
Targeted Therapy in the Treatment of Pediatric Acute Lymphoblastic Leukemia—Therapy and Toxicity Mechanisms

 ,
,
Abstract
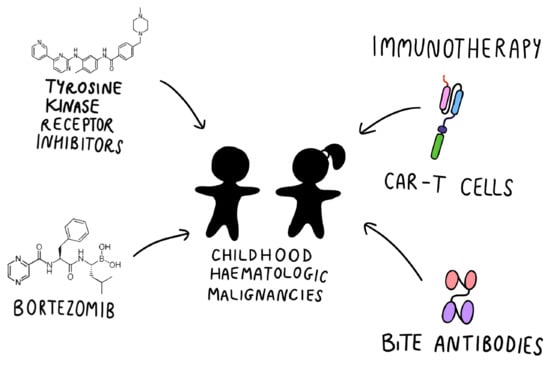
:
1. Introduction
1.1. Conventional Therapy
1.2. Minimal Residual Disease
1.3. Cytogenetic and Molecular Aberrations with Prognostic Impact
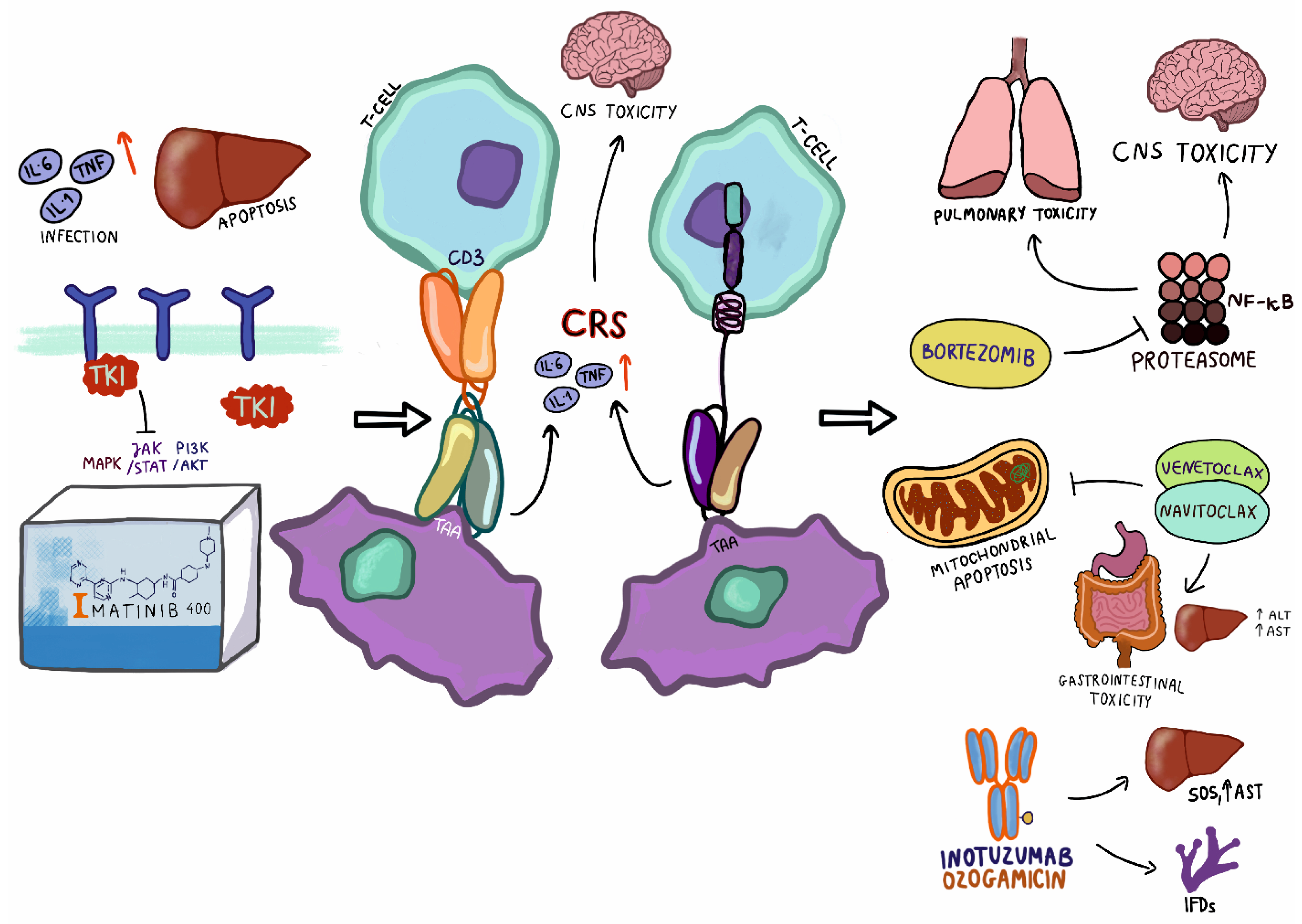
2. Tyrosine Kinase Inhibitors
3. Immunotherapy
3.1. Blinatumomab
3.2. CART-T Cell
4. Other Approaches
4.1. Inotuzumab Ozogamycin
4.2. Bortezomib
4.3. BCL2 Inhibitors
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Available online:www.cancerresearchuk.org (accessed on 20 May 2021).
- Ward, E.; DeSantis, C.; Robbins, A.; Kohler, B.; Jemal, A. Childhood and adolescent cancer statistics, 2014. CA Cancer J. Clin. 2014, 64, 83–103. [Google Scholar] [CrossRef]
- Vora, A.; Goulden, N.; Mitchell, C.; Hancock, J.; Hough, R.; Rowntree, C.; Moorman, A.V.; Wade, R. Augmented post-remission therapy for a minimal residual disease-defined high-risk subgroup of children and young people with clinical standard-risk and intermediate-risk acute lymphoblastic leukaemia (UKALL 2003): A randomised controlled trial. Lancet Oncol. 2014, 15, 809–818. [Google Scholar] [CrossRef]
- Gupta, S.; Hunger, S.P. Recent trends in the results of studies conducted by the Children’s Oncology Group acute lymphoblastic leukemia committee and implications for emerging cooperative trial groups in low- and middle-income countries. Pediatric Hematol. Oncol. J. 2020, 5, 151–155. [Google Scholar] [CrossRef]
- Möricke, A.; Zimmermann, M.; Valsecchi, M.G.; Stanulla, M.; Biondi, A.; Mann, G.; Locatelli, F.; Cazzaniga, G.; Niggli, F.; Aricò, M.; et al. Dexamethasone vs prednisone in induction treatment of pediatric ALL: Results of the randomized trial AIEOP-BFM ALL 2000. Blood 2016, 127, 2101–2112. [Google Scholar] [CrossRef] [PubMed]
- Shimada, A. Hematological malignancies and molecular targeting therapy. Eur. J. Pharmacol. 2019, 862, 172641. [Google Scholar] [CrossRef] [PubMed]
- Schmiegelow, K.; Müller, K.; Mogensen, S.S.; Mogensen, P.R.; Wolthers, B.O.; Stoltze, U.K.; Tuckuviene, R.; Frandsen, T. Non-infectious chemotherapy-associated acute toxicities during childhood acute lymphoblastic leukemia therapy. F1000Research 2017, 6, 444. [Google Scholar] [CrossRef] [Green Version]
- Schmiegelow, K.; Attarbaschi, A.; Barzilai, S.; Escherich, G.; Frandsen, T.L.; Halsey, C.; Hough, R.; Jeha, S.; Kato, M.; Liang, D.C.; et al. Consensus definitions of 14 severe acute toxic effects for childhood lymphoblastic leukaemia treatment: A Delphi consensus. Lancet Oncol. 2016, 17, e231–e239. [Google Scholar] [CrossRef]
- Xue, Y.J.; Wang, Y.; Jia, Y.P.; Zuo, Y.X.; Wu, J.; Lu, A.D.; Zhang, L.P. The role of minimal residual disease in specific subtypes of pediatric acute lymphoblastic leukemia. Int. J. Hematol. 2021, 113, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Pui, C.H.; Pei, D.; Raimondi, S.C.; Coustan-Smith, E.; Jeha, S.; Cheng, C.; Bowman, W.P.; Sandlund, J.T.; Ribeiro, R.C.; Rubnitz, J.E.; et al. Clinical impact of minimal residual disease in children with different subtypes of acute lymphoblastic leukemia treated with Response-Adapted therapy. Leukemia 2017, 31, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Stanulla, M.; Dagdan, E.; Zaliova, M.; Möricke, A.; Palmi, C.; Cazzaniga, G.; Eckert, C.; te Kronnie, G.; Bourquin, J.-P.; Bornhauser, B.; et al. IKZF1plus Defines a New Minimal Residual Disease–Dependent Very-Poor Prognostic Profile in Pediatric B-Cell Precursor Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2018, 36, 1240–1249. [Google Scholar]
- Bader, P.; Kreyenberg, H.; von Stackelberg, A.; Eckert, C.; Salzmann-Manrique, E.; Meisel, R.; Poetschger, U.; Stachel, D.; Schrappe, M.; Alten, J.; et al. Monitoring of minimal residual disease after allogeneic stem-cell transplantation in relapsed childhood acute lymphoblastic leukemia allows for the identification of impending relapse: Results of the ALL-BFM-SCT 2003 trial. J. Clin. Oncol. 2015, 33, 1275–1284. [Google Scholar] [CrossRef] [PubMed]
- Queudeville, M.; Schlegel, P.; Heinz, A.T.; Lenz, T.; Döring, M.; Holzer, U.; Hartmann, U.; Kreyenberg, H.; von Stackelberg, A.; Schrappe, M.; et al. Blinatumomab in pediatric patients with relapsed/refractory B-cell precursor acute lymphoblastic leukemia. Eur. J. Haematol. 2021, 106, 473–483. [Google Scholar] [CrossRef]
- Contreras, C.F.; Higham, C.S.; Behnert, A.; Kim, K.; Stieglitz, E.; Tasian, S.K. Clinical utilization of blinatumomab and inotuzumab immunotherapy in children with relapsed or refractory B-acute lymphoblastic leukemia. Pediatric Blood Cancer 2021, 68, e28718. [Google Scholar] [CrossRef]
- Kansagra, A.J.; Frey, N.V.; Bar, M.; Laetsch, T.W.; Carpenter, P.A.; Savani, B.N.; Heslop, H.E.; Bollard, C.M.; Komanduri, K.V.; Gastineau, D.A.; et al. Clinical utilization of Chimeric Antigen Receptor T-cells (CAR-T) in B-cell acute lymphoblastic leukemia (ALL)-an expert opinion from the European Society for Blood and Marrow Transplantation (EBMT) and the American Society for Blood and Marrow Transplantation (ASBMT). Bone Marrow Transplant. 2019, 25, e76–e85. [Google Scholar]
- Pui, C.H.; Nichols, K.E.; Yang, J.J. Somatic and germline genomics in paediatric acute lymphoblastic leukaemia. Nat. Rev. Clin. Oncol. 2019, 16, 227–240. [Google Scholar] [CrossRef]
- Lejman, M.; Włodarczyk, M.; Styka, B.; Pastorczak, A.; Zawitkowska, J.; Taha, J.; Sędek, Ł.; Skonieczka, K.; Braun, M.; Haus, O.; et al. Advantages and Limitations of SNP Array in the Molecular Characterization of Pediatric T-Cell Acute Lymphoblastic Leukemia. Front. Oncol. 2020, 10, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Lejman, M.; Włodarczyk, M.; Zaucha-Prażmo, A.; Zawitkowska, J. Use of microarrays and mlpa for integrating diagnostics and personalizing treatment—case report of a patient with ph-like acute b-cell lymphoblastic leukemia. Ann. Agric. Environ. Med. 2020, 27, 713–716. [Google Scholar] [CrossRef] [PubMed]
- Mullighan, C.; Goorha, S.; Radtke, I.; Miller, C.B.; Coustan-Smith, E.; Dalton, J.D.; Girtman, K.; Mathew, S.; Ma, J.; Pounds, S.B.; et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature 2007, 446, 758–764. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Inaba, H.; Pui, C.-H. Advances in the Diagnosis and Treatment of Pediatric Acute Lymphoblastic Leukemia. J. Clin. Med. 2021, 10, 1926. [Google Scholar] [CrossRef]
- Pui, C.H. Precision medicine in acute lymphoblastic leukemia. Front. Med. 2020, 14, 689–700. [Google Scholar] [CrossRef] [PubMed]
- Toft, N.; Birgens, H.; Abrahamsson, J.; Griškevičius, L.; Hallböök, H.; Heyman, M.; Klausen, T.W.; Jónsson, Ó.G.; Palk, K.; Pruunsild, K.; et al. Results of NOPHO ALL2008 treatment for patients aged 1–45 years with acute lymphoblastic leukemia. Leukemia 2018, 32, 606–615. [Google Scholar] [CrossRef]
- Stary, J.; Zimmermann, M.; Campbell, M.; Castillo, L.; Dibar, E.; Donska, S.; Gonzalez, A.; Izraeli, S.; Janic, D.; Jazbec, J.; et al. Intensive Chemotherapy for Childhood Acute Lymphoblastic Leukemia: Results of the Randomized Intercontinental Trial ALL IC-BFM 2002. J. Clin. Oncol. 2014, 32, 174–184. [Google Scholar] [CrossRef] [Green Version]
- Biondi, A.; Gandemer, V.; De Lorenzo, P.; Cario, G.; Campbell, M.; Castor, A.; Pieters, R.; Baruchel, A.; Vora, A.J.; Leoni, V.; et al. Imatinib treatment of paediatric Philadelphia chromosome-positive acute lymphoblastic leukaemia (EsPhALL2010): A prospective, intergroup, open-label, single-arm clinical trial. Lancet Haematol. 2018, 5, e641–e652. [Google Scholar] [CrossRef] [Green Version]
- Zawitkowska, J.; Lejman, M.; Płonowski, M.; Bulsa, J.; Szczepański, T.; Romiszewski, M.; Mizia-Malarz, A.; Derwich, K.; Karolczyk, G.; Ociepa, T.; et al. Clinical Outcome in Pediatric Patients with Philadelphia Chromosome Positive ALL Treated with Tyrosine Kinase Inhibitors Plus Chemotherapy-The Experience of a Polish Pediatric Leukemia and Lymphoma Study Group. Cancers 2020, 12, 3751. [Google Scholar] [CrossRef]
- Roberts, K.G.; Li, Y.; Payne-Turner, D.; Harvey, R.C.; Yang, Y.-L.; Pei, D.; McCastlain, K.; Ding, L.; Lu, C.; Song, G.; et al. Targetable Kinase-Activating Lesions in Ph-like Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2014, 371, 1005–1015. [Google Scholar] [CrossRef] [Green Version]
- Pui, C.-H.; Rebora, P.; Schrappe, M.; Attarbaschi, A.; Baruchel, A.; Basso, G.; Cavé, H.; Elitzur, S.; Koh, K.; Liu, H.C.; et al. Ponte di Legno Childhood ALL Working Group. Outcome of children with hypodiploid acute lymphoblastic leukemia: A retrospective multinational study. J. Clin. Oncol. 2019, 37, 770–779. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Flores, E.; Comeaux, E.Q.; Kim, K.L.; Melnik, E.; Beckman, K.; Davis, K.L.; Wu, K.; Akutagawa, J.; Bridges, O.; Marino, R.; et al. Bcl-2 is a therapeutic target for hypodiploid B-lineage acute lymphoblastic leukemia. Cancer Res. 2019, 79, 2339–2351. [Google Scholar] [CrossRef] [Green Version]
- Paul, M.K.; Mukhopadhyay, A.K. Tyrosine kinase—Role and significance in Cancer. Int. J. Med. Sci. 2004, 1, 101–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Natoli, C.; Perrucci, B.; Perrotti, F.; Falchi, L.; Iacobelli, S. Tyrosine kinase inhibitors. Curr. Cancer Drug Targets 2010, 10, 462–483. [Google Scholar] [CrossRef] [PubMed]
- Schultz, K.R.; Bowman, W.P.; Aledo, A.; Slayton, W.B.; Sather, H.; Devidas, M.; Wang, C.; Davies, S.M.; Gaynon, P.S.; Trigg, M.; et al. Improved early event-free survival with imatinib in Philadelphia chromosome-positive acute lymphoblastic leukemia: A children’s oncology group study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2009, 27, 5175–5181. [Google Scholar] [CrossRef]
- Schultz, K.R.; Carroll, A.; Heerema, N.A.; Bowman, W.P.; Aledo, A.; Slayton, W.B.; Sather, H.; Devidas, M.; Zheng, H.W.; Davies, S.M.; et al. Long-term follow-up of imatinib in pediatric Philadelphia chromosome-positive acute lymphoblastic leukemia: Children’s Oncology Group Study AALL0031. Leukemia 2014, 28, 1467–1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milojkovic, D.; Apperley, J. Mechanisms of Resistance to Imatinib and Second-Generation Tyrosine Inhibitors in Chronic Myeloid Leukemia. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 7519–7527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, S.; Chen, X.; Cai, J.; Yu, J.; Gao, J.; Hu, S.; Zhai, X.; Liang, C.; Ju, X.; Jiang, H.; et al. Effect of Dasatinib vs Imatinib in the Treatment of Pediatric Philadelphia Chromosome–Positive Acute Lymphoblastic Leukemia: A Randomized Clinical Trial. JAMA Oncol. 2020, 6, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.E.; Saglio, G.; Kantarjian, H.M.; Baccarani, M.; Mayer, J.; Boqué, C.; Shah, N.P.; Chuah, C.; Casanova, L.; Bradley-Garelik, B.; et al. Final 5-Year Study Results of DASISION: The Dasatinib Versus Imatinib Study in Treatment-Naïve Chronic Myeloid Leukemia Patients Trial. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2016, 34, 2333–2340. [Google Scholar] [CrossRef]
- Tokarski, J.S.; Newitt, J.A.; Chang, C.Y.; Cheng, J.D.; Wittekind, M.; Kiefer, S.E.; Kish, K.; Lee, F.Y.; Borzillerri, R.; Lombardo, L.J.; et al. The structure of Dasatinib (BMS-354825) bound to activated ABL kinase domain elucidates its inhibitory activity against imatinib-resistant ABL mutants. Cancer Res. 2006, 66, 5790–5797. [Google Scholar] [CrossRef] [Green Version]
- Annesley, C.E.; Brown, P. Novel agents for the treatment of childhood acute leukemia. Ther. Adv. Hematol. 2015, 6, 61–79. [Google Scholar] [CrossRef] [Green Version]
- Cortes, J.E.; Kim, D.W.; Pinilla-Ibarz, J.; le Coutre, P.D.; Paquette, R.; Chuah, C.; Nicolini, F.E.; Apperley, J.F.; Khoury, H.J.; Talpaz, M.; et al. Ponatinib efficacy and safety in Philadelphia chromosome-positive leukemia: Final 5-year results of the phase 2 PACE trial. Blood 2018, 132, 393–404. [Google Scholar] [CrossRef]
- Jabbour, E.; Short, N.J.; Ravandi, F.; Huang, X.; Daver, N.; DiNardo, C.D.; Konopleva, M.; Pemmaraju, N.; Wierda, W.; Garcia-Manero, G.; et al. Combination of hyper-CVAD with ponatinib as first-line therapy for patients with Philadelphia chromosome-positive acute lymphoblastic leukaemia: Long-term follow-up of a single-centre, phase 2 study. Lancet Haematol. 2018, 5, e618–e627. [Google Scholar] [CrossRef]
- Millot, F.; Suttorp, M.; Versluys, A.B.; Kalwak, K.; Nelken, B.; Ducassou, S.; Bertrand, Y.; Baruchel, A. Ponatinib in childhood Philadelphia chromosome–positive leukaemias: An international registry of childhood chronic myeloid leukaemia study. Eur. J. Cancer 2020, 136, 107–112. [Google Scholar] [CrossRef]
- Den Boer, M.L.; van Slegtenhorst, M.; De Menezes, R.X.; Cheok, M.H.; Buijs-Gladdines, J.G.; Peters, S.T.; Van Zutven, L.J.; Beverloo, H.B.; Van der Spek, P.J.; Escherich, G.; et al. A Subtype of Childhood Acute Lymphoblastic Leukaemia with Poor Treatment Outcome: A Genome-Wide Classification Study. Lancet Oncol. 2009, 10, 125–134. [Google Scholar] [CrossRef] [Green Version]
- Shiraz, P.; Payne, K.J.; Muffly, L. The Current Genomic and Molecular Landscape of Philadelphia-like Acute Lymphoblastic Leukemia. Int. J. Mol. Sci. 2020, 21, 2193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwab, C.; Ryan, S.L.; Chilton, L.; Elliott, A.; Murray, J.; Richardson, S.; Wragg, C.; Moppett, J.; Cummins, M.; Tunstall, O.; et al. EBF1-PDGFRB fusion in pediatric B-cell precursor acute lymphoblastic leukemia (BCP-ALL): Genetic profile and clinical implications. Blood 2016, 127, 2214–2218. [Google Scholar] [CrossRef]
- Weston, B.W.; Hayden, M.A.; Roberts, K.G.; Bowyer, S.; Hsu, J.; Fedoriw, G.; Rao, K.W.; Mullighan, C.G. Tyrosine kinase inhibitor therapy induces remission in a patient with refractory EBF1-PDGFRB-positive acute lymphoblastic leukemia. J. Clin. Oncol. 2013, 31, e413–e416. [Google Scholar] [CrossRef] [PubMed]
- Tanasi, I.; Ba, I.; Sirvent, N.; Braun, T.; Cuccuini, W.; Ballerini, P.; Duployez, N.; Tanguy-Schmidt, A.; Erômeerˆerôme Tamburini, J.; Ebastien Maury, S.; et al. Letters to Blood Efficacy of Tyrosine Kinase Inhibitors in Ph-like Acute Lymphoblastic Leukemia Harboring ABL-Class Rearrangements. Blood 2019, 134, 1351–1355. [Google Scholar] [CrossRef]
- Den Boer, M.L.; Cario, G.; Moorman, A.V.; Boer, J.M.; de Groot-Kruseman, H.A.; Fiocco, M.; Escherich, G.; Imamura, T.; Yeoh, J.; Sutton, R.; et al. Outcomes of Paediatric Patients with B-Cell Acute Lymphocytic Leukaemia with ABL-Class Fusion in the Pre-Tyrosine-Kinase Inhibitor Era: A Multicentre, Retrospective, Cohort Study. Lancet Haematol. 2021, 8, e55–e66. [Google Scholar] [CrossRef]
- Roberts, K.G.; Yang, Y.L.; Payne-Turner, D.; Lin, W.; Files, J.K.; Dickerson, K.; Gu, Z.; Taunton, J.; Janke, L.J.; Chen, T.; et al. Oncogenic Role and Therapeutic Targeting of ABL-Class and JAK-STAT Activating Kinase Alterations in Ph-like ALL. Blood Adv. 2017, 1, 1657–1671. [Google Scholar]
- Reshmi, S.C.; Harvey, R.C.; Roberts, K.G.; Stonerock, E.; Smith, A.; Jenkins, H.; Chen, I.M.; Valentine, M.; Liu, Y.; Li, Y.; et al. Targetable kinase gene fusions in high-risk B-ALL: A study from the Children’s Oncology Group. Blood 2017, 129, 3352–3361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullighan, C.G.; Zhang, J.; Harvey, R.C.; Collins-Underwood, J.R.; Schulman, B.A.; Phillips, L.A.; Tasian, S.K.; Loh, M.L.; Su, X.; Liu, W.; et al. JAK mutations in high-risk childhood acute lymphoblastic leukemia. Proc. Natl. Acad. Sci. USA 2009, 106, 9414–9418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verstovsek, S.; Kantarjian, H.; Mesa, R.A.; Pardanani, A.D.; Cortes-Franco, J.; Thomas, D.A.; Estrov, Z.; Fridman, J.S.; Bradley, E.C.; Erickson-Viitanen, S.; et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N. Engl. J. Med. 2010, 363, 1117–1127. [Google Scholar] [CrossRef] [Green Version]
- Pemmaraju, N.; Kantarjian, H.; Kadia, T.; Cortes, J.; Borthakur, G.; Newberry, K.; Garcia-Manero, G.; Ravandi, F.; Jabbour, E.; Dellasala, S.; et al. A phase I/II study of the Janus kinase (JAK)1 and 2 inhibitor ruxolitinib in patients with relapsed or refractory acute myeloid leukemia. Clin. Lymphoma Myeloma Leuk. 2015, 15, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Tasian, S.K.; Assad, A.; Hunter, D.S.; Du, Y.; Loh, M.L. A phase 2 study of ruxolitinib with chemotherapy in children with philadelphia chromosome-like acute lymphoblastic leukemia (INCB18424-269/AALL1521): Fose-finding results from the Part 1 safety phase. Blood 2018, 132, 555. [Google Scholar]
- Hijiya, N.; Maschan, A.; Rizzari, C.; Shimada, H.; Dufour, C.; Goto, H.; Kang, H.J.; Guinipero, T.; Karakas, Z.; Bautista, F.; et al. Phase 2 study of nilotinib in pediatric patients with Philadelphia chromosome-positive chronic myeloid leukemia. Blood 2019, 134, 2036–2045. [Google Scholar] [CrossRef]
- Hijiya, N.; Zwaan, C.M.; Rizzari, C.; Foa, R.; Abbink, F.; Lancaster, D.; Landman-Parker, J.; Millot, F.; Moppett, J.; Nelken, B.; et al. Pharmacokinetics of nilotinib in pediatric patients with Philadelphia chromosome–positive chronic myeloid leukemia or acute lymphoblastic leukemia. Clin. Cancer Res. 2020, 26, 812–820. [Google Scholar] [CrossRef]
- Nezhad, M.S.; Yazdanifar, M.; Abdollahpour-Alitappeh, M.; Sattari, A.; Seifalian, A.; Bagheri, N. Strengthening the CAR-T Cell Therapeutic Application using CRISPR/Cas9 Technology. Biotechnol. Bioeng. 2021, bit.27882. [Google Scholar] [CrossRef]
- Von Stackelberg, A.; Locatelli, F.; Zugmaier, G.; Handgretinger, R.; Trippett, T.M.; Rizzari, C.; Bader, P.; O’Brien, L.M.; Brethon, B.; Bhojwani, D. Phase I/Phase II Study of Blinatumomab in Pediatric Patients With Relapsed/Refractory Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2016, 34, 4381–4389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Locatelli, F.; Zugmaier, G.; Mergen, N.; Bader, P.; Jeha, S.; Schlegel, P.-G.; Bourquin, J.-P.; Handgretinger, R.; Brethon, B.; Rossig, C.; et al. Blinatumomab in pediatric patients with relapsed/refractory acute lymphoblastic leukemia: Results of the RIALTO trial, an expanded access study. Blood Cancer J. 2020, 10, 77. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Liu, M.; Ren, F.; Meng, X.; Yu, J. The landscape of bispecific T cell engager in cancer treatment. Biomark. Res. 2021, 9, 38. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, F.; Zugmaier, G.; Rizzari, C.; Morris, J.D.; Gruhn, B.; Klingebiel, T.; Parasole, R.; Linderkamp, C.; Flotho, C.; Petit, A.; et al. Effect of Blinatumomab vs Chemotherapy on Event-Free Survival Among Children With High-risk First-Relapse B-Cell Acute Lymphoblastic Leukemia A Randomized Clinical Trial. JAMA 2021, 325, 843. [Google Scholar] [CrossRef]
- Brown, P.A.; Ji, L.; Xu, X.; Devidas, M.; Hogan, L.E.; Borowitz, M.J.; Raetz, E.A.; Zugmaier, G.; Sharon, E.; Bernhardt, M.B.; et al. Effect of Postreinduction Therapy Consolidation With Blinatumomab vs Chemotherapy on Disease-Free Survival in Children, Adolescents, and Young Adults With First Relapse of B-Cell Acute Lymphoblastic Leukemia: A Randomized Clinical Trial. JAMA 2021, 325, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Vairy, S.; Garcia, J.L.; Teira, P.; Bittencourt, H. CTL019 (Tisagenlecleucel): CAR-T therapy for relapsed and refractory B-cell acute lymphoblastic leukemia. Drug Des. Dev. Ther. 2018, 12, 3885–3898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inaba, H.; Pui, C.H. Immunotherapy in pediatric acute lymphoblastic leukemia. Cancer Metastasis Rev. 2019, 38, 595–610. [Google Scholar] [CrossRef] [PubMed]
- Cook, J.; Litzow, M. Advances in Supportive Care for Acute Lymphoblastic Leukemia. Curr. Hematol. Malig. Rep. 2020, 15, 276–293. [Google Scholar] [CrossRef]
- Grigor, E.; Fergusson, D.A.; Haggar, F.; Kekre, N.; Atkins, H.; Shorr, R.; Holt, R.A.; Hutton, B.; Ramsay, T.; Seftel, M.; et al. Efficacy and safety of chimeric antigen receptor T-cell (CAR-T) therapy in patients with haematological and solid malignancies: Protocol for a systematic review and meta-analysis. BMJ Open 2017, 7, e019321. [Google Scholar] [CrossRef]
- Makita, S.; Yoshimura, K.; Tobinai, K. Clinical development of anti-CD19 chimeric antigen receptor T-cell therapy for B-cell non-Hodgkin lymphoma. Cancer Sci. 2017, 108, 1109–1118. [Google Scholar] [CrossRef] [Green Version]
- Bonifant, C.L.; Jackson, H.J.J.; Brentjens, R.J.; Curran, K.J. Toxicity and management in CAR T-cell therapy. Mol. Ther.-Oncolytics 2016, 3, 16011. [Google Scholar] [CrossRef] [PubMed]
- Gracia Borrega, J.; Godel, P.; Ruger, A.A.; Onur, O.; Shimabukuro-Vornhagen, A.; Kochanek, M.; Boll, B. In the Eye of the Storm: Immune-mediated Toxicities Associated With CAR-T Cell Therapy. HemaSphere 2019, 3, e191. [Google Scholar] [CrossRef] [PubMed]
- Molina, J.C.; Shah, N.N. CAR T cells better than BiTEs. Blood Adv. 2021, 5, 602–606. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Wang, Y.; Li, H.; Song, X.; Qi, K.; Cheng, H.; Cao, J.; Shi, M.; Yan, Z.; Jing, G.; Pan, B.; et al. Kinetics of immune reconstitution after anti-CD19 chimeric antigen receptor T cell therapy in relapsed or refractory acute lymphoblastic leukemia patients. Int. J. Lab. Hematol. 2021, 43, 250–258. [Google Scholar] [CrossRef]
- Moghanloo, E.; Mollanoori, H.; Talebi, M.; Pashangzadeh, S.; Faraji, F.; Hadjilooei, F.; Mahmoodzadeh, H. Remote controlling of CAR-T cells and toxicity management: Molecular switches and next generation CARs. Transl. Oncol. 2021, 14, 101070. [Google Scholar] [CrossRef]
- Sentman, M.L.; Murad, J.M.; Cook, W.J.; Wu, M.R.; Reder, J.; Baumeister, S.H.; Dranoff, G.; Fanger, M.W.; Sentman, C.L. Mechanisms of Acute Toxicity in NKG2D Chimeric Antigen Receptor T Cell-Treated Mice. J. Immunol. 2016, 197, 4674–4685. [Google Scholar] [CrossRef] [Green Version]
- Staedtke, V.; Bai, R.Y.; Kim, K.; Darvas, M.; Davila, M.L.; Riggins, G.J.; Rothman, P.B.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B.; et al. Disruption of a self-amplifying catecholamine loop reduces cytokine release syndrome. Nature 2018, 564, 273–277. [Google Scholar] [CrossRef]
- Mackall, C.L.; Miklos, D.B. CNS endothelial cell activation emerges as a driver of CAR T cell –associated neurotoxicity. Cancer Discov. 2017, 7, 1371–1373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, K.R.; Migliorini, D.; Perkey, E.; Yost, K.E.; Bhaduri, A.; Bagga, P.; Haris, M.; Wilson, N.E.; Liu, F.; Gabunia, K.; et al. Single-Cell Analyses Identify Brain Mural Cells Expressing CD19 as Potential Off-Tumor Targets for CAR-T Immunotherapies. Cell 2020, 183, 126–142. [Google Scholar] [CrossRef] [PubMed]
- Kamal, M.; Joseph, J.; Greenbaum, U.; Hicklen, R.; Kebriaei, P.; Srour, S.A.; Wang, X.S. Patient-Reported Outcomes for Cancer Patients with Hematological Malignancies Undergoing Chimeric Antigen Receptor T Cell Therapy: A Systematic Review. Transplant. Cell. Ther. 2021, 27, 390.e1–390.e7. [Google Scholar] [CrossRef] [PubMed]
- Sterner, R.M.; Sakemura, R.; Cox, M.J.; Yang, N.; Khadka, R.H.; Forsman, C.L.; Hansen, M.J.; Jin, F.; Ayasoufi, K.; Hefazi, M.; et al. GM-CSF inhibition reduces cytokine release syndrome and neuroinflammation but enhances CAR-T cell function in xenografts. Blood 2019, 133, 697–709. [Google Scholar] [CrossRef] [Green Version]
- Dahl, J.; Marx, K.; Jabbour, E. Inotuzumab ozogamicin in the treatment of acute lymphoblastic leukemia. Expert Rev. Hematol. 2016, 9, 329–334. [Google Scholar] [CrossRef]
- Bhojwani, D.; Sposto, R.; Shah, N.N.; Rodriguez, V.; Yuan, C.; Stetler-Stevenson, M.; O’Brien, M.M.; McNeer, J.L.; Quereshi, A.; Cabannes, A.; et al. Inotuzumab ozogamicin in pediatric patients with relapsed/refractory acute lymphoblastic leukemia. Leukemia 2019, 33, 884–892. [Google Scholar] [CrossRef]
- Brivio, E.; Locatelli, F.; Lopez-Yurda, M.; Malone, A.; Díaz-de-Heredia, C.; Bielorai, B.; Rossig, C.; van der Velden, V.; Ammerlaan, A.; Thano, A.; et al. A phase 1 study of inotuzumab ozogamicin in pediatric relapsed/refractory acute lymphoblastic leukemia (ITCC-059 study). Blood 2021, 137, 1582–1590. [Google Scholar] [CrossRef]
- Kyriakidis, I.; Vasileiou, E.; Rossig, C.; Roilides, E.; Groll, A.H.; Tragiannidis, A. Invasive Fungal Diseases in Children with Hematological Malignancies Treated with Therapies That Target Cell Surface Antigens: Monoclonal Antibodies, Immune Checkpoint Inhibitors and CAR T-Cell Therapies. J. Fungi 2021, 7, 186. [Google Scholar] [CrossRef]
- Zahid, M.F. The role of bortezomib in the treatment of acute lymphoblastic leukemia. Future Oncol. 2016, 12, 1861–1864. [Google Scholar] [CrossRef] [PubMed]
- Horton, T.M.; Gannavarapu, A.; Blaney, S.M.; D’Argenio, D.Z.; Plon, S.E.; Berg, S.L. Bortezomib interactions with chemotherapy agents in acute leukemia in vitro. Cancer Chemother. Pharmacol. 2006, 58, 13–23. [Google Scholar] [CrossRef]
- Miyakoshi, S.; Kami, M.; Yuji, K.; Matsumura, T.; Takatoku, M.; Sasaki, M.; Narimatsu, H.; Fujii, T.; Kawabata, M.; Taniguchi, S.; et al. Severe pulmonary complications in Japanese patients after bortezomib treatment for refractory multiple myeloma. Blood 2006, 107, 3492–3494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Argyriou, A.A.; Iconomou, G.; Kalofonos, H.P. Bortezomib-induced peripheral neuropathy in multiple myeloma: A comprehensive review of the literature. Blood 2008, 112, 1593–1599. [Google Scholar] [CrossRef] [Green Version]
- Bertaina, A.; Vinti, L.; Strocchio, L.; Gaspari, S.; Caruso, R.; Algeri, M.; Coletti, V.; Gurnari, C.; Romano, M.; Cefalo, M.G.; et al. The combination of bortezomib with chemotherapy to treat relapsed/refractory acute lymphoblastic leukaemia of childhood. Br. J. Haematol. 2017, 176, 629–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- August, K.J.; Guest, E.M.; Lewing, K.; Hays, J.A.; Gamis, A.S. Treatment of children with relapsed and refractory acute lymphoblastic leukemia with mitoxantrone, vincristine, pegaspargase, dexamethasone, and bortezomib. Pediatric Blood Cancer 2020, 67, e28062. [Google Scholar] [CrossRef]
- Iguchi, A.; Cho, Y.; Sugiyama, M.; Terashita, Y.; Ariga, T.; Hosoya, Y.; Hirabayashi, S.; Manabe, A.; Hara, K.; Aiba, T.; et al. Bortezomib combined with standard induction chemotherapy in Japanese children with refractory acute lymphoblastic leukemia. Int. J. Hematol. 2017, 106, 291–298. [Google Scholar] [CrossRef]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef]
- Tse, C.; Shoemaker, A.R.; Adickes, J.; Anderson, M.G.; Chen, J.; Jin, S.; Johnson, E.F.; Marsh, K.C.; Mitten, M.J.; Nimmer, P.; et al. ABT-263: A potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008, 68, 3421–3428. [Google Scholar] [CrossRef] [Green Version]
- Suryani, S.; Carol, H.; Chonghaile, T.N.; Frismantas, V.; Sarmah, C.; High, L.; Bornhauser, B.; Cowley, M.J.; Szymanska, B.; Evans, K.; et al. Cell and molecular determinants of in vivo efficacy of the BH3 mimetic ABT-263 against pediatric acute lymphoblastic leukemia xenografts. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 4520–4531. [Google Scholar] [CrossRef] [Green Version]
- Khaw, S.L.; Suryani, S.; Evans, K.; Richmond, J.; Robbins, A.; Kurmasheva, R.T.; Billups, C.A.; Erickson, S.W.; Guo, Y.; Houghton, P.J.; et al. Venetoclax responses of pediatric ALL xenografts reveal sensitivity of MLL-rearranged leukemia. Blood 2016, 128, 1382–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leonard, J.T.; Rowley, J.S.; Eide, C.A.; Traer, E.; Hayes-Lattin, B.; Loriaux, M.; Spurgeon, S.E.; Druker, B.J.; Tyner, J.W.; Chang, B.H. Targeting BCL-2 and ABL/LYN in Philadelphia chromosome-positive acute lymphoblastic leukemia. Sci. Transl. Med. 2016, 8, 354ra114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Nimmer, P.M.; Tahir, S.K.; Chen, J.; Fryer, R.M.; Hahn, K.R.; Iciek, L.A.; Morgan, S.J.; Nasarre, M.C.; Nelson, R.; et al. Bcl-2 family proteins are essential for platelet survival. Cell Death Differ. 2007, 14, 943–951. [Google Scholar] [CrossRef]
- Karol, S.E.; Cooper, T.M.; Bittencourt, H.; Gore, L.; O’Brien, M.M.; Fraser, C.; Gambart, M.; Cario, G.; Zwaan, C.M.; Bourquin, J.-P.; et al. Safety, Efficacy, and PK of the BCL2 Inhibitor Venetoclax in Combination with Chemotherapy in Pediatric and Young Adult Patients with Relapsed/Refractory Acute Myeloid Leukemia and Acute Lymphoblastic Leukemia: Phase 1 Study. Blood 2019, 134 (Suppl. 1), 2649. [Google Scholar] [CrossRef]
- Lacayo, N.J.; Pullarkat, V.A.; Stock, W.; Jabbour, E.; Bajel, A.; Rubnitz, J.; Leonard, J.; Mullighan, C.G.; Khaw, S.L.; Vear, S.I.; et al. Safety and Efficacy of Venetoclax in Combination with Navitoclax in Adult and Pediatric Relapsed/Refractory Acute Lymphoblastic Leukemia and Lymphoblastic Lymphoma. Blood 2019, 134 (Suppl. 1), 285. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Drug | Molecular Target | Common Toxicities | Uncommon Toxicities | References |
|---|---|---|---|---|
| Imatinib | BCR-ABL1 | Infections, mostly bacterial | Osteonecrosis | |
| c-KIT | Gastrointestinal toxicity | Joint pain Rash/skin problems | [25,26,31,32,33] | |
| PDGFR | Superficial edema Nausea Muscle cramps Musculoskeletal pain | Headache | ||
| Dasatinib | BCR-ABL1c-KIT PDGFR SRC family FGFR1 EGFR | Pleural effusions Myelosuppression | Bleeding (GI 1, PR 2) Hemorrhage | [31,35] |
| Nilotinib | BCR-ABL1 | AEs 3 related to bilirubin increases Headache Pyrexia | Nasopharyngitis Fatigue Abdominal pain Nausea Vomiting | [35,54,55] |
| c-KIT | [6,51] | |||
| PDGFR | ||||
| Ponatinib | BCR-ABL1 T315I | Platelet toxicities Nausea Abdominal pain | Infection Leucopenia Neutropenia | [45] |
| Ruxolitinib | JAK1 JAK2 | Infection (pneumonia, sepsis) Thrombocytopenia Neutropenia | Stroke Cerebral edema Fatigue Mucositis | [52,53] |
| Immunotherapy | Molecular Target | Common Toxicities | Uncommon Toxicities | References |
|---|---|---|---|---|
| Chimeric Antigen Receptor (CART-T) T cells | CD19 | B cell aplasia Cytokine release syndrome Infections Immune effector cell-associated neurotoxicity syndrome | Febrile neutropenia Tumor lysis syndrome | [72,75,76] |
| Blinatumomab (BiTE antibody) | CD3 and CD19 | Anemia Nausea Neurotoxicities (headache, tremor, dizziness, and higher-grade confusion) | Cytokine release syndrome | [57,58] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lejman, M.; Kuśmierczuk, K.; Bednarz, K.; Ostapińska, K.; Zawitkowska, J. Targeted Therapy in the Treatment of Pediatric Acute Lymphoblastic Leukemia—Therapy and Toxicity Mechanisms. Int. J. Mol. Sci. 2021, 22, 9827. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22189827
Lejman M, Kuśmierczuk K, Bednarz K, Ostapińska K, Zawitkowska J. Targeted Therapy in the Treatment of Pediatric Acute Lymphoblastic Leukemia—Therapy and Toxicity Mechanisms. International Journal of Molecular Sciences. 2021; 22(18):9827. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22189827
Chicago/Turabian StyleLejman, Monika, Kinga Kuśmierczuk, Kinga Bednarz, Katarzyna Ostapińska, and Joanna Zawitkowska. 2021. "Targeted Therapy in the Treatment of Pediatric Acute Lymphoblastic Leukemia—Therapy and Toxicity Mechanisms" International Journal of Molecular Sciences 22, no. 18: 9827. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22189827