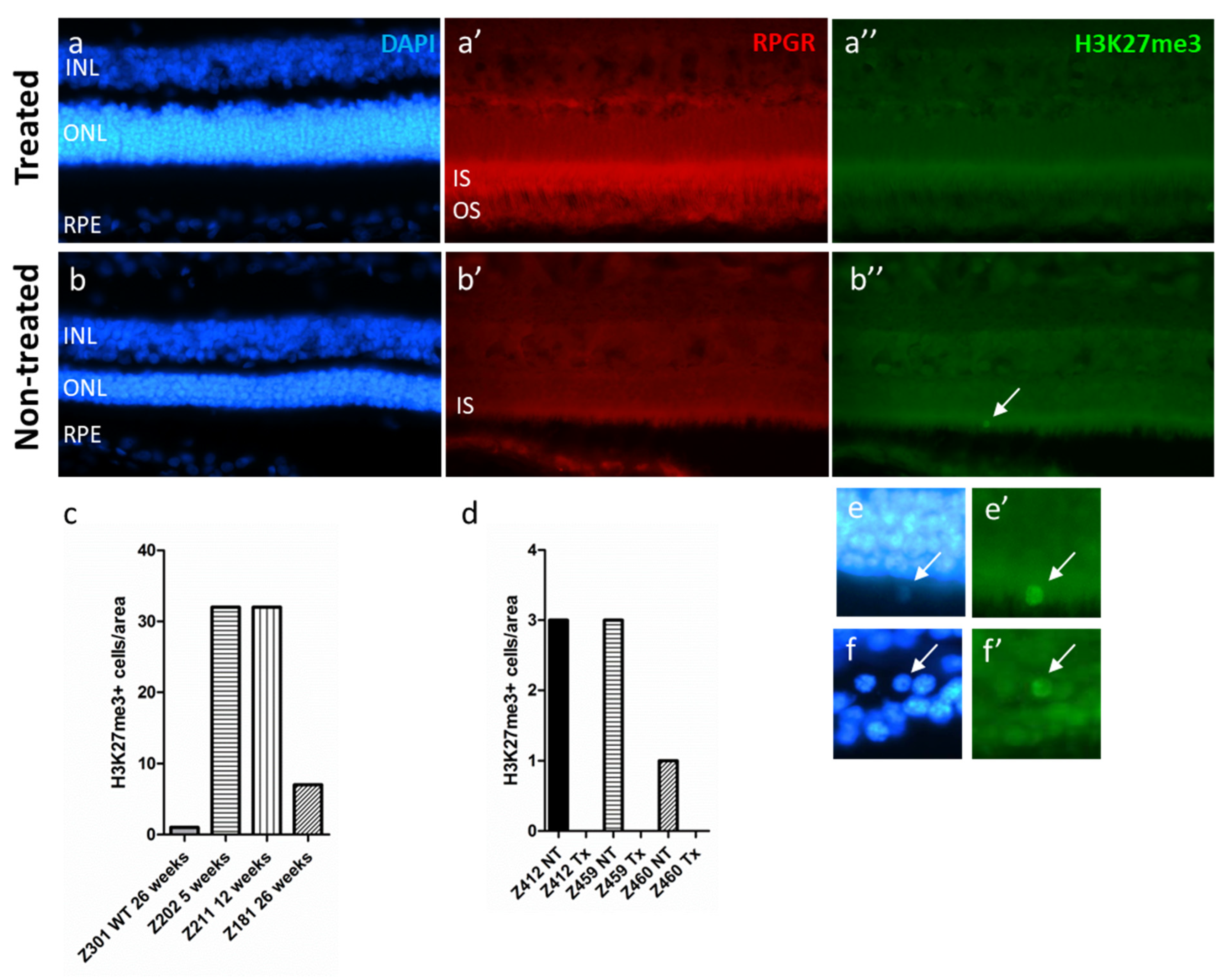
2.6. The H3K27me3 Mark Is Absent in RPGR-Mutant Dogs after Gene Augmentation Therapy Treatment
3. Discussion
4. Materials and Methods
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zencak, D.; Schouwey, K.; Chen, D.; Ekström, P.; Tanger, E.; Bremner, R.; van Lohuizen, M.; Arsenijevic, Y. Retinal degeneration depends on Bmi1 function and reactivation of cell cycle proteins. Proc. Natl. Acad. Sci. USA 2013, 110, E593–E601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, R.; Tsukada, Y.; Zhang, Y. Role of Bmi-1 and Ring1A in H2A Ubiquitylation and Hox Gene Silencing. Mol. Cell 2005, 20, 845–854. [Google Scholar] [CrossRef]
- Cao, R.; Wang, L.; Wang, H.; Xia, L.; Erdjument-Bromage, H.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of Histone H3 Lysine 27 Methylation in Polycomb-Group Silencing. Science 2002, 298, 1039–1043. [Google Scholar] [CrossRef] [Green Version]
- Rao, R.C.; Tchedre, K.T.; Malik, M.T.A.; Coleman, N.; Fang, Y.; Marquez, V.E.; Chen, N.F. Dynamic Patterns of Histone Lysine Methylation in the Developing Retina. Investig. Opthalmology Vis. Sci. 2010, 51, 6784–6792. [Google Scholar] [CrossRef] [Green Version]
- Barhoum, R.; Navarrete, G.M.; Corrochano, S.; Germain, F.; Fernandez-Sanchez, L.; de la Rosa, E.; de la Villa, P.; Cuenca, N. Functional and structural modifications during retinal degeneration in the rd10 mouse. Neuroscience 2008, 155, 698–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, B.; Hawes, N.; Pardue, M.; German, A.; Hurd, R.; Davisson, M.; Nusinowitz, S.; Rengarajan, K.; Boyd, A.; Sidney, S.; et al. Two mouse retinal degenerations caused by missense mutations in the β-subunit of rod cGMP phosphodiesterase gene. Vis. Res. 2007, 47, 624–633. [Google Scholar] [CrossRef] [Green Version]
- Portera-Cailliau, C.; Sung, C.H.; Nathans, J.; Adler, R. Apoptotic photoreceptor cell death in mouse models of retinitis pigmentosa. Proc. Natl. Acad. Sci. USA 1994, 91, 974–978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahaboglu, A.; Paquet-Durand, O.; Dietter, J.; Dengler, K.; Bernhard-Kurz, S.; Ekström, P.A.; Hitzmann, B.; Ueffing, M. Retinitis pigmentosa: Rapid neurodegeneration is governed by slow cell death mechanisms. Cell Death Dis. 2013, 4, e488. [Google Scholar] [CrossRef] [PubMed]
- Paquet-Durand, F.; Sahaboglu, A.; Dietter, J.; Paquet-Durand, O.; Hitzmann, B.; Ueffing, M.; Ekström, P.A.R. How Long Does a Photoreceptor Cell Take to Die? Implications for the Causative Cell Death Mechanisms. Adv. Exp. Med. Biol. 2014, 801, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Farber, D.B.; Lolley, R.N. Cyclic Guanosine Monophosphate: Elevation in Degenerating Photoreceptor Cells of the C3H Mouse Retina. Science 1974, 186, 449–451. [Google Scholar] [CrossRef] [PubMed]
- Konze, K.D.; Ma, A.; Li, F.; Barsyte-Lovejoy, D.; Parton, T.; MacNevin, C.J.; Liu, F.; Gao, C.; Huang, X.-P.; Kuznetsova, E.; et al. An Orally Bioavailable Chemical Probe of the Lysine Methyltransferases EZH2 and EZH1. ACS Chem. Biol. 2013, 8, 1324–1334. [Google Scholar] [CrossRef]
- Takeshima, H.; Wakabayashi, M.; Hattori, N.; Yamashita, S.; Ushijima, T. Identification of coexistence of DNA methylation and H3K27me3 specifically in cancer cells as a promising target for epigenetic therapy. Carcinogenesis 2015, 36, 192–201. [Google Scholar] [CrossRef] [Green Version]
- Beryozkin, A.; Matsevich, C.; Obolensky, A.; Kostic, C.; Arsenijevic, Y.; Wolfrum, U.; Banin, E.; Sharon, D. A new mouse model for retinal degeneration due to Fam161a deficiency. Sci. Rep. 2021, 11, 2030. [Google Scholar] [CrossRef] [PubMed]
- Thompson, D.A.; Iannaccone, A.; Ali, R.R.; Arshavsky, V.Y.; Audo, I.; Bainbridge, J.W.B.; Besirli, C.G.; Birch, D.G.; Branham, K.E.; Cideciyan, A.V.; et al. Advancing Clinical Trials for Inherited Retinal Diseases: Recommendations from the Second Monaciano Symposium. Transl. Vis. Sci. Technol. 2020, 9, 2. [Google Scholar] [CrossRef] [PubMed]
- Bainbridge, J.W.; Mehat, M.S.; Sundaram, V.; Robbie, S.J.; Barker, S.E.; Ripamonti, C.; Georgiadis, A.; Mowat, F.; Beattie, S.G.; Gardner, P.; et al. Long-Term Effect of Gene Therapy on Leber’s Congenital Amaurosis. N. Engl. J. Med. 2015, 372, 1887–1897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardiner, K.L.; Cideciyan, A.V.; Swider, M.; Dufour, V.L.; Sumaroka, A.; Komáromy, A.M.; Hauswirth, W.W.; Iwabe, S.; Jacobson, S.G.; Beltran, W.A.; et al. Long-Term Structural Outcomes of Late-Stage RPE65 Gene Therapy. Mol. Ther. 2020, 28, 266–278. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, S.; Cideciyan, A.V.; Roman, A.J.; Sumaroka, A.; Schwartz, S.B.; Heon, E.; Hauswirth, W. Improvement and Decline in Vision with Gene Therapy in Childhood Blindness. N. Engl. J. Med. 2015, 372, 1920–1926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beltran, W.A.; Hammond, P.; Acland, G.M.; Aguirre, G.D. A Frameshift Mutation inRPGRExon ORF15 Causes Photoreceptor Degeneration and Inner Retina Remodeling in a Model of X-Linked Retinitis Pigmentosa. Investig. Opthalmology Vis. Sci. 2006, 47, 1669–1681. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Acland, G.M.; Wu, W.X.; Johnson, J.L.; Pearce-Kelling, S.; Tulloch, B.; Vervoort, R.; Wright, A.F.; Aguirre, G.D. Different RPGR exon ORF15 mutations in Canids provide insights into photoreceptor cell degeneration. Hum. Mol. Genet. 2002, 11, 993–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, W.-T.; Dyka, F.M.; Dinculescu, A.; Li, J.; Zhu, P.; Chiodo, V.A.; Boye, S.L.; Conlon, T.J.; Erger, K.; Cossette, T.; et al. Stability and Safety of an AAV Vector for Treating RPGR-ORF15 X-Linked Retinitis Pigmentosa. Hum. Gene Ther. 2015, 26, 593–602. [Google Scholar] [CrossRef] [Green Version]
- Beltran, W.A.; Cideciyan, A.V.; Iwabe, S.; Swider, M.; Kosyk, M.S.; McDaid, K.; Martynyuk, I.; Ying, G.-S.; Shaffer, J.; Deng, W.-T.; et al. Successful arrest of photoreceptor and vision loss expands the therapeutic window of retinal gene therapy to later stages of disease. Proc. Natl. Acad. Sci. USA 2015, 112, E5844–E5853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshino, A.; Ratnapriya, R.; Brooks, M.J.; Chaitankar, V.; Wilken, M.S.; Zhang, C.; Starostik, M.; Gieser, L.; La Torre, A.; Nishio, M.; et al. Molecular Anatomy of the Developing Human Retina. Dev. Cell 2017, 43, 763–779.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, S.; Xiao, L.; Liu, Y.; Wang, Y.; Cheng, L.; Zhang, J.; Yan, N.; Chen, D. DZNep inhibits H3K27me3 deposition and delays retinal degeneration in the rd1 mice. Cell Death Dis. 2018, 9, 310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahaboglu, A.; Barth, M.; Secer, E.; del Amo, E.M.; Urtti, A.; Arsenijevic, Y.; Zrenner, E.; Paquet-Durand, F. Olaparib significantly delays photoreceptor loss in a model for hereditary retinal degeneration. Sci. Rep. 2016, 6, 39537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samardzija, M.; Corna, A.; Gomez-Sintes, R.; Jarboui, M.A.; Armento, A.; Roger, J.E.; Petridou, E.; Haq, W.; Paquet-Durand, F.; Zrenner, E.; et al. HDAC inhibition ameliorates cone survival in retinitis pigmentosa mice. Cell Death Differ. 2021, 28, 1317–1332. [Google Scholar] [CrossRef]
- Arrowsmith, C.; E Audia, J.; Austin, C.; Baell, J.; Bennett, J.; Blagg, J.; Bountra, C.; E Brennan, P.; Brown, P.J.; E Bunnage, M.; et al. The promise and peril of chemical probes. Nat. Chem. Biol. 2015, 11, 536–541, Erratum in 2015, 11, 887. [Google Scholar] [CrossRef] [Green Version]
- Iida, A.; Iwagawa, T.; Baba, Y.; Satoh, S.; Mochizuki, Y.; Nakauchi, H.; Furukawa, T.; Koseki, H.; Murakami, A.; Watanabe, S. Roles of histone H3K27 trimethylase Ezh2 in retinal proliferation and differentiation. Dev. Neurobiol. 2015, 75, 947–960. [Google Scholar] [CrossRef]
- Zhang, J.; Taylor, R.J.; La Torre, A.; Wilken, M.S.; Cox, K.E.; Reh, T.A.; Vetter, M.L. Ezh2 maintains retinal progenitor proliferation, transcriptional integrity, and the timing of late differentiation. Dev. Biol. 2015, 403, 128–138. [Google Scholar] [CrossRef] [Green Version]
- Yan, N.; Cheng, L.; Cho, K.; Malik, M.T.A.; Xiao, L.; Guo, C.; Yu, H.; Zhu, R.; Rao, R.C.; Chen, D.F. Postnatal onset of retinal degeneration by loss of embryonic Ezh2 repression of Six1. Sci. Rep. 2016, 6, 33887. [Google Scholar] [CrossRef] [Green Version]
- Arango-Gonzalez, B.; Trifunovic, D.; Sahaboglu, A.; Kranz, K.; Michalakis, S.; Farinelli, P.; Koch, S.; Koch, F.; Cottet, S.; Janssen-Bienhold, U.; et al. Identification of a Common Non-Apoptotic Cell Death Mechanism in Hereditary Retinal Degeneration. PLoS ONE 2014, 9, e112142. [Google Scholar] [CrossRef] [PubMed]
- Maes, M.; Schlamp, C.L.; Nickells, R.W. BAX to basics: How the BCL2 gene family controls the death of retinal ganglion cells. Prog. Retin. Eye Res. 2017, 57, 1–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porciatti, V.; Chou, T.-H. Modeling Retinal Ganglion Cell Dysfunction in Optic Neuropathies. Cells 2021, 10, 1398. [Google Scholar] [CrossRef] [PubMed]
- Romano, G.L.; Amato, R.; Lazzara, F.; Porciatti, V.; Chou, T.-H.; Drago, F.; Bucolo, C. P2X7 receptor antagonism preserves retinal ganglion cells in glaucomatous mice. Biochem. Pharmacol. 2020, 180, 114199. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Brommer, B.; Tian, X.; Krishnan, A.; Meer, M.; Wang, C.; Vera, D.L.; Zeng, Q.; Yu, D.; Bonkowski, M.S.; et al. Reprogramming to recover youthful epigenetic information and restore vision. Nat. Cell Biol. 2020, 588, 124–129. [Google Scholar] [CrossRef]
- Li, J.; Hart, R.; Mallimo, E.M.; Swerdel, M.R.; Kusnecov, A.W.; Herrup, K. EZH2-mediated H3K27 trimethylation mediates neurodegeneration in ataxia-telangiectasia. Nat. Neurosci. 2013, 16, 1745–1753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, X.; Chen, J.; Li, J.; Kofler, J.; Herrup, K. Neurons in Vulnerable Regions of the Alzheimer’s Disease Brain Display Reduced ATM Signaling. Eneuro 2016, 3. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease Form | Animal Model | Age | H3K27me3h |
|---|---|---|---|
| Recessive RP | Rd1 (mouse) | PN12 | +++ |
| Rd10 (mouse) | PN20 | +++ | |
| Rho−/− (mouse) | PN30 | ++ | |
| Rcd1 (dog) | PN28 | ++ | |
| Dominant RP | RhoP23H (mouse) | PN15 | +++ |
| RhoS334ter (rat) | PN12 | +++ | |
| Ciliopathy | Fam161atm1b/tm1b (mouse) | PN30 | ++ |
| BBS10 (mouse) | PN14 | + | |
| XLRPA2 (dog) | 5 weeks | ++ | |
| 26 weeks | + |
| Antibody | Application | Dilution | Supplier (Catalog #) |
|---|---|---|---|
| Anti-H3k27me3 | IHC/WB | 1:400/1:2000 | Millipore (07-449) |
| Anti-H3k27me3 | IHC | 1:1000 | Abcam (AB6002) |
| Anti-H3K9me2 | IHC | 1:500 | Millipore (07-212) |
| Anti-H3K4me3 | IHC | 1:500 | Millipore (07-473) |
| Anti-H3, ct | WB | 1:5000 | Millipore (07-690) |
| Anti-BMI1 | IHC | 1:100 | USBiological (B2185) |
| Anti-BMI1 | WB | 1:2000 | Boster immunoleader (PB9133) |
| Anti-Rhodopsin | IHC/WB | 1:1000/1:1000 | NeoMarkers (MS-1233-P) |
| Anti-recoverin | Western blot | 1:1000 | Chemicon (AB5585) |
| Anti-cGMP | IHC | 1:1500 | H.Steinbusch (MHENS) |
| Anti-CDK4 | IHC | 1:50 | Santa Cruz (SC-601) |
| Anti-RPGR | IHC | 1:100 | Sigma (HPA001593) |
| Anti-EZH2 | IHC | 1:125 | ABGENT (AM1836A) |
| Anti-EZH2 | WB | 1:2000 | Cell Signaling (3147s) |
| Anti-GFP | IHC/WB | 1:1000/1:1000 | Abcam (ab290) |
| Anti-GAPDH | WB | 1:2000 | Chemicon (MAB374) |
| Anti-Glutamine synthase | WB | 1:50 | Millipore (MAB302) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mbefo, M.; Berger, A.; Schouwey, K.; Gérard, X.; Kostic, C.; Beryozkin, A.; Sharon, D.; Dolfuss, H.; Munier, F.; Tran, H.V.; et al. Enhancer of Zeste Homolog 2 (EZH2) Contributes to Rod Photoreceptor Death Process in Several Forms of Retinal Degeneration and Its Activity Can Serve as a Biomarker for Therapy Efficacy. Int. J. Mol. Sci. 2021, 22, 9331. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22179331
Mbefo M, Berger A, Schouwey K, Gérard X, Kostic C, Beryozkin A, Sharon D, Dolfuss H, Munier F, Tran HV, et al. Enhancer of Zeste Homolog 2 (EZH2) Contributes to Rod Photoreceptor Death Process in Several Forms of Retinal Degeneration and Its Activity Can Serve as a Biomarker for Therapy Efficacy. International Journal of Molecular Sciences. 2021; 22(17):9331. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22179331
Chicago/Turabian StyleMbefo, Martial, Adeline Berger, Karine Schouwey, Xavier Gérard, Corinne Kostic, Avigail Beryozkin, Dror Sharon, Hélène Dolfuss, Francis Munier, Hoai Viet Tran, and et al. 2021. "Enhancer of Zeste Homolog 2 (EZH2) Contributes to Rod Photoreceptor Death Process in Several Forms of Retinal Degeneration and Its Activity Can Serve as a Biomarker for Therapy Efficacy" International Journal of Molecular Sciences 22, no. 17: 9331. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22179331