
Glucocorticoid Resistance: Interference between the Glucocorticoid Receptor and the MAPK Signalling Pathways

 , ,
, , 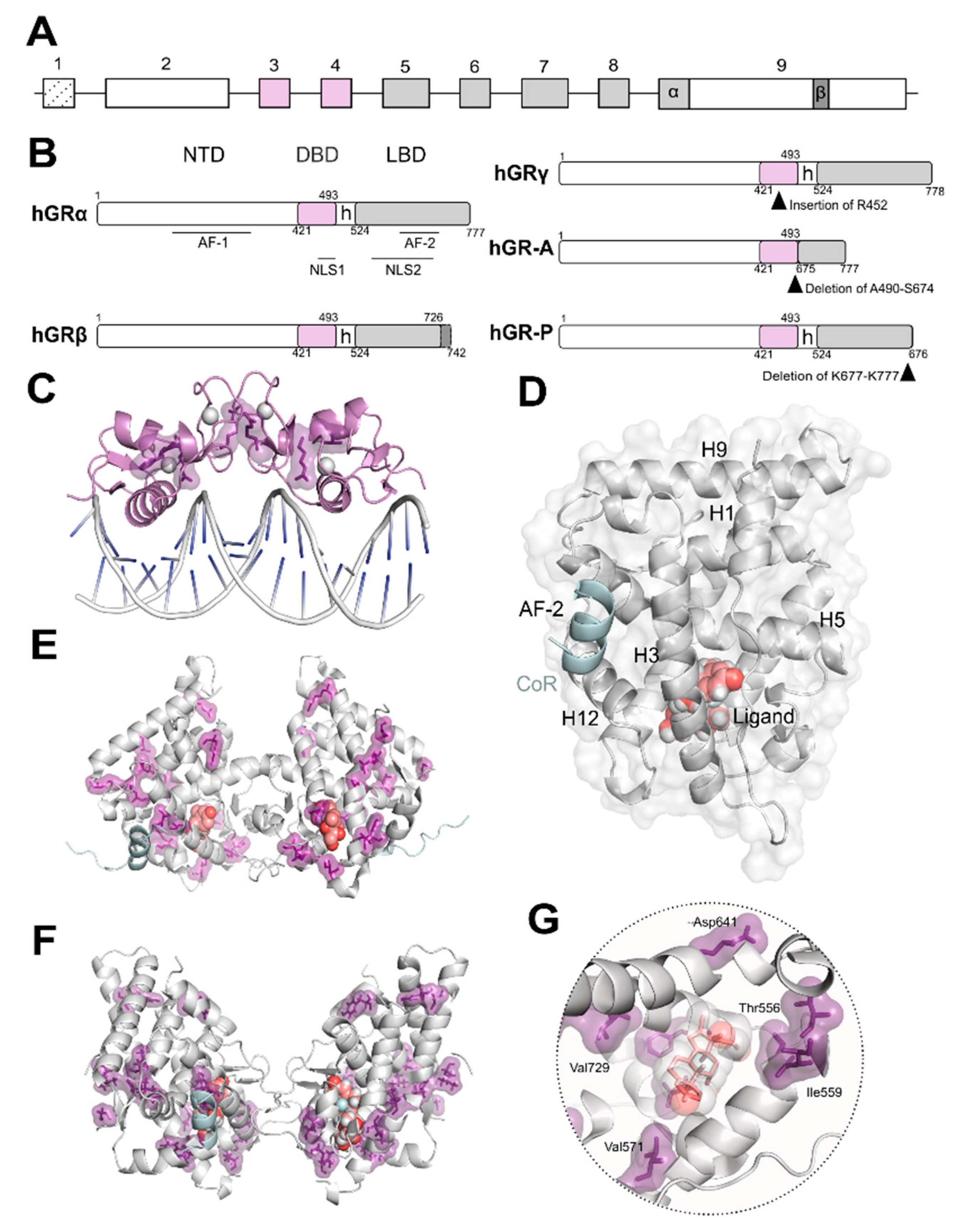{kind=link}
{kind=link}
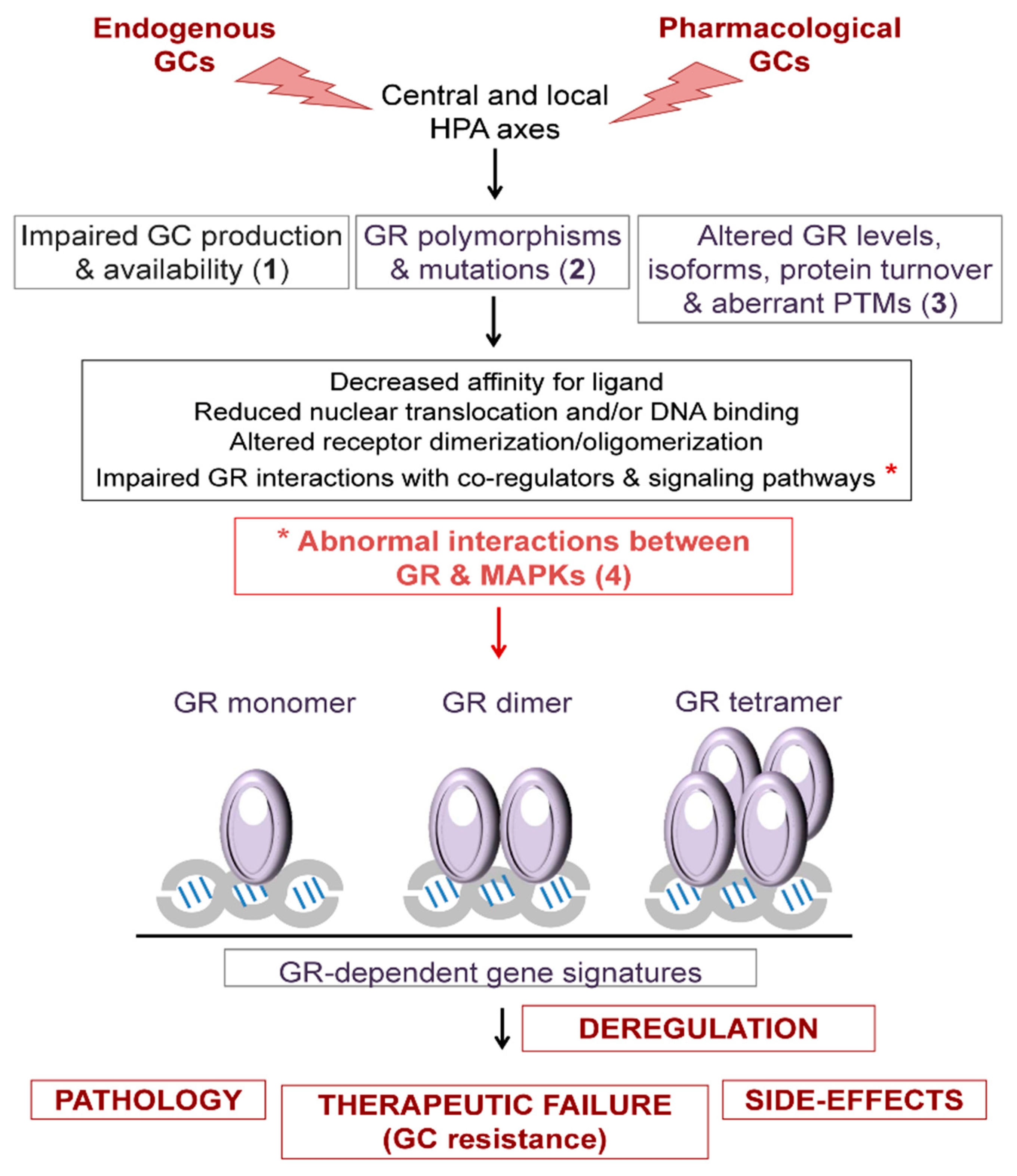{kind=link}
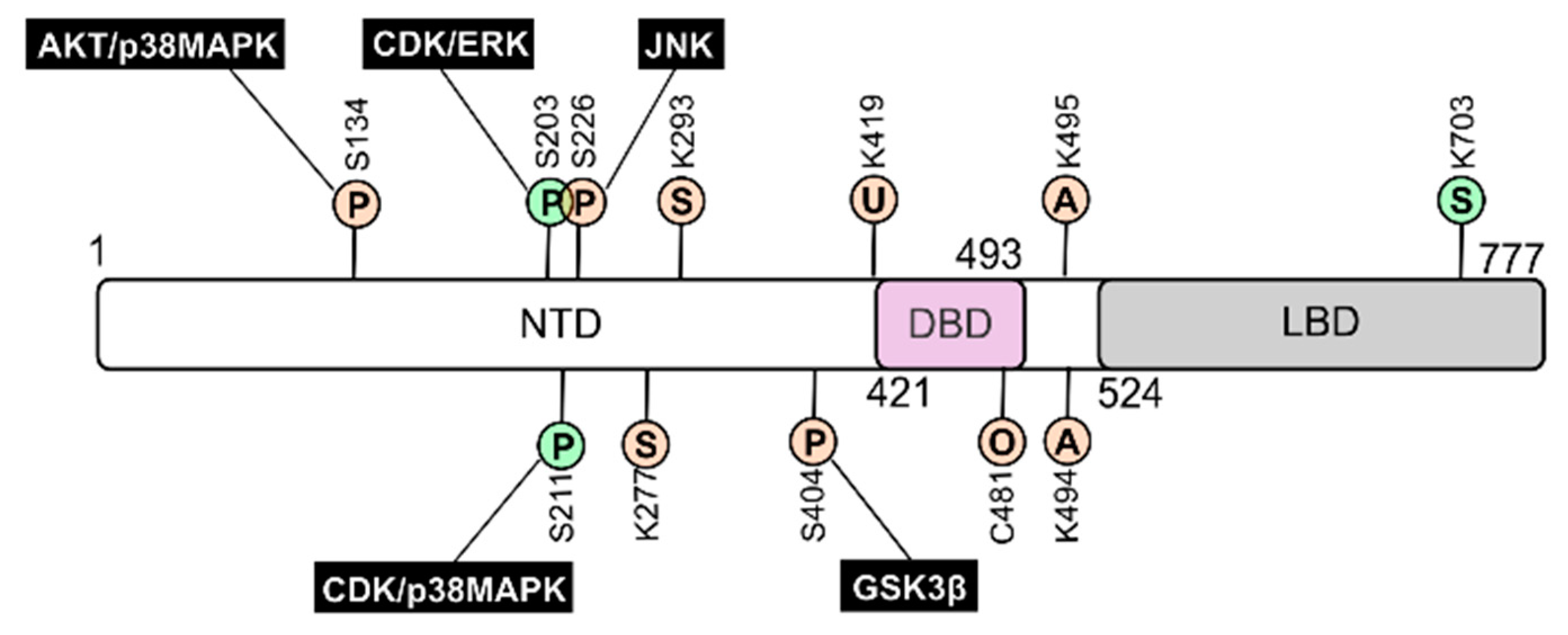{kind=link}
Abstract
:1. Introduction
2. Structure and Function of the Glucocorticoid Receptor (GR)
3. Mutual Interference between GR and Mitogen-Activated Protein Kinases (MAPKs)
4. Overview of the Mechanisms That Regulate Glucocorticoid (GC) Sensitivity
4.1. GC Production and Availability
4.2. GR Polymorphisms and Mutations
4.3. GR Levels, Isoforms, Protein Turnover, and Post-Translational Modifications
5. GC Resistance due to the Crosstalk between GR and MAPK Signalling
5.1. Respiratory Diseases
5.2. Leukemias
5.3. Skin Diseases
5.4. Autoimmune Diseases
6. Conclusions and Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACTH | Adrenocorticotropic hormone |
| AF | Activation function |
| ALL | Acute lymphoblastic leukaemia |
| COPD | Chronic obstructive pulmonary disease |
| CRH | Corticotropin-releasing hormone |
| DBD | DNA-binding domain |
| DUSP | Dual-Specific Phosphatase |
| EM | Electron microscopy |
| ERK | Extracellular Signal-Regulated kinases |
| GCs | Glucocorticoids |
| GILZ | Glucocorticoid induced leucine zipper |
| GR | GC receptor |
| GRE | GC response element |
| GSK | Glycogen synthase kinase |
| HPA | Hypothalamic-pituitary-adrenal axis |
| HSD11B | 11 beta hydroxysteroid dehydrogenase |
| IBD | Inflammatory bowel diseases |
| IL | Interleukin |
| JNK | c-Jun N-terminal kinase |
| LBD | Ligand-binding domain |
| MAPK | Mitogen-Activated Protein Kinase |
| mTOR | Mammalian target of rapamycin |
| NLS | Nuclear localization signal |
| NR | Nuclear receptor |
| PBMC | Peripheral blood mononuclear cells |
| PTM | Post-translational modification |
| TF | Transcription factor |
| TNF-α | Tumor necrosis factor α |
References
- Granner, D.K.; Wang, J.C.; Yamamoto, K.R. Regulatory Actions of Glucocorticoid Hormones: From Organisms to Mechanisms. Adv. Exp. Med. Biol. 2015, 872, 3–31. [Google Scholar]
- Cain, D.W.; Cidlowski, J.A. Specificity and Sensitivity of Glucocorticoid Signaling in Health and Disease. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 545–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whirledge, S.; DeFranco, D.B. Glucocorticoid Signaling in Health and Disease: Insights from Tissue-Specific GR Knockout Mice. Endocrinology 2018, 159, 46–61. [Google Scholar] [CrossRef] [PubMed]
- Timmermans, S.; Souffriau, J.; Libert, C. A General Introduction to Glucocorticoid Biology. Front. Immunol. 2019, 10, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Nicolaides, N.C.; Charmandari, E. Novel Insights into the Molecular Mechanisms Underlying Generalized Glucocorticoid Resistance and Hypersensitivity Syndromes. Hormones 2017, 16, 124–138. [Google Scholar] [PubMed] [Green Version]
- Quax, R.A.; Manenschijn, L.; Koper, J.W.; Hazes, J.M.; Lamberts, S.W.J.; Van Rossum, E.F.C.; Feelders, R.A. Glucocorticoid Sensitivity in Health and Disease. Nat. Rev. Endocrinol. 2013, 9, 670–686. [Google Scholar] [CrossRef]
- Talabér, G.; Jondal, M.; Okret, S. Extra-Adrenal Glucocorticoid Synthesis: Immune Regulation and Aspects on Local Organ Homeostasis. Mol. Cell. Endocrinol. 2013, 380, 89–98. [Google Scholar] [CrossRef]
- Slominski, R.M.; Tuckey, R.C.; Manna, P.R.; Jetten, A.M.; Postlethwaite, A.; Raman, C.; Slominski, A.T. Extra-Adrenal Glucocorticoid Biosynthesis: Implications for Autoimmune and Inflammatory Disorders. Genes Immun. 2020, 21, 150–168. [Google Scholar] [CrossRef]
- Merk, V.M.; Phan, T.S.; Brunner, T. Regulation of Tissue Immune Responses by Local Glucocorticoids at Epithelial Barriers and Their Impact on Interorgan Crosstalk. Front. Immunol. 2021, 12, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Vandewalle, J.; Luypaert, A.; De Bosscher, K.; Libert, C. Therapeutic Mechanisms of Glucocorticoids. Trends Endocrinol. Metab. 2018, 29, 42–54. [Google Scholar] [CrossRef]
- Meijer, O.C.; Pereira, A.M. Three Percent Annually on Systemic Glucocorticoids: Facts, Worries and Perspectives. Eur. J. Endocrinol. 2019, 181, C23–C28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitellius, G.; Lombes, M. Genetics in Endocrinology: Glucocorticoid Resistance Syndrome. Eur. J. Endocrinol. 2020, 182, R15–R27. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J.; Adcock, I.M. Glucocorticoid Resistance in Inflammatory Diseases. Lancet 2009, 373, 1905–1917. [Google Scholar] [CrossRef]
- Rodriguez, J.M.; Monsalves-Alvarez, M.; Henriquez, S.; Llanos, M.N.; Troncoso, R. Glucocorticoid Resistance in Chronic Diseases. Steroids 2016, 115, 182–192. [Google Scholar] [CrossRef]
- Newton, R.; Shah, S.; Altonsy, M.O.; Gerber, A.N. Glucocorticoid and Cytokine Crosstalk: Feedback, Feedforward, and Co-Regulatory Interactions Determine Repression or Resistance. J. Biol. Chem. 2017, 292, 7163–7172. [Google Scholar] [CrossRef] [Green Version]
- Kadmiel, M.; Cidlowski, J.A. Glucocorticoid Receptor Signaling in Health and Disease. Trends Pharmacol. Sci. 2013, 34, 518–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandevyver, S.; Dejager, L.; Libert, C. Comprehensive Overview of the Structure and Regulation of the Glucocorticoid Receptor. Endocr. Rev. 2014, 35, 671–693. [Google Scholar] [CrossRef] [Green Version]
- Weikum, E.R.; Knuesel, M.T.; Ortlund, E.A.; Yamamoto, K.R. Glucocorticoid Receptor Control of Transcription: Precision and Plasticity via Allostery. Nat. Rev. Mol. Cell Biol. 2017, 18, 159–174. [Google Scholar] [CrossRef]
- Gomez-Sanchez, E.; Gomez-Sanchez, C.E. The Multifaceted Mineralocorticoid Receptor. Compr. Physiol. 2014, 4, 965–994. [Google Scholar]
- Jaisser, F.; Farman, N. Emerging Roles of the Mineralocorticoid Receptor in Pathology: Toward New Paradigms in Clinical Pharmacology. Pharmacol. Rev. 2016, 68, 49–75. [Google Scholar] [CrossRef] [Green Version]
- Gathercole, L.L.; Lavery, G.G.; Morgan, S.A.; Cooper, M.S.; Sinclair, A.J.; Tomlinson, J.W.; Stewart, P.M. 11β-Hydroxysteroid Dehydrogenase 1: Translational and Therapeutic Aspects. Endocr. Rev. 2013, 34, 525–555. [Google Scholar] [CrossRef] [Green Version]
- Chapman, K.; Holmes, M.; Seckl, J. 11β-Hydroxysteroid Dehydrogenases Intracellular Gate-Keepers of Tissue Glucocorticoid Action. Physiol. Rev. 2013, 93, 1139–1206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duma, D.; Cidlowski, J.A.; Cidlowski, J.A. Generating Diversity in Glucocorticoid Receptor Signaling: Mechanisms, Receptor Isoforms, and Post-Translational Modifications. Horm. Mol. Biol. Clin. Investig. 2010, 3, 319–328. [Google Scholar] [CrossRef]
- Oakley, R.H.; Ramamoorthy, S.; Foley, J.F.; Busada, J.T.; Lu, N.Z.; Cidlowski, J.A. Glucocorticoid Receptor Isoform-Specific Regulation of Development, Circadian Rhythm, and Inflammation in Mice. FASEB J. 2018, 32, 5258–5271. [Google Scholar] [CrossRef]
- De Bosscher, K.; Desmet, S.J.; Clarisse, D.; Estébanez-Perpiña, E.; Brunsveld, L. Nuclear Receptor Crosstalk—Defining the Mechanisms for Therapeutic Innovation. Nat. Rev. Endocrinol. 2020, 16, 363–377. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Chen, J.; Wei, W. Dimerization of Glucocorticoid Receptors and Its Role in Inflammation and Immune Responses. Pharmacol. Res. 2021, 166, 105334. [Google Scholar] [CrossRef]
- Presman, D.M.; Hager, G.L. More than Meets the Dimer: What Is the Quaternary Structure of the Glucocorticoid Receptor? Transcription 2017, 8, 32–39. [Google Scholar] [CrossRef] [Green Version]
- Frank, F.; Okafor, C.D.; Ortlund, E.A. The First Crystal Structure of a DNA-Free Nuclear Receptor DNA Binding Domain Sheds Light on DNA-Driven Allostery in the Glucocorticoid Receptor. Sci. Rep. 2018, 8, 13497. [Google Scholar] [CrossRef]
- Bianchetti, L.; Wassmer, B.; Defosset, A.; Smertina, A.; Tiberti, M.L.; Stote, R.H.; Dejaegere, A. Alternative Dimerization Interfaces in the Glucocorticoid Receptor-α Ligand Binding Domain. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 1810–1825. [Google Scholar] [CrossRef] [PubMed]
- Presman, D.M.; Ganguly, S.; Schiltz, R.L.; Johnson, T.A.; Karpova, T.S.; Hager, G.L. DNA Binding Triggers Tetramerization of the Glucocorticoid Receptor in Live Cells. Proc. Natl. Acad. Sci. USA 2016, 113, 8236–8241. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Yi, P.; Hamilton, R.A.; Shen, H.; Chen, M.; Foulds, C.E.; Mancini, M.A.; Ludtke, S.J.; Wang, Z.; O’Malley, B.W. Structural Insights of Transcriptionally Active, Full-Length Androgen Receptor Coactivator Complexes. Mol. Cell 2020, 79, 812–823.e4. [Google Scholar] [CrossRef] [PubMed]
- Kirschke, E.; Goswami, D.; Southworth, D.; Griffin, P.R.; Agard, D.A. Glucocorticoid Receptor Function Regulated by Coordinated Action of the Hsp90 and Hsp70 Chaperone Cycles. Cell 2014, 157, 1685–1697. [Google Scholar] [CrossRef] [Green Version]
- Ratman, D.; Vanden Berghe, W.; Dejager, L.; Libert, C.; Tavernier, J.; Beck, I.M.; De Bosscher, K. How Glucocorticoid Receptors Modulate the Activity of Other Transcription Factors: A Scope beyond Tethering. Mol. Cell. Endocrinol. 2013, 380, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Petta, I.; Dejager, L.; Ballegeer, M.; Lievens, S.; Tavernier, J.; De Bosscher, K.; Libert, C. The Interactome of the Glucocorticoid Receptor and Its Influence on the Actions of Glucocorticoids in Combatting Inflammatory and Infectious Diseases. Microbiol. Mol. Biol. Rev. 2016, 80, 495–522. [Google Scholar] [CrossRef] [Green Version]
- Quatrini, L.; Ugolini, S. New Insights into the Cell- and Tissue-Specificity of Glucocorticoid Actions. Cell. Mol. Immunol. 2021, 18, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Sacta, M.A.; Chinenov, Y.; Rogatsky, I. Glucocorticoid Signaling: An Update from a Genomic Perspective. Annu. Rev. Physiol. 2016, 78, 155–180. [Google Scholar] [CrossRef]
- Franco, L.M.; Gadkari, M.; Howe, K.N.; Sun, J.; Kardava, L.; Kumar, P.; Kumari, S.; Hu, Z.; Fraser, I.D.C.; Moir, S.; et al. Immune Regulation by Glucocorticoids Can Be Linked to Cell Type–Dependent Transcriptional Responses. J. Exp. Med. 2019, 216, 384–406. [Google Scholar] [CrossRef] [Green Version]
- Desmet, S.J.; De Bosscher, K. Glucocorticoid Receptors: Finding the Middle Ground. J. Clin. Investig. 2017, 127, 1136–1145. [Google Scholar] [CrossRef]
- Escoter-Torres, L.; Caratti, G.; Mechtidou, A.; Tuckermann, J.; Uhlenhaut, N.H.; Vettorazzi, S. Fighting the Fire: Mechanisms of Inflammatory Gene Regulation by the Glucocorticoid Receptor. Front. Immunol. 2019, 10, 1–17. [Google Scholar] [CrossRef]
- Surjit, M.; Ganti, K.P.; Mukherji, A.; Ye, T.; Hua, G.; Metzger, D.; Li, M.; Chambon, P. Widespread Negative Response Elements Mediate Direct Repression by Agonist-Liganded Glucocorticoid Receptor. Cell 2011, 145, 224–241. [Google Scholar] [CrossRef] [Green Version]
- Cain, D.W.; Cidlowski, J.A. Immune Regulation by Glucocorticoids. Nat. Rev. Immunol. 2017, 17, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Johnson, G.L.; Lapadat, R. Mitogen-Activated Protein Kinase Pathways Mediated by ERK, JNK, and P38 Protein Kinases. Science 2002, 298, 1911–1912. [Google Scholar] [CrossRef] [Green Version]
- Kyriakis, J.M.; Avruch, J. Mammalian MAPK Signal Transduction Pathways Activated by Stress and Inflammation: A 10-Year Update. Physiol. Rev. 2012, 92, 689–737. [Google Scholar] [CrossRef] [Green Version]
- Lang, R.; Raffi, F.A.M. Dual-Specificity Phosphatases in Immunity and Infection: An Update. Int. J. Mol. Sci. 2019, 20, 2710. [Google Scholar] [CrossRef] [Green Version]
- Hoppstädter, J.; Ammit, A.J. Role of Dual-Specificity Phosphatase 1 in Glucocorticoid-Driven Antiinflammatory Responses. Front. Immunol. 2019, 10, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Beck, I.M.E.; Vanden Berghe, W.; Vermeulen, L.; Yamamoto, K.R.; Haegeman, G.; De Bosscher, K. Crosstalk in Inflammation: The Interplay of Glucocorticoid Receptor-Based Mechanisms and Kinases and Phosphatases. Endocr. Rev. 2009, 30, 830–882. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Tilló, E.; Comalada, M.; Xaus, J.; Farrera, C.; Valledor, A.F.; Caelles, C.; Lloberas, J.; Celada, A. JNK1 Is Required for the Induction of Mkp1 Expression in Macrophages during Proliferation and Lipopolysaccharide-Dependent Activation. J. Biol. Chem. 2007, 282, 12566–12573. [Google Scholar] [CrossRef] [Green Version]
- Casals-Casas, C.; Álvarez, E.; Serra, M.; de la Torre, C.; Farrera, C.; Sánchez-Tilló, E.; Caelles, C.; Lloberas, J.; Celada, A. CREB and AP-1 Activation Regulates MKP-1 Induction by LPS or M-CSF and Their Kinetics Correlate with Macrophage Activation versus Proliferation. Eur. J. Immunol. 2009, 39, 1902–1913. [Google Scholar] [CrossRef]
- Chi, H.; Barry, S.P.; Roth, R.J.; Wu, J.J.; Jones, E.A.; Bennett, A.M.; Flavell, R.A. Dynamic Regulation of Pro- and Anti-Inflammatory Cytokines by MAPK Phosphatase 1 (MKP-1) in Innate Immune Responses. Proc. Natl. Acad. Sci. USA 2006, 103, 2274–2279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salojin, K.V.; Owusu, I.B.; Millerchip, K.A.; Potter, M.; Platt, K.A.; Oravecz, T. Essential Role of MAPK Phosphatase-1 in the Negative Control of Innate Immune Responses. J. Immunol. 2006, 176, 1899–1907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caelles, C.; Bruna, A.; Morales, M.; González-Sancho, J.M.; González, M.V.; Jiménez, B.; Muñoz, A. Glucocorticoid Receptor Antagonism of AP-1 Activity by Inhibition of MAPK Family. In Recent Advances in Glucocorticoid Receptor Action. Ernst Schering Research Foundation Workshop; Cato, A.C.B., Schäcke, H., Asadullah, K., Eds.; Springer: Berlin/Heidelberg, Germany, 2002; pp. 131–152. [Google Scholar]
- Ayroldi, E.; Cannarile, L.; Migliorati, G.; Nocentini, G.; Delfino, D.V.; Riccardi, C. Mechanisms of the Anti-Inflammatory Effects of Glucocorticoids: Genomic and Nongenomic Interference with MAPK Signaling Pathways. FASEB J. 2012, 26, 4805–4820. [Google Scholar] [CrossRef] [PubMed]
- Caelles, C.; González-Sancho, J.M.; Muñoz, A. Nuclear Hormone Receptor Antagonism with AP-1 by Inhibition of the JNK Pathway. Genes Dev. 1997, 11, 3351–3364. [Google Scholar] [CrossRef] [Green Version]
- Swantek, J.L.; Cobb, M.H.; Geppert, T.D. Jun N-Terminal Kinase/Stress-Activated Protein Kinase (JNK/SAPK) Is Required for Lipopolysaccharide Stimulation of Tumor Necrosis Factor Alpha (TNF-Alpha) Translation: Glucocorticoids Inhibit TNF-Alpha Translation by Blocking JNK/SAPK. Mol. Cell. Biol. 1997, 17, 6274–6282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kassel, O.; Sancono, A.; Krätzschmar, J.; Kreft, B.; Stassen, M.; Cato, A.C.B. Glucocorticoids Inhibit MAP Kinase via Increased Expression and Decreased Degradation of MKP-1. EMBO J. 2001, 20, 7108–7116. [Google Scholar] [CrossRef] [Green Version]
- Lasa, M.; Abraham, S.M.; Boucheron, C.; Saklatvala, J.; Clark, A.R. Dexamethasone Causes Sustained Expression of Mitogen-Activated Protein Kinase (MAPK) Phosphatase 1 and Phosphatase-Mediated Inhibition of MAPK P38. Mol. Cell. Biol. 2002, 22, 7802–7811. [Google Scholar] [CrossRef] [Green Version]
- González, M.V.; Jiménez, B.; Berciano, M.T.; González-Sancho, J.M.; Caelles, C.; Lafarga, M.; Muñoz, A. Glucocorticoids Antagonize AP-1 by Inhibiting the Activation/Phosphorylation of JNK without Affecting Its Subcellular Distribution. J. Cell Biol. 2000, 150, 1199–1207. [Google Scholar] [CrossRef] [Green Version]
- Bruna, A.; Nicolàs, M.; Muñoz, A.; Kyriakis, J.M.; Caelles, C. Glucocorticoid Receptor-JNK Interaction Mediates Inhibition of the JNK Pathway by Glucocorticoids. EMBO J. 2003, 22, 6035–6044. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.; King, E.M.; Chandrasekhar, A.; Newton, R. Roles for the Mitogen-Activated Protein Kinase (MAPK) Phosphatase, DUSP1, in Feedback Control of Inflammatory Gene Expression and Repression by Dexamethasone. J. Biol. Chem. 2014, 289, 13667–13679. [Google Scholar] [CrossRef] [Green Version]
- Ayroldi, E.; Zollo, O.; Macchiarulo, A.; Di Marco, B.; Marchetti, C.; Riccardi, C. Glucocorticoid-Induced Leucine Zipper Inhibits the Raf-Extracellular Signal-Regulated Kinase Pathway by Binding to Raf-1. Mol. Cell. Biol. 2002, 22, 7929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkinson, L.; Verhoog, N.J.D.; Louw, A. Disease-and Treatment-Associated Acquired Glucocorticoid Resistance. Endocr. Connect. 2018, 7, R328–R349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martins, C.S.; de Castro, M. Generalized and Tissue Specific Glucocorticoid Resistance. Mol. Cell. Endocrinol. 2021, 530, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Mariotti, A. The Effects of Chronic Stress on Health: New Insights into the Molecular Mechanisms of Brain–Body Communication. Futur. Sci. OA 2015, 1, 3. [Google Scholar] [CrossRef] [Green Version]
- Ferraù, F.; Korbonits, M. Metabolic Syndrome in Cushing’s Syndrome Patients. Front. Horm. Res. 2018, 49, 85–103. [Google Scholar]
- Zhang, D.; Bergsneider, M.; Wang, M.B.; Heaney, A.P.; Zhang, D.; Bergsneider, M.; Wang, M.B.; Heaney, A.P. Targeting the ERK Pathway for the Treatment of Cushing’s Disease. Oncotarget 2016, 7, 69149–69158. [Google Scholar] [CrossRef] [Green Version]
- Charmandari, E.; Ichijo, T.; Jubiz, W.; Baid, S.; Zachman, K.; Chrousos, G.P.; Kino, T. A Novel Point Mutation in the Amino Terminal Domain of the Human Glucocorticoid Receptor (HGR) Gene Enhancing HGR-Mediated Gene Expression. J. Clin. Endocrinol. Metab. 2008, 93, 4963–4968. [Google Scholar] [CrossRef] [Green Version]
- Santen, R.J.; Jewell, C.M.; Yue, W.; Heitjan, D.F.; Raff, H.; Katen, K.S.; Cidlowski, J.A. Glucocorticoid Receptor Mutations and Hypersensitivity to Endogenous and Exogenous Glucocorticoids. J. Clin. Endocrinol. Metab. 2018, 103, 3630–3639. [Google Scholar] [CrossRef] [PubMed]
- Hurt, D.E.; Suzuki, S.; Mayama, T.; Charmandari, E.; Kino, T. Structural Analysis on the Pathologic Mutant Glucocorticoid Receptor Ligand-Binding Domains. Mol. Endocrinol. 2016, 30, 173–188. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Li, P.; Zhang, Q.; Lv, H.; Liu, J.; Si, J. Interleukin-1β Regulates the Expression of Glucocorticoid Receptor Isoforms in Nasal Polyps in Vitro via P38 MAPK and JNK Signal Transduction Pathways. J. Inflamm. 2015, 12, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallace, A.D.; Cidlowski, J.A. Proteasome-Mediated Glucocorticoid Receptor Degradation Restricts Transcriptional Signaling by Glucocorticoids. J. Biol. Chem. 2001, 276, 42714–42721. [Google Scholar] [CrossRef] [Green Version]
- Wallace, A.D.; Cao, Y.; Chandramouleeswaran, S.; Cidlowski, J.A. Lysine 419 Targets Human Glucocorticoid Receptor for Proteasomal Degradation. Steroids 2010, 75, 1016–1023. [Google Scholar] [CrossRef] [Green Version]
- Galliher-Beckley, A.J.; Williams, J.G.; Cidlowski, J.A. Ligand-Independent Phosphorylation of the Glucocorticoid Receptor Integrates Cellular Stress Pathways with Nuclear Receptor Signaling. Mol. Cell. Biol. 2011, 31, 4663–4675. [Google Scholar] [CrossRef] [Green Version]
- Piovan, E.; Yu, J.; Tosello, V.; Herranz, D.; Ambesi-Impiombato, A.; DaSilva, A.C.; Sanchez-Martin, M.; Perez-Garcia, A.; Rigo, I.; Castillo, M.; et al. Direct Reversal of Glucocorticoid Resistance by AKT Inhibition in Acute Lymphoblastic Leukemia. Cancer Cell 2013, 24, 766–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ismaili, N.; Garabedian, M.J. Modulation of Glucocorticoid Receptor Function via Phosphorylation. Ann. N. Y. Acad. Sci. 2004, 1024, 86–101. [Google Scholar] [CrossRef]
- Itoh, M.; Adachi, M.; Yasui, H.; Takekawa, M.; Tanaka, H.; Imai, K. Nuclear Export of Glucocorticoid Receptor Is Enhanced by C-Jun N-Terminal Kinase-Mediated Phosphorylation. Mol. Endocrinol. 2002, 16, 2382–2392. [Google Scholar] [CrossRef] [PubMed]
- Galliher-Beckley, A.J.; Williams, J.G.; Collins, J.B.; Cidlowski, J.A. Glycogen Synthase Kinase 3β-Mediated Serine Phosphorylation of the Human Glucocorticoid Receptor Redirects Gene Expression Profiles. Mol. Cell. Biol. 2008, 28, 7309–7322. [Google Scholar] [CrossRef] [Green Version]
- Ito, K.; Yamamura, S.; E-Quaye, S.E.; Cosio, B.; Ito, M.; Barnes, P.J.; Adcock, I.M. Histone Deacetylase 2–Mediated Deacetylation of the Glucocorticoid Receptor Enables NF-ΚB Suppression. J. Exp. Med. 2006, 203, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Nader, N.; Chrousos, G.P.; Kino, T. Circadian Rhythm Transcription Factor CLOCK Regulates the Transcriptional Activity of the Glucocorticoid Receptor by Acetylating Its Hinge Region Lysine Cluster: Potential Physiological Implications. FASEB J. 2009, 23, 1572–1583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kassi, E.; Papavassiliou, A.G. Glucose Can Promote a Glucocorticoid Resistance State. J. Cell. Mol. Med. 2012, 16, 1146–1149. [Google Scholar] [CrossRef]
- Druker, J.; Liberman, A.C.; Antunica-Noguerol, M.; Gerez, J.; Paez-Pereda, M.; Rein, T.; Iñiguez-Lluhí, J.A.; Holsboer, F.; Arzt, E. RSUME Enhances Glucocorticoid Receptor SUMOylation and Transcriptional Activity. Mol. Cell. Biol. 2013, 33, 2116–2127. [Google Scholar] [CrossRef] [Green Version]
- Paakinaho, V.; Kaikkonen, S.; Makkonen, H.; Benes, V.; Palvimo, J.J. SUMOylation Regulates the Chromatin Occupancy and Anti-Proliferative Gene Programs of Glucocorticoid Receptor. Nucleic Acids Res. 2014, 42, 1575–1592. [Google Scholar] [CrossRef] [Green Version]
- Davies, L.; Karthikeyan, N.; Lynch, J.T.; Sial, E.A.; Gkourtsa, A.; Demonacos, C.; Krstic-Demonacos, M. Cross Talk of Signaling Pathways in the Regulation of the Glucocorticoid Receptor Function. Mol. Endocrinol. 2008, 22, 1331–1344. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, K.; Tanaka, H.; Ogawa, H.; Makino, Y.; Eguchi, H.; Hayashi, S.I.; Yoshikawa, N.; Poellinger, L.; Umesono, K.; Makino, I. Redox-Dependent Regulation of Nuclear Import of the Glucocorticoid Receptor. J. Biol. Chem. 1999, 274, 10363–10371. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, Y.; Ito, K.; Kanda, A.; Tomoda, K.; Mercado, N.; Barnes, P.J. Impaired Dual-Specificity Protein Phosphatase DUSP4 Reduces Corticosteroid Sensitivity. Mol. Pharmacol. 2017, 91, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Khorasanizadeh, M.H.; Eskian, M.; Gelfand, E.W.; Rezaei, N. Mitogen-Activated Protein Kinases as Therapeutic Targets for Asthma. Pharmacol. Ther. 2017, 174, 112–126. [Google Scholar] [CrossRef]
- Barnes, P.J. Kinases as Novel Therapeutic Targets in Asthma and Chronic Obstructive Pulmonary Disease. Pharmacol. Rev. 2016, 68, 788–815. [Google Scholar] [CrossRef] [Green Version]
- Khorasani, N.; Baker, J.; Johnson, M.; Chung, K.F.; Bhavsar, P.K. Reversal of Corticosteroid Insensitivity by P38 MAPK Inhibition in Peripheral Blood Mononuclear Cells from COPD. Int. J. COPD 2015, 10, 283–291. [Google Scholar]
- Abraham, S.M.; Lawrence, T.; Kleiman, A.; Warden, P.; Medghalchi, M.; Tuckermann, J.; Saklatvala, J.; Clark, A.R. Antiinflammatory Effects of Dexamethasone Are Partly Dependent on Induction of Dual Specificity Phosphatase 1. J. Exp. Med. 2006, 203, 1883–1889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maneechotesuwan, K.; Yao, X.; Ito, K.; Jazrawi, E.; Usmani, O.S.; Adcock, I.M.; Barnes, P.J. Suppression of GATA-3 Nuclear Import and Phosphorylation: A Novel Mechanism of Corticosteroid Action in Allergic Disease. PLoS Med. 2009, 6, e1000076. [Google Scholar] [CrossRef] [Green Version]
- Mei, D.; Tan, W.S.D.; Wong, W.S.F. Pharmacological Strategies to Regain Steroid Sensitivity in Severe Asthma and COPD. Curr. Opin. Pharmacol. 2019, 46, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Mitani, A.; Ito, K.; Vuppusetty, C.; Barnes, P.J.; Mercado, N. Restoration of Corticosteroid Sensitivity in Chronic Obstructive Pulmonary Disease by Inhibition of Mammalian Target of Rapamycin. Am. J. Respir. Crit. Care Med. 2016, 193, 143–153. [Google Scholar] [CrossRef] [Green Version]
- Li, L.B.; Goleva, E.; Hall, C.F.; Ou, L.S.; Leung, D.Y.M. Superantigen-Induced Corticosteroid Resistance of Human T Cells Occurs through Activation of the Mitogen-Activated Protein Kinase Kinase/Extracellular Signal-Regulated Kinase (MEK-ERK) Pathway. J. Allergy Clin. Immunol. 2004, 114, 1059–1069. [Google Scholar] [CrossRef] [PubMed]
- Pelaia, C.; Vatrella, A.; Sciacqua, A.; Terracciano, R.; Pelaia, G. Role of P38-Mitogen-Activated Protein Kinase in COPD: Pathobiological Implications and Therapeutic Perspectives. Expert Rev. Respir. Med. 2020, 14, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Pniewska, E.; Pawliczak, R. The Involvement of Phospholipases A2 in Asthma and Chronic Obstructive Pulmonary Disease. Mediators Inflamm. 2013, 2013, 793505–793516. [Google Scholar] [CrossRef] [Green Version]
- Kitsiouli, E.; Nakos, G.; Lekka, M.E. Phospholipase A2 Subclasses in Acute Respiratory Distress Syndrome. Biochim. Biophys. Acta 2009, 1792, 941–953. [Google Scholar] [CrossRef] [Green Version]
- Garza, A.S.; Miller, A.L.; Johnson, B.H.; Thompson, E.B. Converting Cell Lines Representing Hematological Malignancies from Glucocorticoid-Resistant to Glucocorticoid-Sensitive: Signaling Pathway Interactions. Leuk. Res. 2009, 33, 717–727. [Google Scholar] [CrossRef]
- Lin, K.T.; Wang, L.H. New Dimension of Glucocorticoids in Cancer Treatment. Steroids 2016, 111, 84–88. [Google Scholar] [CrossRef] [Green Version]
- Bhadri, V.A.; Trahair, T.N.; Lock, R.B. Glucocorticoid Resistance in Paediatric Acute Lymphoblastic Leukaemia. J. Paediatr. Child Health. 2012, 48, 634–640. [Google Scholar] [CrossRef]
- Olivas-Aguirre, M.; Torres-López, L.; Pottosin, I.; Dobrovinskaya, O. Overcoming Glucocorticoid Resistance in Acute Lymphoblastic Leukemia: Repurposed Drugs Can Improve the Protocol. Front. Oncol. 2021, 11, 647. [Google Scholar] [CrossRef]
- Renner, K.; Amberger, A.; Konwalinka, G.; Kofler, R.; Gnaiger, E. Changes of Mitochondrial Respiration, Mitochondrial Content and Cell Size after Induction of Apoptosis in Leukemia Cells. Biochim. Biophys. Acta Mol. Cell Res. 2003, 1642, 115–123. [Google Scholar] [CrossRef] [Green Version]
- Tissing, W.J.E.; Den Boer, M.L.; Meijerink, J.P.P.; Menezes, R.X.; Swagemakers, S.; Van Der Spek, P.J.; Sallan, S.E.; Armstrong, S.A.; Pieters, R. Genomewide Identification of Prednisolone-Responsive Genes in Acute Lymphoblastic Leukemia Cells. Blood 2007, 109, 3929–3935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webb, M.S.; Miller, A.L.; Howard, T.L.; Johnson, B.H.; Chumakov, S.; Fofanov, Y.; Nguyen-Vu, T.; Lin, C.Y.; Thompson, E.B. Sequential Gene Regulatory Events Leading to Glucocorticoid-Evoked Apoptosis of CEM Human Leukemic Cells:Interactions of MAPK, MYC and Glucocorticoid Pathways. Mol. Cell. Endocrinol. 2018, 471, 118–130. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.L.; Webb, M.S.; Copik, A.J.; Wang, Y.; Johnson, B.H.; Kumar, R.; Thompson, E.B. P38 Mitogen-Activated Protein Kinase (MAPK) Is a Key Mediator in Glucocorticoid-Induced Apoptosis of Lymphoid Cells: Correlation between P38 MAPK Activation and Site-Specific Phosphorylation of the Human Glucocorticoid Receptor at Serine 211. Mol. Endocrinol. 2005, 19, 1569–1583. [Google Scholar] [CrossRef] [Green Version]
- Jones, C.L.; Gearheart, C.M.; Fosmire, S.; Delgado-Martin, C.; Evensen, N.A.; Bride, K.; Waanders, A.J.; Pais, F.; Wang, J.; Bhatla, T.; et al. MAPK Signaling Cascades Mediate Distinct Glucocorticoid Resistance Mechanisms in Pediatric Leukemia. Blood 2015, 126, 2202–2212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Planey, S.L.; Abrams, M.T.; Robertson, N.M.; Litwack, G. Role of Apical Caspases and Glucocorticoid-Regulated Genes in Glucocorticoid-Induced Apoptosis of Pre-B Leukemic Cells. Cancer Res. 2003, 63, 172–178. [Google Scholar]
- Abrams, M.T.; Robertson, N.M.; Litwack, G.; Wickstrom, E. Evaluation of Glucocorticoid Sensitivity in 697 Pre-B Acute Lymphoblastic Leukemia Cells after Overexpression or Silencing of MAP Kinase Phosphatase-1. J. Cancer Res. Clin. Oncol. 2005, 131, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ge, J.; Li, Q.; Guo, X.; Gu, L.; Ma, Z.G.; Li, X.H.; Zhu, Y.P. Low-Dose Anisomycin Sensitizes Glucocorticoid-Resistant T-Acute Lymphoblastic Leukemia CEM-C1 Cells to Dexamethasone-Induced Apoptosis through Activation of Glucocorticoid Receptor and P38-MAPK/JNK. Leuk. Lymphoma 2014, 55, 2179–2188. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, L.; Evans, C.A.; Matheson, E.; Minto, L.; Keilty, C.; Sanichar, M.; Case, M.; Schwab, C.; Williamson, D.; Rainer, J.; et al. Quantitative Proteomic Analysis Reveals Maturation as a Mechanism Underlying Glucocorticoid Resistance in B Lineage ALL and Re-Sensitization by JNK Inhibition. Br. J. Haematol. 2015, 171, 595–605. [Google Scholar] [CrossRef] [Green Version]
- Sevilla, L.M.; Pérez, P. Roles of the Glucocorticoid and Mineralocorticoid Receptors in Skin Pathophysiology. Int. J. Mol. Sci. 2018, 19, 1906. [Google Scholar] [CrossRef] [Green Version]
- Leung, D. Superantigens, Steroid Insensitivity and Innate Immunity in Atopic Eczema. Acta Derm.-Venereol. Suppl. 2005, 215, 11–15. [Google Scholar] [CrossRef]
- Taheri, A.; Cantrell, J.; Feldman, S.R. Tachyphylaxis to Topical Glucocorticoids; What Is the Evidence? Dermatol. Online J. 2013, 19, 7. [Google Scholar]
- Czarnowicki, T.; Linkner, R.V.; Suárez-Fariñas, M.; Ingber, A.; Lebwohl, M. An Investigator-Initiated, Double-Blind, Vehicle-Controlled Pilot Study: Assessment for Tachyphylaxis to Topically Occluded Halobetasol 0.05% Ointment in the Treatment of Psoriasis. J. Am. Acad. Dermatol. 2014, 71, 954–959.e1. [Google Scholar] [CrossRef] [PubMed]
- Chebotaev, D.V.; Yemelyanov, A.Y.; Lavker, R.M.; Budunova, I.V. Epithelial Cells in the Hair Follicle Bulge Do Not Contribute to Epidermal Regeneration after Glucocorticoid-Induced Cutaneous Atrophy. J. Investig. Dermatol. 2007, 127, 2749–2758. [Google Scholar] [CrossRef] [Green Version]
- Kishibe, M.; Baida, G.; Bhalla, P.; Lavker, R.M.; Schlosser, B.; Iinuma, S.; Yoshida, S.; Dudley, J.T.; Budunova, I. Important Role of Kallikrein 6 for the Development of Keratinocyte Proliferative Resistance to Topical Glucocorticoids. Oncotarget 2016, 7, 69479–69488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sevilla, L.M.; Bayo, P.; Latorre, V.; Sanchis, A.; Pérez, P. Glucocorticoid Receptor Regulates Overlapping and Differential Gene Subsets in Developing and Adult Skin. Mol. Endocrinol. 2010, 24, 2166–2178. [Google Scholar] [CrossRef] [Green Version]
- Sevilla, L.M.; Latorre, V.; Sanchis, A.; Pérez, P. Epidermal Inactivation of the Glucocorticoid Receptor Triggers Skin Barrier Defects and Cutaneous Inflammation. J. Investig. Dermatol. 2013, 133, 361–370. [Google Scholar] [CrossRef] [Green Version]
- Bigas, J.; Sevilla, L.M.; Carceller, E.; Boix, J.; Pérez, P. Epidermal Glucocorticoid and Mineralocorticoid Receptors Act Cooperatively to Regulate Epidermal Development and Counteract Skin Inflammation Article. Cell Death Dis. 2018, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Hua, G.; Ganti, K.P.; Chambon, P. Glucocorticoid-Induced Tethered Transrepression Requires SUMOylation of GR and Formation of a SUMO-SMRT/NCoR1-HDAC3 Repressing Complex. Proc. Natl. Acad. Sci. USA 2016, 113, E635–E643. [Google Scholar] [CrossRef] [Green Version]
- Hua, G.; Paulen, L.; Chambon, P. GR SUMOylation and Formation of an SUMO-SMRT/ NCoR1-HDAC3 Repressing Complex Is Mandatory for GC-Induced IR NGRE-Mediated Transrepression. Proc. Natl. Acad. Sci. USA 2016, 113, E626–E634. [Google Scholar] [CrossRef] [Green Version]
- Hobbs, R.M.; Silva-Vargas, V.; Groves, R.; Watt, F.M. Expression of Activated MEK1 in Differentiating Epidermal Cells Is Sufficient to Generate Hyperproliferative and Inflammatory Skin Lesions. J. Investig. Dermatol. 2004, 123, 503–515. [Google Scholar] [CrossRef] [Green Version]
- Mavropoulos, A.; Rigopoulou, E.I.; Liaskos, C.; Bogdanos, D.P.; Sakkas, L.I. The Role of P38 Mapk in the Aetiopathogenesis of Psoriasis and Psoriatic Arthritis. Clin. Dev. Immunol. 2013, 2013, 569751. [Google Scholar] [CrossRef]
- Hammouda, M.B.; Ford, A.E.; Liu, Y.; Zhang, J.Y. The JNK Signaling Pathway in Inflammatory Skin Disorders and Cancer. Cells 2020, 9, 857. [Google Scholar] [CrossRef] [Green Version]
- Hannen, R.; Udeh-Momoh, C.; Upton, J.; Wright, M.; Michael, A.; Gulati, A.; Rajpopat, S.; Clayton, N.; Halsall, D.; Burrin, J.; et al. Dysfunctional Skin-Derived Glucocorticoid Synthesis Is a Pathogenic Mechanism of Psoriasis. J. Investig. Dermatol. 2017, 137, 1630–1637. [Google Scholar] [CrossRef]
- Sarkar, M.K.; Kaplan, N.; Tsoi, L.C.; Xing, X.; Liang, Y.; Swindell, W.R.; Hoover, P.; Aravind, M.; Baida, G.; Clark, M.; et al. Endogenous Glucocorticoid Deficiency in Psoriasis Promotes Inflammation and Abnormal Differentiation. J. Investig. Dermatol. 2017, 137, 1474–1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jozic, I.; Stojadinovic, O.; Kirsner, R.S.; Tomic-Canic, M. Stressing the Steroids in Skin: Paradox or Fine-Tuning. J. Investig. Dermatol. 2014, 134, 2869–2872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terao, M.; Katayama, I. Local Cortisol/Corticosterone Activation in Skin Physiology and Pathology. J. Dermatol. Sci. 2016, 84, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Nikolakis, G.; Stratakis, C.A.; Kanaki, T.; Slominski, A.; Zouboulis, C.C. Skin Steroidogenesis in Health and Disease. Rev. Endocr. Metab. Disord. 2016, 17, 247–258. [Google Scholar] [CrossRef]
- Segrelles, C.; Ruiz, S.; Perez, P.; Murga, C.; Santos, M.; Budunova, I.V.; Martínez, J.; Larcher, F.; Slaga, T.J.; Gutkind, J.S.; et al. Functional Roles of Akt Signaling in Mouse Skin Tumorigenesis. Oncogene 2002, 21, 53–64. [Google Scholar] [CrossRef] [Green Version]
- Budunova, I.V.; Carbajal, S.; Kang, H., II; Viaje, A.; Slaga, T.J. Altered Glucocorticoid Receptor Expression and Function during Mouse Skin Carcinogenesis. Mol. Carcinog. 1997, 18, 177–185. [Google Scholar] [CrossRef]
- Latorre, V.; Sevilla, L.M.; Sanchis, A.; Pérez, P. Selective Ablation of Glucocorticoid Receptor in Mouse Keratinocytes Increases Susceptibility to Skin Tumorigenesis. J. Investig. Dermatol. 2013, 133, 2771–2779. [Google Scholar] [CrossRef] [Green Version]
- Leis, H.; Page, A.; Ramírez, A.; Bravo, A.; Segrelles, C.; Paramio, J.; Barettino, D.; Jorcano, J.L.; Pérez, P. Glucocorticoid Receptor Counteracts Tumorigenic Activity of Akt in Skin through Interference with the Phosphatidylinositol 3-Kinase Signaling Pathway. Mol. Endocrinol. 2004, 18, 303–311. [Google Scholar] [CrossRef] [Green Version]
- Straub, R.H. Rheumatoid Arthritis—A Neuroendocrine Immune Disorder: Glucocorticoid Resistance, Relative Glucocorticoid Defi Ciency, Low-Dose Glucocorticoid Therapy, and Insulin Resistance. Arthritis Res. Ther. 2014, 16 (Suppl. 2), I1. [Google Scholar] [CrossRef] [Green Version]
- Dubois-Camacho, K.; Ottum, P.A.; Franco-Muñoz, D.; De La Fuente, M.; Torres-Riquelme, A.; Díaz-Jiménez, D.; Olivares-Morales, M.; Astudillo, G.; Quera, R.; Hermoso, M.A. Glucocorticosteroid Therapy in Inflammatory Bowel Diseases: From Clinical Practice to Molecular Biology. World J. Gastroenterol. 2017, 23, 6628–6638. [Google Scholar]
- Ralph, J.A.; Morand, E.F. MAPK Phosphatases as Novel Targets for Rheumatoid Arthritis. Expert Opin. Ther. Targets 2008, 12, 795–808. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Kao, W.; Yang, Y.H.; Gu, R.; Harris, J.; Fingerle-Rowson, G.; Bucala, R.; Ngo, D.; Beaulieu, E.; Morand, E.F. Macrophage Migration Inhibitory Factor Inhibits the Antiinflammatory Effects of Glucocorticoids via Glucocorticoid-Induced Leucine Zipper. Arthritis Rheumatol. 2014, 66, 2059–2070. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Fan, H.; Ngo, D.; Beaulieu, E.; Leung, P.; Lo, C.Y.; Burgess, R.; van der Zwan, Y.G.; White, S.J.; Khachigian, L.M.; et al. GILZ Overexpression Inhibits Endothelial Cell Adhesive Function through Regulation of NF-ΚB and MAPK Activity. J. Immunol. 2013, 191, 424–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Iudicibus, S.; Franca, R.; Martelossi, S.; Ventura, A.; Decorti, G. Molecular Mechanism of Glucocorticoid Resistance in Inflammatory Bowel Disease. World J. Gastroenterol. 2011, 17, 1095–1108. [Google Scholar] [CrossRef]
- Lorén, V.; Cabré, E.; Ojanguren, I.; Domènech, E.; Pedrosa, E.; García-Jaraquemada, A.; Mañosa, M.; Manyé, J. Interleukin-10 Enhances the Intestinal Epithelial Barrier in the Presence of Corticosteroids through P38 MAPK Activity in Caco-2 Monolayers: A Possible Mechanism for Steroid Responsiveness in Ulcerative Colitis. PLoS ONE 2015, 10, e0130921. [Google Scholar]
- Fischer, A.; Gluth, M.; Weege, F.; Pape, U.F.; Wiedenmann, B.; Baumgart, D.C.; Theuring, F. Glucocorticoids Regulate Barrier Function and Claudin Expression in Intestinal Epithelial Cells via MKP-1. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 306, G218–G228. [Google Scholar] [CrossRef] [Green Version]
- Bantel, H.; Schmitz, M.L.; Raible, A.; Gregor, M.; Schulze-Osthoff, K. Critical Role of Nuclear Factor-κB and Stress-activated Protein Kinases in Steroid Unresponsiveness. FASEB J. 2002, 16, 1–19. [Google Scholar]
- Hommes, D.; Van Den Blink, B.; Plasse, T.; Bartelsman, J.; Xu, C.; Macpherson, B.; Tytgat, G.; Peppelenbosch, M.; Van Deventer, S. Inhibition of Stress-Activated MAP Kinases Induces Clinical Improvement in Moderate to Severe Crohn’s Disease. Gastroenterology 2002, 122, 7–14. [Google Scholar] [CrossRef]
- Ishiguro, Y.; Ohkawara, T.; Sakuraba, H.; Yamagata, K.; Hiraga, H.; Yamaguchi, S.; Fukuda, S.; Munakata, A.; Nakane, A.; Nishihira, J. Macrophage Migration Inhibitory Factor Has a Proinflammatory Activity via the P38 Pathway in Glucocorticoid-Resistant Ulcerative Colitis. Clin. Immunol. 2006, 120, 335–341. [Google Scholar] [CrossRef] [PubMed]
- He, Y.J.; Xu, J.Q.; Sun, M.M.; Fang, X.Z.; Peng, Z.K.; Pan, S.W.; Zhou, T.; Wang, Y.X.; Shang, Y. Glucocorticoid-Induced Leucine Zipper: A Promising Marker for Monitoring and Treating Sepsis. Front. Immunol. 2020, 11, 1–10. [Google Scholar] [CrossRef]
- Maitra, S.R.; Chen, E.; Rosa, D.; Valane, P.D.; El-Maghrabi, M.R.; Brathwaite, C.E.M. Modulations of Signal Transduction Pathways during Sepsis and the Effects of Insulin and Mifepristone. Acad. Emerg. Med. 2003, 10, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ballegeer, M.; Vandewalle, J.; Eggermont, M.; Van Isterdael, G.; Dejager, L.; De Bus, L.; Decruyenaere, J.; Vandenbroucke, R.E.; Libert, C. Overexpression of Gilz Protects Mice Against Lethal Septic Peritonitis. Shock 2019, 52, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Dendoncker, K.; Libert, C. Glucocorticoid Resistance as a Major Drive in Sepsis Pathology. Cytokine Growth Factor Rev. 2017, 35, 85–96. [Google Scholar] [CrossRef]
- Dendoncker, K.; Timmermans, S.; Vandewalle, J.; Eggermont, M.; Lempiäinen, J.; Van Hamme, E.; Dewaele, S.; Vandevyver, S.; Ballegeer, M.; Souffriau, J.; et al. TNF-α Inhibits Glucocorticoid Receptor-Induced Gene Expression by Reshaping the GR Nuclear Cofactor Profile. Proc. Natl. Acad. Sci. USA 2019, 116, 12942–12951. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sevilla, L.M.; Jiménez-Panizo, A.; Alegre-Martí, A.; Estébanez-Perpiñá, E.; Caelles, C.; Pérez, P. Glucocorticoid Resistance: Interference between the Glucocorticoid Receptor and the MAPK Signalling Pathways. Int. J. Mol. Sci. 2021, 22, 10049. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221810049
Sevilla LM, Jiménez-Panizo A, Alegre-Martí A, Estébanez-Perpiñá E, Caelles C, Pérez P. Glucocorticoid Resistance: Interference between the Glucocorticoid Receptor and the MAPK Signalling Pathways. International Journal of Molecular Sciences. 2021; 22(18):10049. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221810049
Chicago/Turabian StyleSevilla, Lisa M., Alba Jiménez-Panizo, Andrea Alegre-Martí, Eva Estébanez-Perpiñá, Carme Caelles, and Paloma Pérez. 2021. "Glucocorticoid Resistance: Interference between the Glucocorticoid Receptor and the MAPK Signalling Pathways" International Journal of Molecular Sciences 22, no. 18: 10049. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221810049