
Disulfiram Sensitizes a Therapeutic-Resistant Glioblastoma to the TGF-β Receptor Inhibitor


Abstract
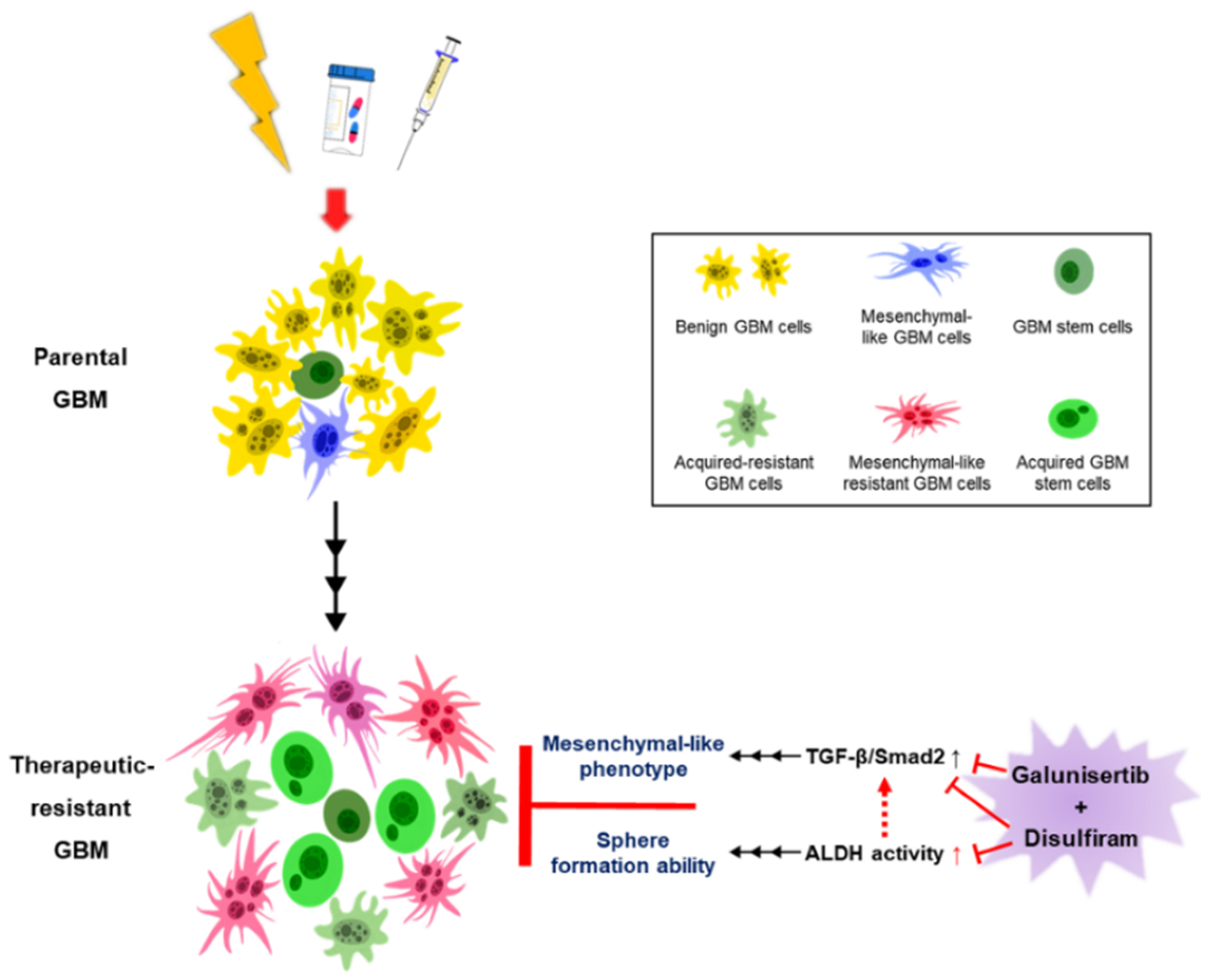
:1. Introduction
2. Results
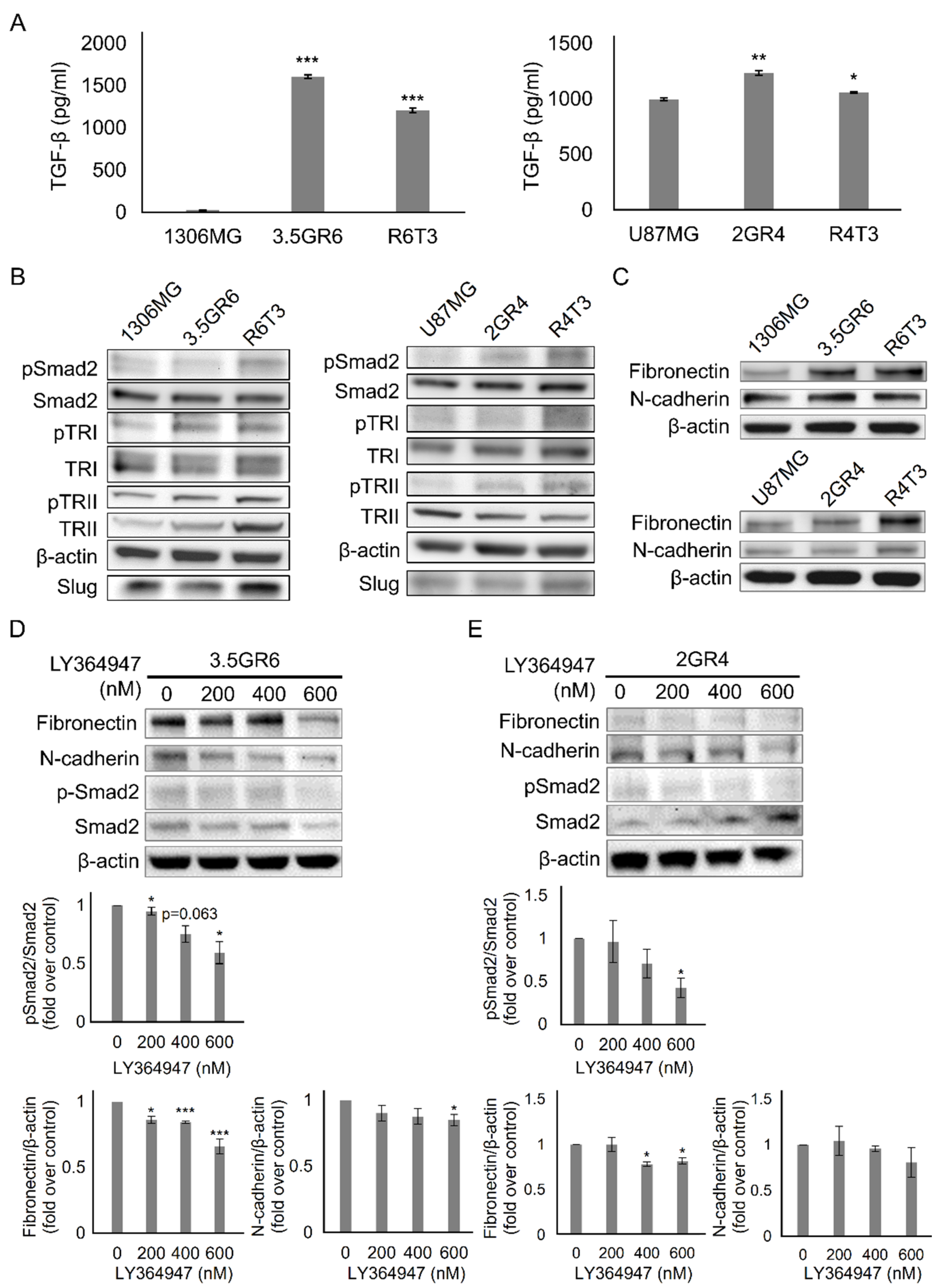
2.1. Developing Therapeutic Resistance in Glioblastoma Promotes TGF-β-Induced Mesenchymal-Like Phenotype
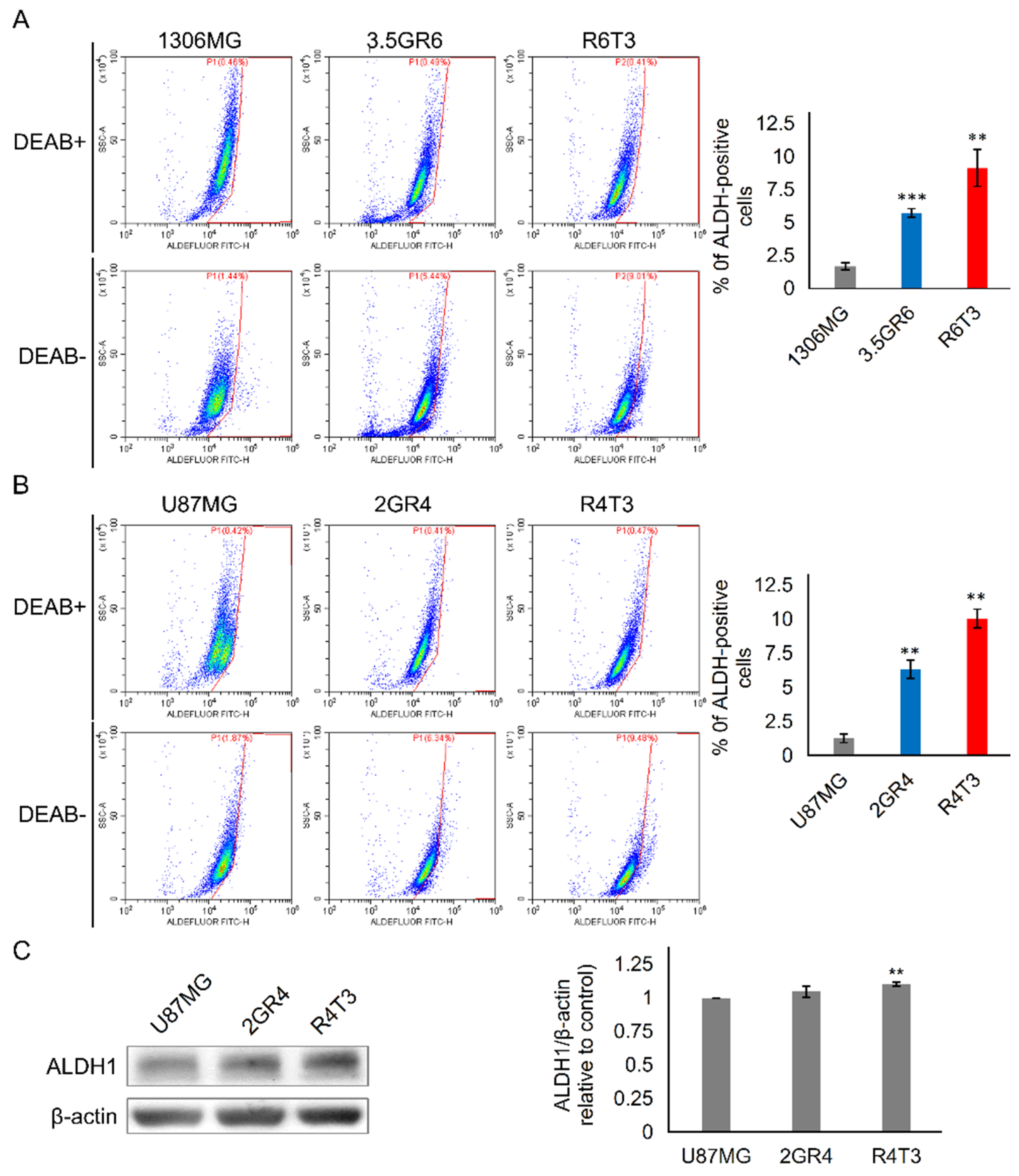
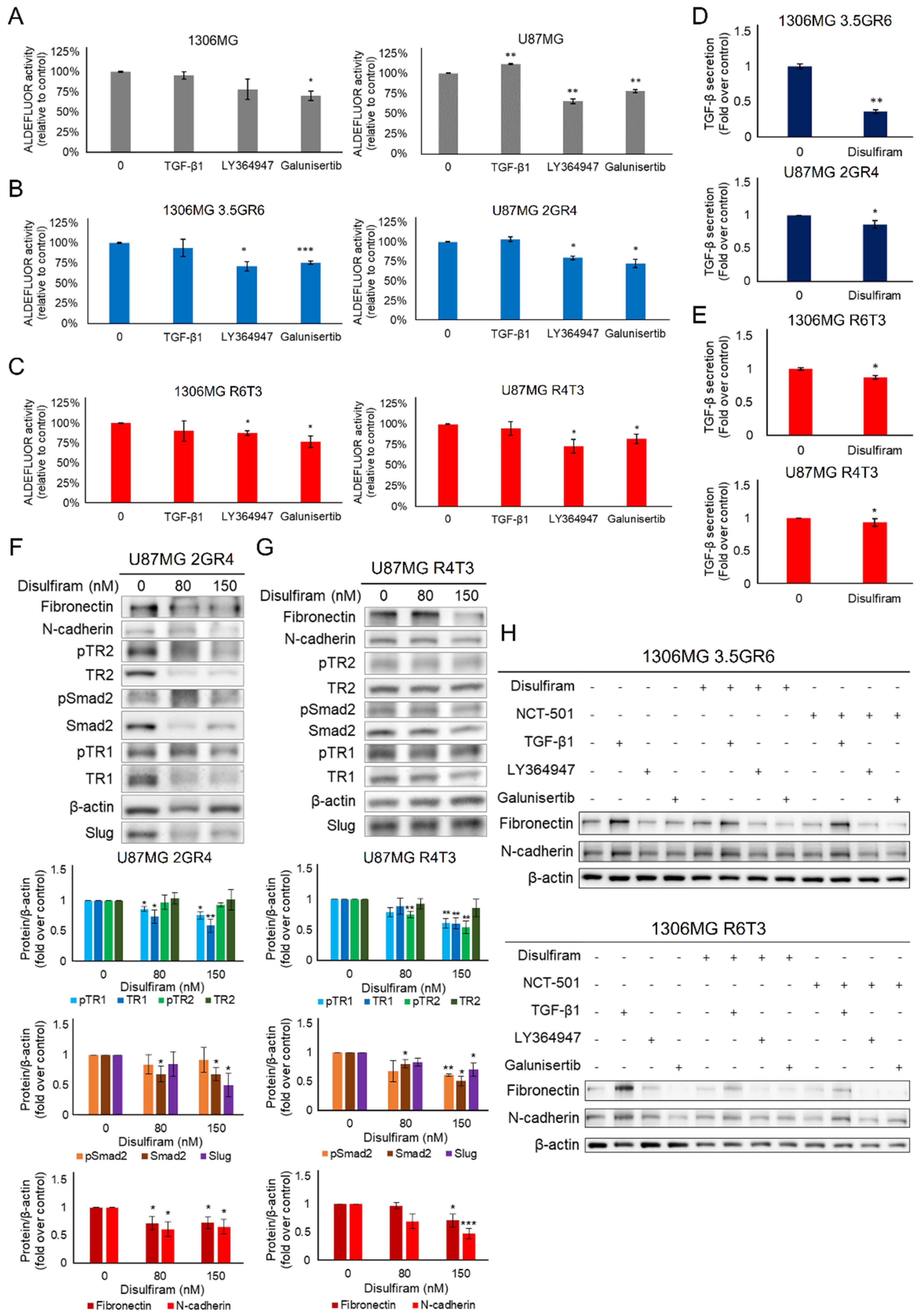
2.2. The Role of ALDH in Therapeutic Resistance
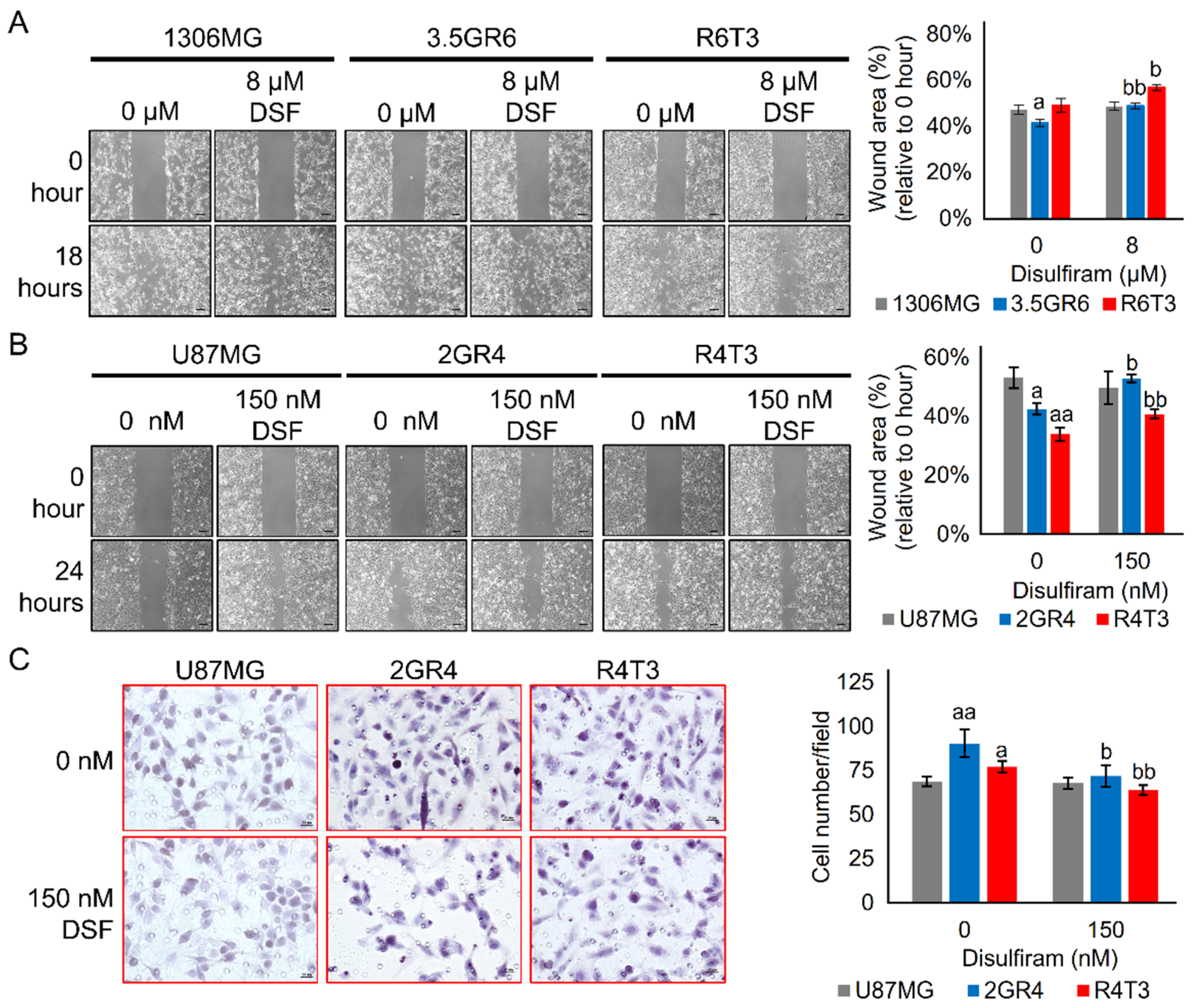
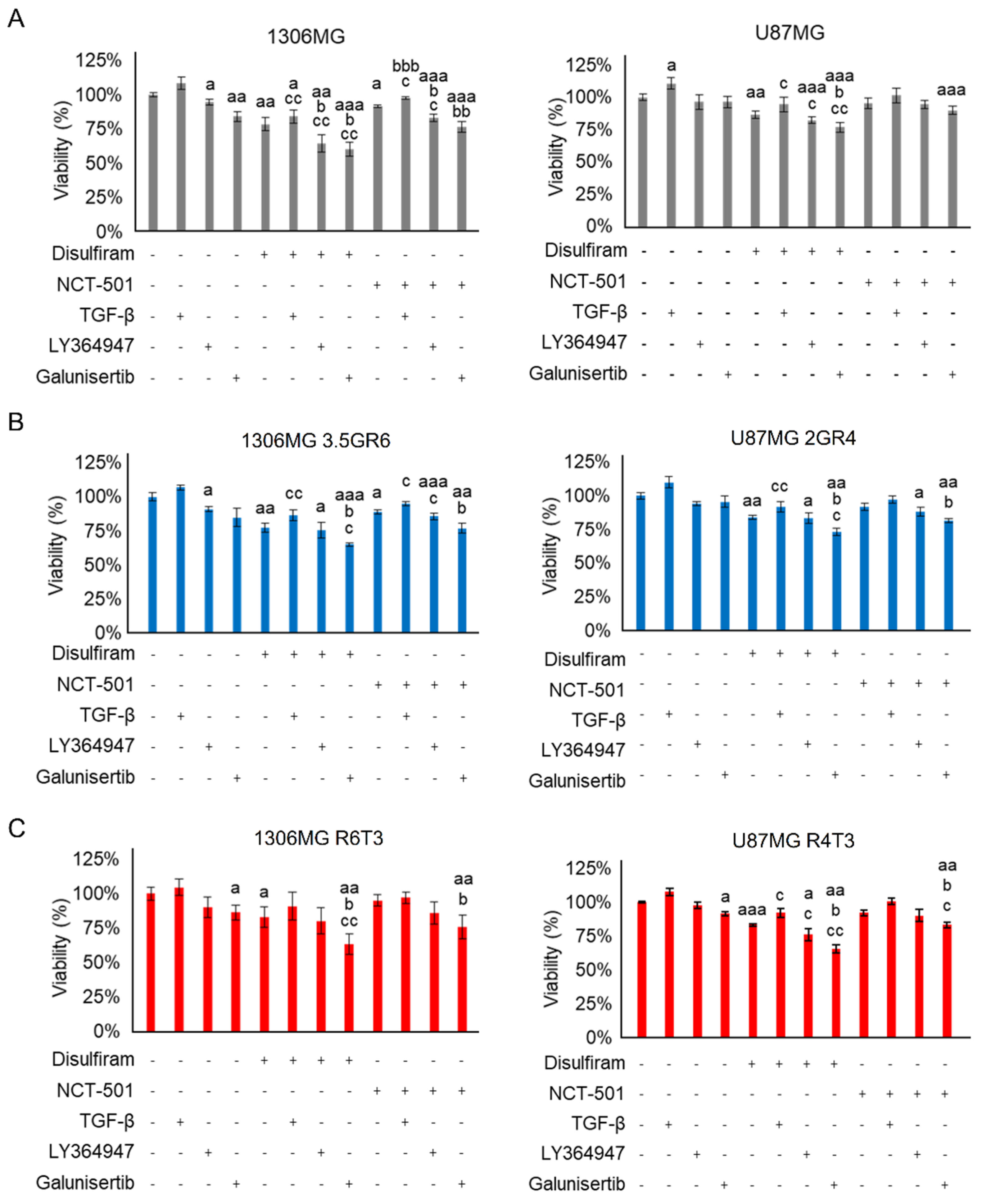
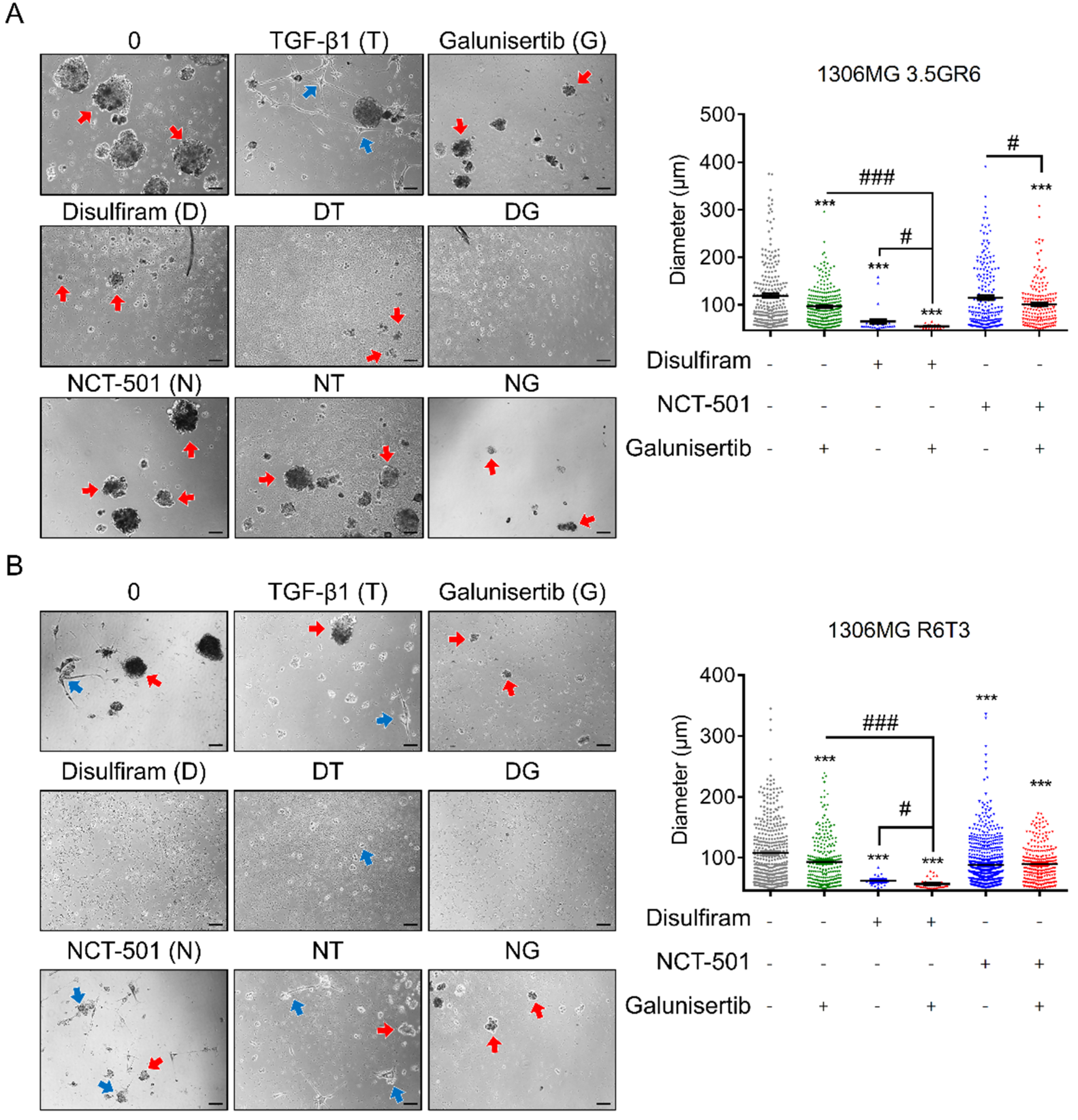
2.3. Combining TGF-β Signaling Inhibition and ALDH Inactivation Inhibits Cell Migration and the Growth of Tumor Spheres
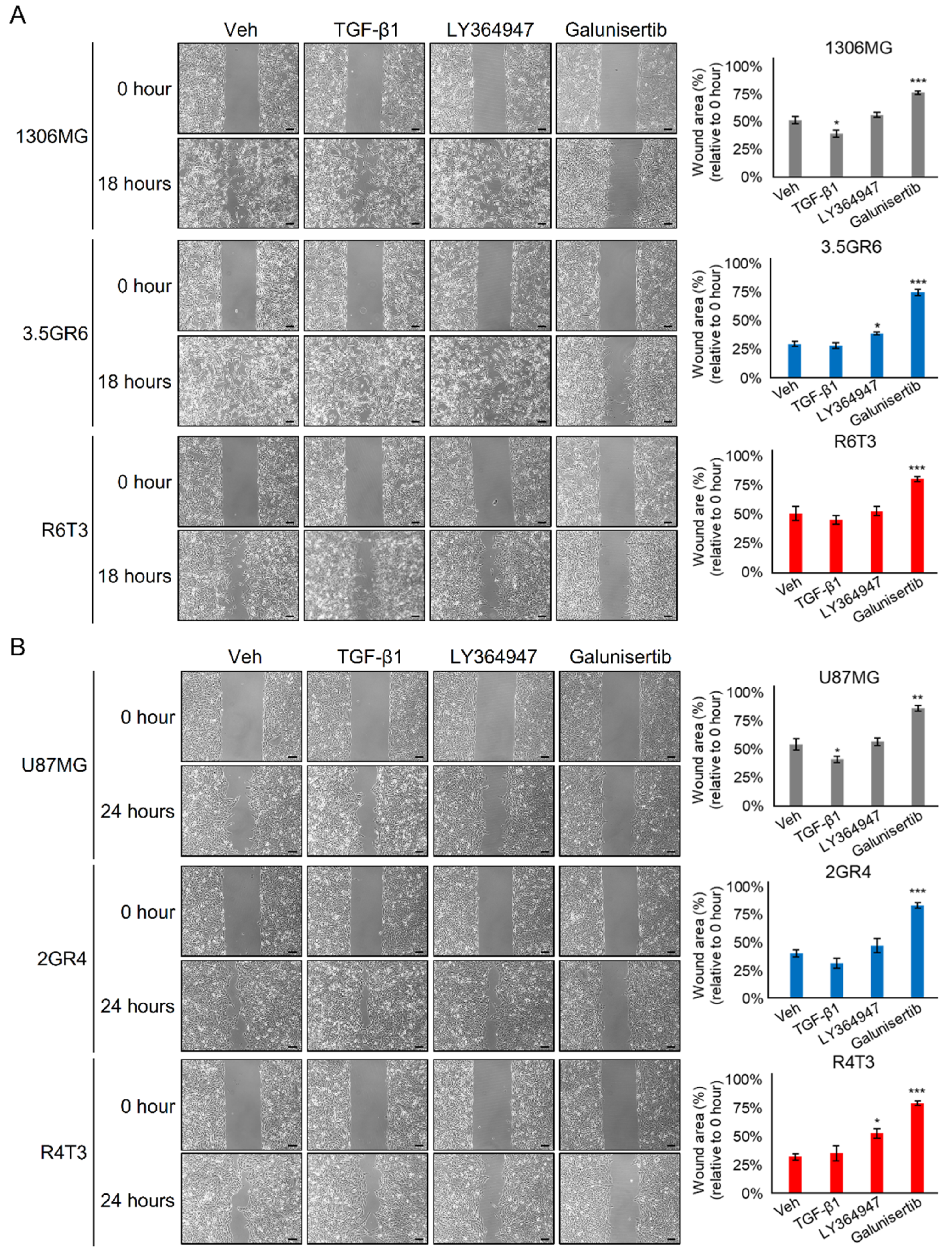
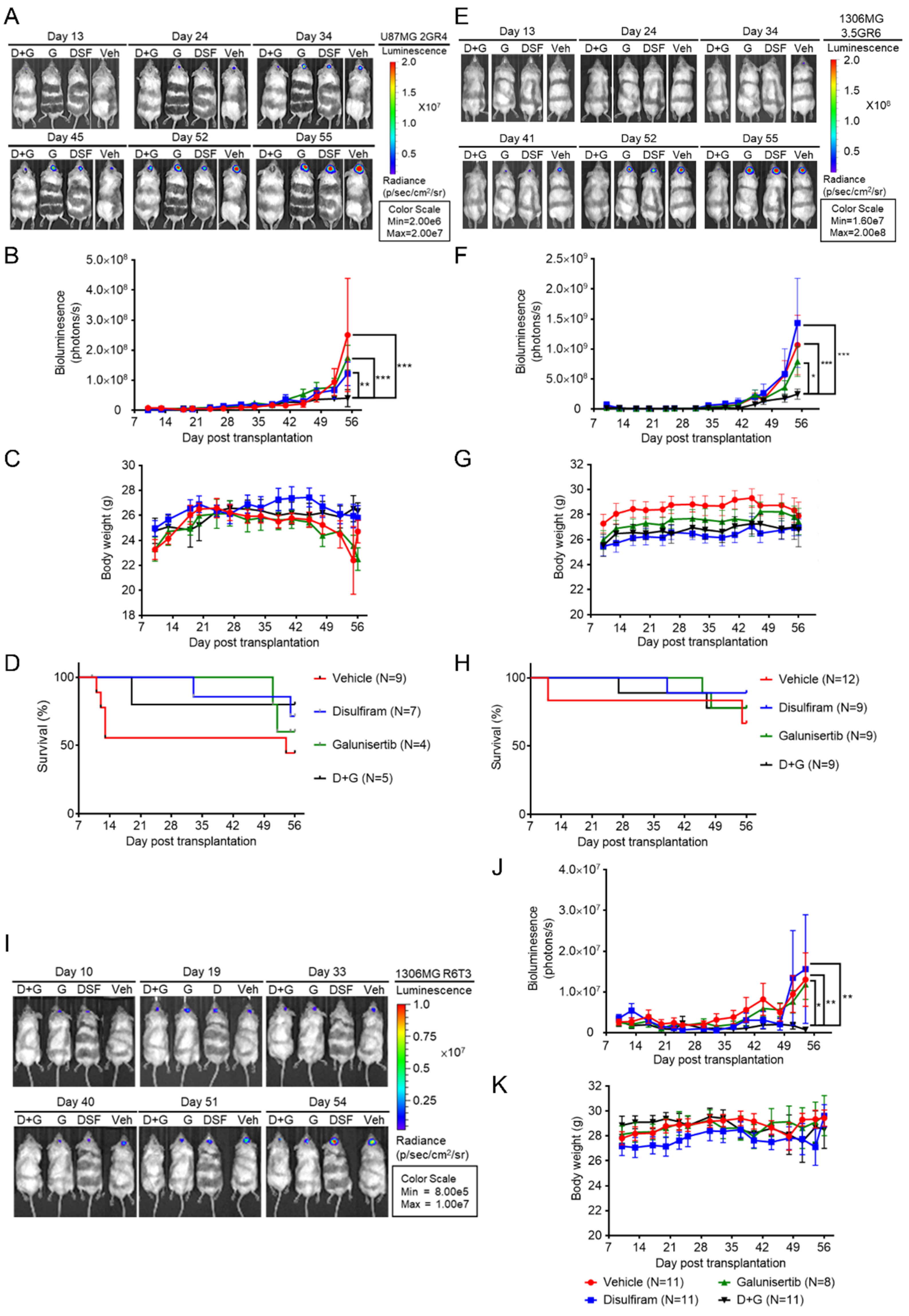
2.4. Disulfiram Sensitizes Resistant GBM to Galunisertib
2.5. The Interplay between TGF-β Signaling and ALDH
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Developing Therapeutic-Resistant GBM Cell Lines
4.3. Enzyme-Linked Immunosorbent Assay (ELISA)
4.4. MTT Assay
4.5. ALDEFLUOR Assay
4.6. Trypan Blue Exclusion Assay for Cell Viability
4.7. Combination Index Calculation and Determination of Combined Effects
4.8. Wound Healing Assay
4.9. Transwell Assay
4.10. Tumor Sphere Formation Assay
4.11. Western Blotting
4.12. Animals
4.13. Orthotopic Xenograft Animal Model and Bioluminescence Imaging
4.14. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stylli, S.S. Novel Treatment Strategies for Glioblastoma. Cancers 2020, 12, 2883. [Google Scholar] [CrossRef]
- Hein, A.L.; Ouellette, M.M.; Yan, Y. Radiation-induced signaling pathways that promote cancer cell survival (review). Int. J. Oncol. 2014, 45, 1813–1819. [Google Scholar] [CrossRef] [Green Version]
- Van Vulpen, M.; Kal, H.B.; Taphoorn, M.J.; El-Sharouni, S.Y. Changes in blood-brain barrier permeability induced by radiotherapy: Implications for timing of chemotherapy? (Review). Oncol. Rep. 2002, 9, 683–688. [Google Scholar] [CrossRef] [Green Version]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [Green Version]
- Osuka, S.; Van Meir, E.G. Overcoming therapeutic resistance in glioblastoma: The way forward. J. Clin. Investig. 2017, 127, 415–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zong, H.; Parada, L.F.; Baker, S.J. Cell of origin for malignant gliomas and its implication in therapeutic development. Cold Spring Harb. Perspect. Biol. 2015, 7, a020610. [Google Scholar] [CrossRef] [Green Version]
- Filbin, M.G.; Stiles, C.D. Of Brains and Blood: Developmental Origins of Glioma Diversity? Cancer Cell 2015, 28, 403–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Feng, Y.; Kennedy, D. Multidrug-resistant cancer cells and cancer stem cells hijack cellular systems to circumvent systemic therapies, can natural products reverse this? Cell. Mol. Life Sci. 2017, 74, 777–801. [Google Scholar] [CrossRef] [PubMed]
- Prieto-Vila, M.; Takahashi, R.U.; Usuba, W.; Kohama, I.; Ochiya, T. Drug Resistance Driven by Cancer Stem Cells and Their Niche. Int. J. Mol. Sci. 2017, 18, 2574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paula, A.C.; Lopes, C. Implications of Different Cancer Stem Cell Phenotypes in Breast Cancer. Anticancer Res. 2017, 37, 2173–2183. [Google Scholar] [CrossRef] [Green Version]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.; Rich, J.N. Cancer stem cells in glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roth, P.; Silginer, M.; Goodman, S.L.; Hasenbach, K.; Thies, S.; Maurer, G.; Schraml, P.; Tabatabai, G.; Moch, H.; Tritschler, I.; et al. Integrin control of the transforming growth factor-beta pathway in glioblastoma. Brain 2013, 136, 564–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drabsch, Y.; ten Dijke, P. TGF-beta signalling and its role in cancer progression and metastasis. Cancer Metastasis Rev. 2012, 31, 553–568. [Google Scholar] [CrossRef] [PubMed]
- Nana, A.W.; Yang, P.M.; Lin, H.Y. Overview of Transforming Growth Factor beta Superfamily Involvement in Glioblastoma Initiation and Progression. Asian Pac. J. Cancer Prev. 2015, 16, 6813–6823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruna, A.; Darken, R.S.; Rojo, F.; Ocana, A.; Penuelas, S.; Arias, A.; Paris, R.; Tortosa, A.; Mora, J.; Baselga, J.; et al. High TGFbeta-Smad activity confers poor prognosis in glioma patients and promotes cell proliferation depending on the methylation of the PDGF-B gene. Cancer Cell 2007, 11, 147–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Kleber, S.; Rohrich, M.; Timke, C.; Han, N.; Tuettenberg, J.; Martin-Villalba, A.; Debus, J.; Peschke, P.; Wirkner, U.; et al. Blockade of TGF-beta signaling by the TGFbetaR-I kinase inhibitor LY2109761 enhances radiation response and prolongs survival in glioblastoma. Cancer Res. 2011, 71, 7155–7167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhat, K.P.L.; Balasubramaniyan, V.; Vaillant, B.; Ezhilarasan, R.; Hummelink, K.; Hollingsworth, F.; Wani, K.; Heathcock, L.; James, J.D.; Goodman, L.D.; et al. Mesenchymal differentiation mediated by NF-κB promotes radiation resistance in glioblastoma. Cancer Cell 2013, 24, 331–346. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Cruzado, L.; Tornin, J.; Santos, L.; Rodriguez, A.; Garcia-Castro, J.; Moris, F.; Rodriguez, R. Aldh1 Expression and Activity Increase During Tumor Evolution in Sarcoma Cancer Stem Cell Populations. Sci. Rep. 2016, 6, 27878. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Chen, T.; Zhu, J.; Zhang, H.; Jiang, H.; Sun, H. High ALDH activity defines ovarian cancer stem-like cells with enhanced invasiveness and EMT progress which are responsible for tumor invasion. Biochem. Biophys. Res. Commun. 2018, 495, 1081–1088. [Google Scholar] [CrossRef]
- Gu, S.; Nguyen, B.N.; Rao, S.; Li, S.; Shetty, K.; Rashid, A.; Shukla, V.; Deng, C.X.; Mishra, L.; Mishra, B. Alcohol, stem cells and cancer. Genes Cancer 2017, 8, 695–700. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Torres, M.; Allan, A.L. Aldehyde dehydrogenase as a marker and functional mediator of metastasis in solid tumors. Clin. Exp. Metastasis 2016, 33, 97–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Januchowski, R.; Wojtowicz, K.; Zabel, M. The role of aldehyde dehydrogenase (ALDH) in cancer drug resistance. Biomed. Pharm. 2013, 67, 669–680. [Google Scholar] [CrossRef]
- Croker, A.K.; Allan, A.L. Inhibition of aldehyde dehydrogenase (ALDH) activity reduces chemotherapy and radiation resistance of stem-like ALDHhiCD44(+) human breast cancer cells. Breast Cancer Res. Treat. 2012, 133, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Wu, C.L.; Yeh, I.C.; Wu, S.N.; Sze, C.I.; Gean, P.W. Cilostazol eliminates radiation-resistant glioblastoma by re-evoking big conductance calcium-activated potassium channel activity. Am. J. Cancer Res. 2021, 11, 1148–1169. [Google Scholar] [CrossRef] [PubMed]
- Abbruzzese, C.; Matteoni, S.; Signore, M.; Cardone, L.; Nath, K.; Glickson, J.D.; Paggi, M.G. Drug repurposing for the treatment of glioblastoma multiforme. J. Exp. Clin. Cancer Res. 2017, 36, 169. [Google Scholar] [CrossRef]
- Koppaka, V.; Thompson, D.C.; Chen, Y.; Ellermann, M.; Nicolaou, K.C.; Juvonen, R.O.; Petersen, D.; Deitrich, R.A.; Hurley, T.D.; Vasiliou, V. Aldehyde dehydrogenase inhibitors: A comprehensive review of the pharmacology, mechanism of action, substrate specificity, and clinical application. Pharmacol. Rev. 2012, 64, 520–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tesson, M.; Anselmi, G.; Bell, C.; Mairs, R. Cell cycle specific radiosensitisation by the disulfiram and copper complex. Oncotarget 2017, 8, 65900–65916. [Google Scholar] [CrossRef]
- Karamanakos, P.N.; Trafalis, D.T.; Papachristou, D.J.; Panteli, E.S.; Papavasilopoulou, M.; Karatzas, A.; Kardamakis, D.; Nasioulas, G.; Marselos, M. Evidence for the efficacy of disulfiram and copper combination in glioblastoma multiforme—A propos of a case. J. BUON 2017, 22, 1227–1232. [Google Scholar]
- Wang, Y.; Li, W.; Patel, S.S.; Cong, J.; Zhang, N.; Sabbatino, F.; Liu, X.; Qi, Y.; Huang, P.; Lee, H.; et al. Blocking the formation of radiation-induced breast cancer stem cells. Oncotarget 2014, 5, 3743–3755. [Google Scholar] [CrossRef] [Green Version]
- David, C.J.; Massague, J. Contextual determinants of TGFbeta action in development, immunity and cancer. Nat. Rev. Mol. Cell Biol. 2018, 19, 419–435. [Google Scholar] [CrossRef]
- Iwadate, Y. Epithelial-mesenchymal transition in glioblastoma progression. Oncol. Lett. 2016, 11, 1615–1620. [Google Scholar] [CrossRef] [Green Version]
- Katsuno, Y.; Lamouille, S.; Derynck, R. TGF-beta signaling and epithelial-mesenchymal transition in cancer progression. Curr. Opin. Oncol. 2013, 25, 76–84. [Google Scholar] [CrossRef]
- van den Hoogen, C.; van der Horst, G.; Cheung, H.; Buijs, J.T.; Pelger, R.C.; van der Pluijm, G. The aldehyde dehydrogenase enzyme 7A1 is functionally involved in prostate cancer bone metastasis. Clin. Exp. Metastasis 2011, 28, 615–625. [Google Scholar] [CrossRef] [Green Version]
- Shuang, Z.Y.; Wu, W.C.; Xu, J.; Lin, G.; Liu, Y.C.; Lao, X.M.; Zheng, L.; Li, S. Transforming growth factor-beta1-induced epithelial-mesenchymal transition generates ALDH-positive cells with stem cell properties in cholangiocarcinoma. Cancer Lett. 2014, 354, 320–328. [Google Scholar] [CrossRef]
- Hoshino, Y.; Nishida, J.; Katsuno, Y.; Koinuma, D.; Aoki, T.; Kokudo, N.; Miyazono, K.; Ehata, S. Smad4 Decreases the Population of Pancreatic Cancer-Initiating Cells through Transcriptional Repression of ALDH1A1. Am. J. Pathol. 2015, 185, 1457–1470. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jiang, Y.; Tian, T.; Hori, Y.; Wada, N.; Ikeda, J.; Morii, E. Inhibitory effect of Nodal on the expression of aldehyde dehydrogenase 1 in endometrioid adenocarcinoma of uterus. Biochem. Biophys. Res. Commun. 2013, 440, 731–736. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, J.P.; Cooper, D.A.; Schank, J.R.; Lyle, M.A.; Gaval-Cruz, M.; Ogbonmwan, Y.E.; Pozdeyev, N.; Freeman, K.G.; Iuvone, P.M.; Edwards, G.L.; et al. Disulfiram attenuates drug-primed reinstatement of cocaine seeking via inhibition of dopamine beta-hydroxylase. Neuropsychopharmacology 2010, 35, 2440–2449. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Chen, F.; Chen, J.; Chan, S.; He, Y.; Liu, W.; Zhang, G. Disulfiram/Copper Induces Antitumor Activity against Both Nasopharyngeal Cancer Cells and Cancer-Associated Fibroblasts through ROS/MAPK and Ferroptosis Pathways. Cancers 2020, 12, 138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, D.; Wu, G.; Chang, C.; Zhu, F.; Xiao, Y.; Li, Q.; Zhang, T.; Zhang, L. Disulfiram inhibits TGF-β-induced epithelial-mesenchymal transition and stem-like features in breast cancer via ERK/NF-κB/Snail pathway. Oncotarget 2015, 6, 40907–40919. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Li, X.; Ren, Y.; Zhang, X. Disulfiram: A novel repurposed drug for cancer therapy. Cancer Chemother. Pharm. 2021, 87, 159–172. [Google Scholar] [CrossRef]
- Zhang, J.; Pu, K.; Bai, S.; Peng, Y.; Li, F.; Ji, R.; Guo, Q.; Sun, W.; Wang, Y. The anti-alcohol dependency drug disulfiram inhibits the viability and progression of gastric cancer cells by regulating the Wnt and NF-kappaB pathways. J. Int. Med. Res. 2020, 48. [Google Scholar] [CrossRef]
- Xu, Y.; Zhou, Q.; Feng, X.; Dai, Y.; Jiang, Y.; Jiang, W.; Liu, X.; Xing, X.; Wang, Y.; Ni, Y.; et al. Disulfiram/copper markedly induced myeloma cell apoptosis through activation of JNK and intrinsic and extrinsic apoptosis pathways. Biomed. Pharm. 2020, 126, 110048. [Google Scholar] [CrossRef]
- Huang, J.; Chaudhary, R.; Cohen, A.L.; Fink, K.; Goldlust, S.; Boockvar, J.; Chinnaiyan, P.; Wan, L.; Marcus, S.; Campian, J.L. A multicenter phase II study of temozolomide plus disulfiram and copper for recurrent temozolomide-resistant glioblastoma. J. Neuro-Oncol. 2019, 142, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Michelakos, T.; Wang, B.; Shang, Z.; DeLeo, A.B.; Duan, Z.; Hornicek, F.J.; Schwab, J.H.; Wang, X. Targeting cancer stem cells by disulfiram and copper sensitizes radioresistant chondrosarcoma to radiation. Cancer Lett. 2021, 505, 37–48. [Google Scholar] [CrossRef]
- Wang, R.; Shen, J.; Yan, H.; Gao, X.; Dong, T.; Wang, P.; Zhou, J. The Evolving Role of Disulfiram in Radiobiology and the Treatment of Breast Cancer. OncoTargets Ther. 2020, 13, 10441–10446. [Google Scholar] [CrossRef]
- Koh, H.K.; Seo, S.Y.; Kim, J.H.; Kim, H.J.; Chie, E.K.; Kim, S.K.; Kim, I.H. Disulfiram, a Re-positioned Aldehyde Dehydrogenase Inhibitor, Enhances Radiosensitivity of Human Glioblastoma Cells In Vitro. Cancer Res. Treat. 2019, 51, 696–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rolle, F.; Bincoletto, V.; Gazzano, E.; Rolando, B.; Lollo, G.; Stella, B.; Riganti, C.; Arpicco, S. Coencapsulation of disulfiram and doxorubicin in liposomes strongly reverses multidrug resistance in breast cancer cells. Int. J. Pharm. 2020, 580, 119191. [Google Scholar] [CrossRef] [PubMed]
- Jangra, A.; Choi, S.A.; Yang, J.; Koh, E.J.; Phi, J.H.; Lee, J.Y.; Wang, K.C.; Kim, S.K. Disulfiram potentiates the anticancer effect of cisplatin in atypical teratoid/rhabdoid tumors (AT/RT). Cancer Lett. 2020, 486, 38–45. [Google Scholar] [CrossRef]
- Rodon, J.; Carducci, M.A.; Sepulveda-Sanchez, J.M.; Azaro, A.; Calvo, E.; Seoane, J.; Brana, I.; Sicart, E.; Gueorguieva, I.; Cleverly, A.L.; et al. First-in-human dose study of the novel transforming growth factor-beta receptor I kinase inhibitor LY2157299 monohydrate in patients with advanced cancer and glioma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 553–560. [Google Scholar] [CrossRef] [Green Version]
- Wick, A.; Desjardins, A.; Suarez, C.; Forsyth, P.; Gueorguieva, I.; Burkholder, T.; Cleverly, A.L.; Estrem, S.T.; Wang, S.; Lahn, M.M.; et al. Phase 1b/2a study of galunisertib, a small molecule inhibitor of transforming growth factor-beta receptor I, in combination with standard temozolomide-based radiochemotherapy in patients with newly diagnosed malignant glioma. Invesig. New Drugs 2020, 38, 1570–1579. [Google Scholar] [CrossRef] [Green Version]
- Brandes, A.A.; Carpentier, A.F.; Kesari, S.; Sepulveda-Sanchez, J.M.; Wheeler, H.R.; Chinot, O.; Cher, L.; Steinbach, J.P.; Capper, D.; Specenier, P.; et al. A Phase II randomized study of galunisertib monotherapy or galunisertib plus lomustine compared with lomustine monotherapy in patients with recurrent glioblastoma. Neuro Oncol. 2016, 18, 1146–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujii, H.; Honoki, K.; Tsujiuchi, T.; Kido, A.; Yoshitani, K.; Takakura, Y. Sphere-forming stem-like cell populations with drug resistance in human sarcoma cell lines. Int. J. Oncol. 2009, 34, 1381–1386. [Google Scholar] [PubMed]
- Franco, S.S.; Szczesna, K.; Iliou, M.S.; Al-Qahtani, M.; Mobasheri, A.; Kobolák, J.; Dinnyés, A. In vitro models of cancer stem cells and clinical applications. BMC Cancer 2016, 16, 738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gedye, C.; Sirskyj, D.; Lobo, N.C.; Meens, J.; Hyatt, E.; Robinette, M.; Fleshner, N.; Hamilton, R.J.; Kulkarni, G.; Zlotta, A.; et al. Cancer stem cells are underestimated by standard experimental methods in clear cell renal cell carcinoma. Sci. Rep. 2016, 6, 25220. [Google Scholar] [CrossRef] [Green Version]
- Chou, T.C. Preclinical versus clinical drug combination studies. Leuk. Lymphoma 2008, 49, 2059–2080. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disufiram (DSF) (μM) | Galunisertib (G) (μM) | Cytotoxicity of DSF + G | CI | Description | |
|---|---|---|---|---|---|
| 1306MG | 8 | 100 | 0.328 | 1.203 | Moderate antagonism |
| 3.5GR6 | 8 | 100 | 0.359 | 0.895 | Slight synergism |
| R6T3 | 8 | 100 | 0.377 | 0.897 | Slight synergism |
| U87MG | 150 | 100 | 0.274 | 1.333 | Moderate antagonism |
| 2GR4 | 150 | 100 | 0.279 | 0.672 | Synergism |
| R4T3 | 150 | 100 | 0.393 | 0.970 | Nearly additive |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, C.-C.; Wu, C.-L.; Lin, M.-X.; Sze, C.-I.; Gean, P.-W. Disulfiram Sensitizes a Therapeutic-Resistant Glioblastoma to the TGF-β Receptor Inhibitor. Int. J. Mol. Sci. 2021, 22, 10496. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910496
Liu C-C, Wu C-L, Lin M-X, Sze C-I, Gean P-W. Disulfiram Sensitizes a Therapeutic-Resistant Glioblastoma to the TGF-β Receptor Inhibitor. International Journal of Molecular Sciences. 2021; 22(19):10496. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910496
Chicago/Turabian StyleLiu, Chan-Chuan, Cheng-Lin Wu, Meng-Xuan Lin, Chun-I Sze, and Po-Wu Gean. 2021. "Disulfiram Sensitizes a Therapeutic-Resistant Glioblastoma to the TGF-β Receptor Inhibitor" International Journal of Molecular Sciences 22, no. 19: 10496. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910496