
In Silico Modeling of the Influence of Environment on Amyloid Folding Using FOD-M Model



Abstract
:1. Introduction
2. Results
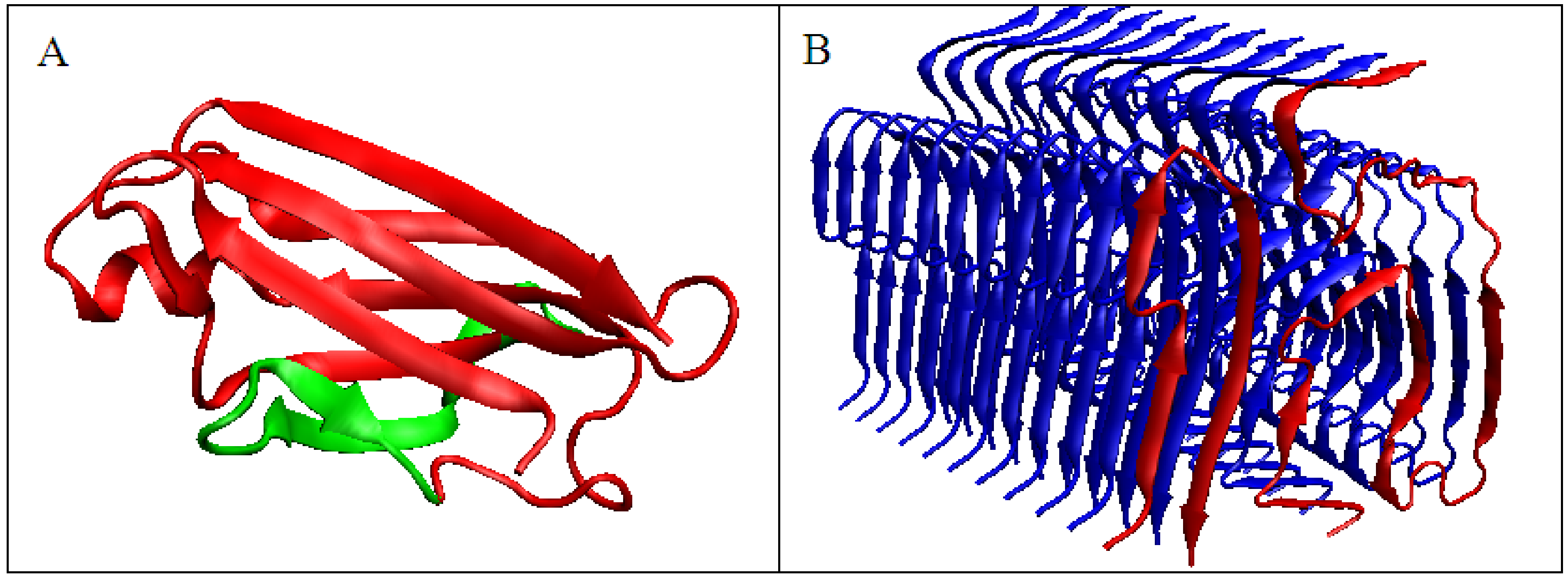
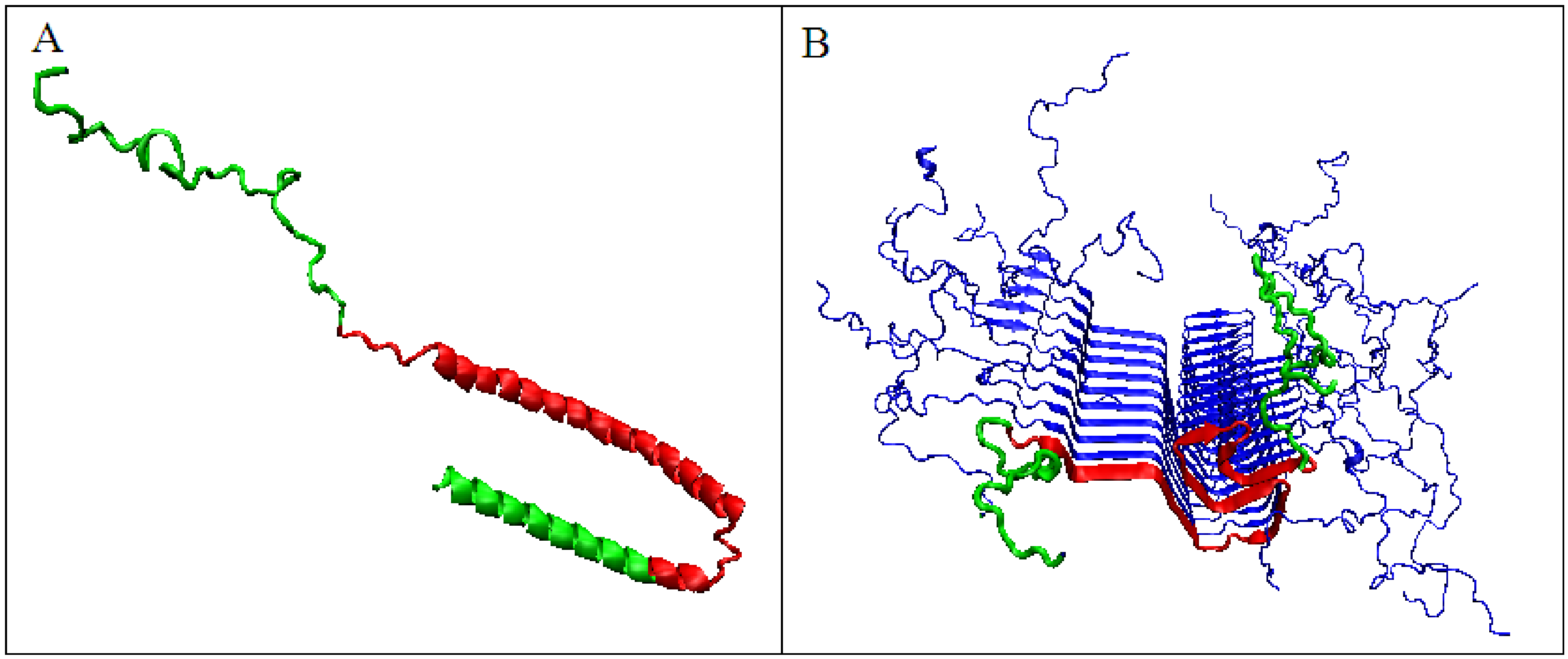
2.1. Comparative Analysis of Amyloid Proteins and Their Native Forms
2.2. Proteins with Increasing K Values Accompanying Amyloid Transformation
2.3. Amyloid Transformation, Accompanied by a Decrease in the Value of the Parameter K
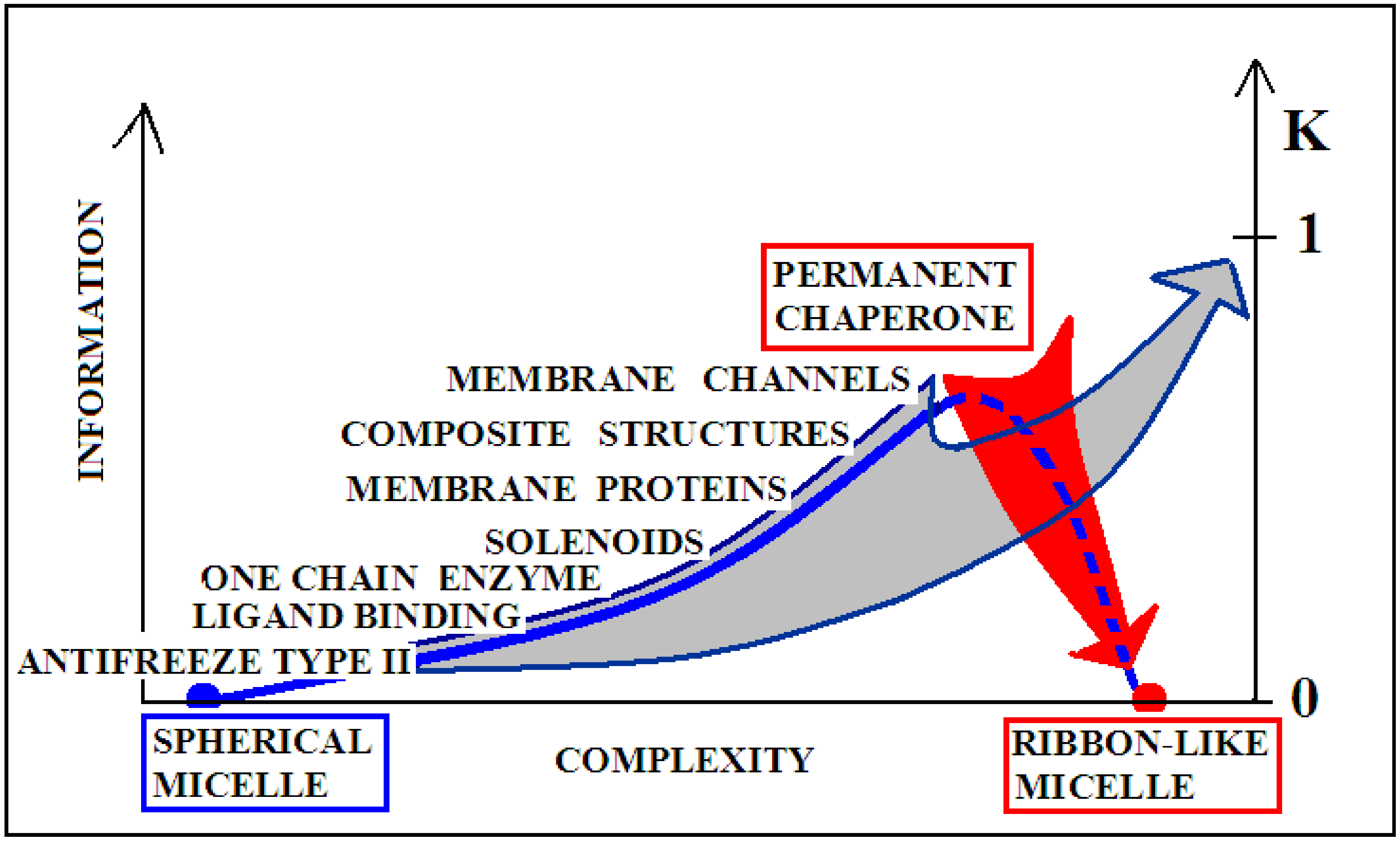
3. Discussion
4. Materials and Methods
4.1. Data
4.2. Description of the FOD-M Model
4.3. Programs Used
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Heemels, M.T. Neurodegenerative diseases. Nature 2016, 539, 179. [Google Scholar] [CrossRef] [Green Version]
- Kovacs, G.G. Concepts and classification of neurodegenerative diseases. Handb. Clin. Neurol. 2017, 145, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Tejera, D.; Heneka, M.T. Microglia in Neurodegenerative Disorders. Methods Mol. Biol. 2019, 2034, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Lendahl, U.; Nilsson, P.; Betsholtz, C. Emerging links between cerebrovascular and neurodegenerative diseases—A special role for pericytes. EMBO Rep. 2019, 20, e48070. [Google Scholar] [CrossRef] [PubMed]
- Gitler, A.D.; Dhillon, P.; Shorter, J. Neurodegenerative disease: Models, mechanisms, and a new hope. Dis. Model Mech. 2017, 10, 499–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuny, G.D. Foreword: Neurodegenerative diseases: Challenges and opportunities. Future Med. Chem. 2012, 4, 1647–1649. [Google Scholar] [CrossRef]
- Miksys, S.L.; Tyndale, R.F. Neurodegenerative diseases: A growing challenge. Clin. Pharmacol. Ther. 2010, 88, 427–430. [Google Scholar] [CrossRef]
- Bassil, F.; Fernagut, P.O.; Bezard, E.; Meissner, W.G. Insulin, IGF-1 and GLP-1 signaling in neurodegenerative disorders: Targets for disease modification? Prog Neurobiol. 2014, 118, 1–18. [Google Scholar] [CrossRef]
- Delacourte, A.; Buée, L. Tau pathology: A marker of neurodegenerative disorders. Curr. Opin. Neurol. 2000, 13, 371–376. [Google Scholar] [CrossRef]
- Christen, Y. Proteins and mutations: A new vision (molecular) of neurodegenerative diseases. J. Soc. Biol. 2002, 196, 85–94. [Google Scholar] [CrossRef]
- Chuang, E.; Hori, A.M.; Hesketh, C.D.; Shorter, J. Amyloid assembly and disassembly. J. Cell Sci. 2018, 131, jcs189928. [Google Scholar] [CrossRef] [Green Version]
- Killian, A.N.; Miller, S.C.; Hines, J.K. Impact of Amyloid Polymorphism on Prion-Chaperone Interactions in Yeast. Viruses 2019, 11, 349. [Google Scholar] [CrossRef] [Green Version]
- Adamcik, J.; Mezzenga, R. Amyloid Polymorphism in the Protein Folding and Aggregation Energy Landscape. Angew. Chem. Int. Ed. 2018, 57, 8370–8382. [Google Scholar] [CrossRef]
- Stanković, I.M.; Niu, S.; Hall, M.B.; Zarić, S.D. Role of aromatic amino acids in amyloid self-assembly. Int. J. Biol. Macromol. 2020, 156, 949–959. [Google Scholar] [CrossRef] [PubMed]
- Wetzel, R.; Shivaprasad, S.; Williams, A.D. Plasticity of amyloid fibrils. Biochemistry 2007, 46, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toyama, B.H.; Weissman, J.S. Amyloid structure: Conformational diversity and consequences. Annu. Rev. Biochem. 2011, 80, 557–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otzen, D.; Riek, R. Functional Amyloids. Cold Spring Harbor Perspect. Biol. 2019, 11, a033860. [Google Scholar] [CrossRef] [PubMed]
- Riek, R.; Eisenberg, D.S. The activities of amyloids from a structural perspective. Nature 2016, 539, 227–235. [Google Scholar] [CrossRef]
- Bi, T.M.; Daggett, V. The Role of alpha-sheet in Amyloid Oligomer Aggregation and Toxicity. Yale J. Biol. Med. 2018, 91, 247–255. [Google Scholar]
- Recasens, A.; Ulusoy, A.; Kahle, P.J.; Di Monte, D.A.; Dehay, B. In vivo models of alpha-synuclein transmission and propagation. Cell Tissue Res. 2018, 373, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Oldfield, C.J.; Dunker, A.K. Intrinsically disordered proteins in human diseases: Introducing the D2 concept. Annu. Rev. Biophys. 2008, 37, 215–246. [Google Scholar] [CrossRef] [PubMed]
- Van der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.; et al. Classification of intrinsically disordered regions and proteins. Chem. Rev. 2014, 114, 6589–6631. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, P.; Uversky, V.N. Intrinsically Disordered Proteins in Chronic Diseases. Biomolecules 2019, 9, 147. [Google Scholar] [CrossRef] [Green Version]
- Uversky, V.N. Intrinsically disordered proteins in overcrowded milieu: Membrane-less organelles, phase separation, and intrinsic disorder. Curr. Opin. Struct. Biol. 2017, 44, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Yruela, I.; Neira, J.L. Intrinsically disordered proteins in biology: One for all, all for one. Arch. Biochem. Biophys. 2020, 684, 108328. [Google Scholar] [CrossRef]
- Kulkarni, V.; Kulkarni, P. Intrinsically disordered proteins and phenotypic switching: Implications in cancer. Prog Mol. Biol. Transl. Sci. 2019, 166, 63–84. [Google Scholar] [CrossRef]
- Serpell, L.C. Alzheimer’s amyloid fibrils: Structure and assembly. Biochim. Biophys. Acta Mol. Basis Dis. 2000, 1502, 16–30. [Google Scholar] [CrossRef] [Green Version]
- Ladner-Keay, C.L.; Griffith, B.J.; Wishart, D.S. Shaking alone induces de novo conversion of recombinant prion proteins to beta-sheet rich oligomers and fibrils. PLoS ONE 2014, 9, e98753. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Xu, L.; Tang, J.Y.; Zuo, Y.Y.; Hu, G. Adsorption of Phospholipids at the Air-Water Surface. Biophys. J. 2019, 117, 1224–1233. [Google Scholar] [CrossRef]
- Sarker, D.K. Architectures and Mechanical Properties of Drugs and Complexes of Surface-Active Compounds at Air-Water and Oil-Water Interfaces. Curr. Drug Discov. Technol. 2019, 16, 11–29. [Google Scholar] [CrossRef]
- Ariga, K.; Hill, J.P. Monolayers at air-water interfaces: From origins-of-life to nanotechnology. Chem. Rec. 2011, 11, 199–211. [Google Scholar] [CrossRef]
- Fan, Y.; Chen, X.; Yang, L.; Cremer, P.S.; Gao, Y.Q. On the structure of water at the aqueous/air interface. J. Phys. Chem. B 2009, 113, 11672–11679. [Google Scholar] [CrossRef]
- Nguyen, C.V.; Nakahara, H.; Phan, C.M. Surface Potential of the Air/Water Interface. J. Oleo Sci. 2020, 69, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Kalinowska, B.; Banach, M.; Konieczny, L.; Roterman, I. Application of Divergence Entropy to Characterize the Structure of the Hydrophobic Core in DNA Interacting Proteins. Entropy 2015, 17, 1477–1507. [Google Scholar] [CrossRef] [Green Version]
- Banach, M.; Stapor, K.; Konieczny, L.; Fabian, P.; Roterman, I. Downhill, Ultrafast and Fast Folding Proteins Revised. Int. J. Mol. Sci. 2020, 21, 7632. [Google Scholar] [CrossRef] [PubMed]
- Banach, M.; Konieczny, L.; Roterman, I. Local discordance. In From Globular Proteins to Amyloids; Roterman-Konieczna, I., Ed.; Elsevier: Amsterdam, The Netherlands; Oxford, UK; Cambridge, MA, USA, 2020; pp. 71–94. [Google Scholar]
- Roterman, I.; Stapor, K.; Fabian, P.; Konieczny, L.; Banach, M. Model of Environmental Membrane Field for Transmembrane Proteins. Int. J. Mol. Sci. 2021, 22, 3619. [Google Scholar] [CrossRef] [PubMed]
- Colvin, M.T.; Silvers, R.; Ni, Q.Z.; Can, T.V.; Sergeyev, I.; Rosay, M.; Donovan, K.J.; Wall, M.B.; Linse, J.S.; Griffin, R.G. Atomic Resolution Structure of Monomorphic Aβ42 Amyloid Fibrils. J. Am. Chem. Soc. 2016, 138, 9663–9674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klabunde, T.; Petrassi, H.M.; Oza, V.B.; Raman, P.; Kelly, J.W.; Sacchettini, J.C. Rational design of potent human transthyretin amyloid disease inhibitors. Nat. Struct. Biol. 2000, 7, 312–321. [Google Scholar] [CrossRef]
- Swuec, P.; Lavatelli, F.; Tasaki, M.; Paissoni, C.; Rognoni, P.; Maritan, M.; Brambilla, F.; Milani, P.; Mauri, P.; Camilloni, C.; et al. Cryo-EM structure of cardiac amyloid fibrils from an immunoglobulin light chain AL amyloidosis patient. Nat. Commun. 2019, 10, 1269. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.B.; Ainsworth, C.F.; Stevens, F.J.; Schiffer, M. Three quaternary structures for a single protein. Proc. Natl. Acad. Sci. USA 1996, 93, 7017–7021. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, M.; Wiese, S.; Adak, V.; Engler, J.; Agarwal, S.; Fritz, G.; Westermark, P.; Zacharias, M.; Fändrich, M. Cryo-EM structure of a transthyretin-derived amyloid fibril from a patient with hereditary ATTR amyloidosis. Nat. Commun. 2019, 10, 5008. [Google Scholar] [CrossRef] [Green Version]
- Fitzpatrick, A.W.P.; Falcon, B.; He, S.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Crowther, R.A.; Ghetti, B.; Goedert, M.; Scheres, S.H.W. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 2017, 547, 185–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulmer, T.S.; Bax, A.; Cole, N.B.; Nussbaum, R.L. Structure and dynamics of micelle-bound human alpha-synuclein. J. Biol. Chem. 2005, 280, 9595–9603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuttle, M.D.; Comellas, G.; Nieuwkoop, A.J.; Covell, D.J.; Berthold, D.A.; Kloepper, K.D.; Courtney, J.M.; Kim, J.K.; Barclay, A.M.; Kendall, A.; et al. Solid-state NMR structure of a pathogenic fibril of full-length human α-synuclein. Nat. Struct. Mol. Biol. 2016, 23, 409–415. [Google Scholar] [CrossRef]
- Xiao, Y.; Ma, B.; McElheny, D.; Parthasarathy, S.; Long, F.; Hoshi, M.; Nussinov, R.; Ishii, Y. Aβ(1–42) fibril structure illuminates self-recognition and replication of amyloid in Alzheimer’s disease. Nat. Struct. Mol. Biol. 2015, 22, 499–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sgourakis, N.S.; Yau, W.-M.; Qiang, W. Modeling an in-register, parallel “iowa” aβ fibril structure using solid-state NMR data from labeled samples with rosetta. Structure 2015, 23, 216–227. [Google Scholar] [CrossRef] [Green Version]
- Schütz, A.K.; Vagt, T.; Huber, M.; Ovchinnikova, O.Y.; Cadalbert, R.; Wall, J.; Güntert, P.; Böckmann, A.; Glockshuber, R.; Meier, B.H. Atomic-resolution three-dimensional structure of amyloid β fibrils bearing the Osaka mutation. Angew. Chem. Int. Ed. 2015, 54, 331–335. [Google Scholar] [CrossRef] [Green Version]
- Roterman, I.; Stapor, K.; Fabian, P.; Konieczny, L. The functional significance of the distribution of hydrophobic residues in bacterial beta-barrel transmembrane proteins. Membranes 2021, 11, 580. [Google Scholar] [CrossRef]
- Van Melckebeke, H.; Wasmer, C.; Lange, A.; Ab, E.; Loquet, A.; Böckmann, A.; Meier, B.H. Atomic-resolution three-dimensional structure of HET-s(218–289) amyloid fibrils by solid-state NMR spectroscopy. J. Am. Chem. Soc. 2010, 132, 13765–13775. [Google Scholar] [CrossRef] [Green Version]
- Schütz, A.K.; Soragni, A.; Hornemann, S.; Aguzzi, A.; Ernst, M.; Böckmann, A.; Meier, B.H. The amyloid-Congo red interface at atomic resolution. Angew. Chem. Int. Ed. 2011, 50, 5956–5960. [Google Scholar] [CrossRef]
- Herrmann, U.S.; Schütz, A.K.; Shirani, H.; Huang, D.; Saban, D.; Nuvolone, M.; Li, B.; Ballmer, B.; Åslund, A.K.O.; Mason, J.J.; et al. Structure-based drug design identifies polythiophenes as antiprion compounds. Sci. Transl. Med. 2015, 7, 299ra123. [Google Scholar] [CrossRef]
- Martinez, D.; Daskalov, A.; Andreas, L.; Bardiaux, B.; Coustou, V.; Stanek, J.; Berbon, M.; Noubhani, M.; Kauffmann, B.; Wall, J.S.; et al. Solid-state MAS NMR structure of the HELLF prion amyloid fibrils. 2018. [Google Scholar] [CrossRef]
- Wang, L.-Q.; Zhao, K.; Yuan, H.-E.; Wang, Q.; Guan, Z.; Tao, J.; Li, X.-N.; Sun, Y.; Yi, C.-W.; Chen, J.; et al. Cryo-EM structure of an amyloid fibril formed by full-length human prion protein. Nat. Struct. Mol. Biol. 2020, 27, 598–602. [Google Scholar] [CrossRef]
- Glynn, C.; Sawaya, M.R.; Ge, P.; Gallagher-Jones, M.; Short, C.W.; Bowman, R.; Apostol, M.; Zhou, Z.H.; Eisenberg, D.S.; Rodriguez, J.A. Cryo-EM structure of a human prion fibril with a hydrophobic, protease-resistant core. Nat. Struct. Mol. Biol. 2020, 27, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Hervas, R.; Rau, M.J.; Park, Y.; Zhang, W.; Murzin, A.G.; Fitzpatrick, J.A.; Scheres, S.H.; Si, K. Cryo-Em structure of a neuronal functional amyloid implicated in memory persistence in drosophila. Science 2020, 367, 1230. [Google Scholar] [CrossRef] [PubMed]
- Bansal, A.; Schmidt, M.; Rennegarbe, M.; Haupt, C.; Liberta, F.; Stecher, S.; Puscalau-Girtu, I.; Biedermann, A.; Fändrich, M. AA amyloid fibrils from diseased tissue are structurally different from in vitro formed SAA fibrils. Nat. Commun. 2021, 12, 1013. [Google Scholar] [CrossRef] [PubMed]
- Murray, D.T.; Kato, M.; Lin, Y.; Thurber, K.R.; Hung, I.; McKnight, S.L.; Tycko, R. Structure of FUS Protein Fibrils and Its Relevance to Self-Assembly and Phase Separation of Low-Complexity Domains. Cell 2017, 171, 615–627. [Google Scholar] [CrossRef] [Green Version]
- James, T.L.; Liu, H.; Ulyanov, N.B.; Farr-Jones, S.; Zhang, H.; Donne, D.G.; Kaneko, K.; Groth, D.; Mehlhorn, I.; Prusiner, S.B.; et al. Solution structure of a 142-residue recombinant prion protein corresponding to the infectious fragment of the scrapie isoform. Proc. Natl. Acad. Sci. USA 1997, 94, 10086–10091. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Antony, L.; Hartmann, R.; Knaus, K.J.; Surewicz, K.; Surewicz, W.K.; Yee, V.C. Conformational diversity in prion protein variants influences intermolecular beta-sheet formation. EMBO J. 2010, 29, 251–262. [Google Scholar] [CrossRef]
- Zahn, R.; Liu, A.; Lührs, T.; Riek, R.; Von Schroetter, C.; López García, F.; Billeter, M.; Calzolai, L.; Wider, G.; Wüthrich, K. NMR solution structure of the human prion protein. Proc. Natl. Acad. Sci. USA 2000, 97, 145–150. [Google Scholar] [CrossRef] [Green Version]
- Whelligan, D.K.; Solanki, S.; Taylor, D.; Thomson, D.W.; Cheung, K.-M.J.; Boxall, K.; Mas-Droux, C.; Barillari, C.; Burns, S.; Grummitt, C.G.; et al. Aminopyrazine inhibitors binding to an unusual inactive conformation of the mitotic kinase Nek2: SAR and structural characterization. J. Med. Chem. 2010, 53, 7682–7698. [Google Scholar] [CrossRef] [PubMed]
- Eichner, T.; Kalverda, A.P.; Thompson, G.S.; Homans, S.W.; Radford, S.E. Conformational conversion during amyloid formation at atomic resolution. Mol. Cell 2011, 41, 161–172. [Google Scholar] [CrossRef]
- Knaus, K.J.; Morillas, M.; Swietnicki, W.; Malone, M.; Surewicz, W.K.; Yee, V.C. Crystal structure of the human prion protein reveals a mechanism for oligomerization. Nat. Struct. Biol. 2001, 8, 770–774. [Google Scholar] [CrossRef]
- Zheng, Z.; Zhang, M.; Wang, Y.; Ma, R.; Guo, C.; Feng, L.; Wu, J.; Yao, H.; Lin, D. Structural basis for the complete resistance of the human prion protein mutant G127V to prion disease. Sci. Rep. 2018, 8, 13211. [Google Scholar] [CrossRef]
- Slapsak, U.; Ilc, G.; Plavec, J. Solution structure of mule deer prion protein with polymorphism S138. 2019. [Google Scholar] [CrossRef]
- Abskharon, R.; Wang, F.; Wohlkonig, A.; Ruan, J.; Soror, S.; Giachin, G.; Pardon, E.; Zou, W.; Legname, G.; Ma, J.; et al. Structural evidence for the critical role of the prion protein hydrophobic region in forming an infectious prion. PLoS Pathog. 2019, 15, e1008139. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, J.; Zhang, X.; Xu, C.; Tu, X. Solution structure of Rap1 BRCT domain from Saccharomyces cerevisiae reveals a novel fold. Biochem. Biophys. Res. Commun. 2011, 404, 1055–1059. [Google Scholar] [CrossRef]
- Sawaya, M.R.; Kraut, J. Loop and subdomain movements in the mechanism of Escherichia coli dihydrofolate reductase: Crystallographic evidence. Biochemistry 1997, 36, 586–603. [Google Scholar] [CrossRef] [PubMed]
- Brzovic, P.S.; Heikaus, C.C.; Kisselev, L.; Vernon, R.; Herbig, E.; Pacheco, D.; Warfield, L.; Littlefield, P.; Baker, D.; Klevit, R.E.; et al. The acidic transcription activator Gcn4 binds the mediator subunit Gal11/Med15 using a simple protein interface forming a fuzzy complex. Mol. Cell 2011, 44, 942–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spraggon, G.; Phillips, C.; Nowak, U.K.; Ponting, C.P.; Saunders, D.; Dobson, C.M.; Stuart, D.I.; Jones, E.Y. The crystal structure of the catalytic domain of human urokinase-type plasminogen activator. Structure 1995, 3, 681–691. [Google Scholar] [CrossRef] [Green Version]
- Mao, H.; Williamson, J.R. Local folding coupled to RNA binding in the yeast ribosomal protein L30. J. Mol. Biol. 1999, 292, 345–359. [Google Scholar] [CrossRef]
- Eigenbrot, C.; Gerber, N. X-ray structure of glial cell-derived neurotrophic factor at 1.9 A resolution and implications for receptor binding. Nat. Struct. Biol. 1997, 4, 435–438. [Google Scholar] [CrossRef]
- Abajian, C.; Yatsunyk, L.A.; Ramirez, B.E.; Rosenzweig, A.C. Yeast cox17 solution structure and Copper(I) binding. J. Biol. Chem. 2004, 279, 53584–53592. [Google Scholar] [CrossRef] [Green Version]
- Zahn, R. The octapeptide repeats in mammalian prion protein constitute a pH-dependent folding and aggregation site. J. Mol. Biol. 2003, 334, 477–488. [Google Scholar] [CrossRef]
- Saviano, G.; Tancredi, T. hPrP180-195 structure. 2006. [Google Scholar] [CrossRef]
- Kozin, S.A.; Lepage, C.; Hui Bon Hoa, G.; Rabesona, H.; Mazur, A.K.; Blond, A.; Cheminant, M.; Haertle, T.; Debey, P.; Rebuffat, S. Solution structure of synthetic 21mer peptide spanning region 135-155 (in human numbering) of sheep prion protein. 2004. [Google Scholar] [CrossRef]
- Saviano, G.; Tancredi, T. hPrP-173-195-D178N solution structure. 2006. [Google Scholar] [CrossRef]
- Da Silva Giotto, M.T.; Hannaert, V.; Vertommen, D.; de Navarro, M.V.; Rider, M.H.; Michels, P.A.M.; Garratt, R.C.; Rigden, D.J. The crystal structure of Trypanosoma brucei enolase: Visualisation of the inhibitory metal binding site III and potential as target for selective, irreversible inhibition. J Mol Biol. 2003, 331, 653–665. [Google Scholar] [CrossRef]
- Megy, S.; Bertho, G.; Kozin, S.A.; Debey, P.; Hui Bon Hoa, G.; Girault, J.-P. Possible role of region 152–156 in the structural duality of a peptide fragment from sheep prion protein. Protein Sci. 2004, 13, 3151–3160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Stubbs, G. Molecular dynamics in refinement against fiber diffraction data. Acta Crystallogr. Sect. A Found. Crystallogr. 1993, 49, 504–513. [Google Scholar] [CrossRef]
- Nelson, R.; Sawaya, M.R.; Balbirnie, M.; Madsen, A.Ø.; Riekel, C.; Grothe, R.; Eisenberg, D. Structure of the cross-beta spine of amyloid-like fibrils. Nature 2005, 435, 773–778. [Google Scholar] [CrossRef] [Green Version]
- Sawaya, M.R.; Rodriguez, J.; Cascio, D.; Collazo, M.J.; Shi, D.; Reyes, F.E.; Hattne, J.; Gonen, T.; Eisenberg, D.S. Ab initio structure determination from prion nanocrystals at atomic resolution by MicroED. Proc. Natl. Acad. Sci. USA 2016, 113, 11232–11236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hattne, J.; Shi, D.; Glynn, C.; Zee, C.-Z.; Gallagher-Jones, M.; Martynowycz, M.W.; Rodriguez, J.A.; Gonen, T. Analysis of Global and Site-Specific Radiation Damage in Cryo-EM. Structure 2018, 26, 759–766. [Google Scholar] [CrossRef] [Green Version]
- Apostol, M.I.; Sawaya, M.R.; Cascio, D.; Eisenberg, D. Crystallographic studies of prion protein (PrP) segments suggest how structural changes encoded by polymorphism at residue 129 modulate susceptibility to human prion disease. J. Biol. Chem. 2010, 285, 29671–29675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apostol, M.I.; Perry, K.; Surewicz, W.K. Crystal structure of a human prion protein fragment reveals a motif for oligomer formation. J. Am. Chem. Soc. 2013, 135, 10202–10205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, L.; Lee, S.-J.; Yee, V.C. Crystal Structures of Polymorphic Prion Protein β1 Peptides Reveal Variable Steric Zipper Conformations. Biochemistry 2015, 54, 3640–3648. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Leinala, E.; Jia, Z. Structure of type III antifreeze protein at 277 K. Acta Crystallogr. D Biol. Crystallogr. 1998, 54, 700–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graether, S.P.; Kuiper, M.J.; Gagné, S.M.; Walker, V.K.; Jia, Z.; Sykes, B.D.; Davies, P.L. Beta-helix structure and ice-binding properties of a hyperactive antifreeze protein from an insect. Nature 2000, 406, 325–328. [Google Scholar] [CrossRef] [PubMed]
- Jia, Z.; DeLuca, C.I.; Chao, H.; Davies, P.L. Structural basis for the binding of a globular antifreeze protein to ice. Nature 1996, 384, 285–288. [Google Scholar] [CrossRef]
- Bertolucci, C.M.; Guibao, C.; Zhang, C.; Zheng, J. FAT-LD2 Double labeled construct with free LD4 peptide. 2012. [Google Scholar] [CrossRef]
- Sborgi, L.; Verma, A.; Munoz, V.; De Alba, E. High resolution NMR structure of gpW (W protein of bacteriophage lambda) at acidic pH. 2011. [Google Scholar] [CrossRef]
- Ferguson, N.; Sharpe, T.D.; Schartau, P.J.; Sato, S.; Allen, M.D.; Johnson, C.M.; Rutherford, T.J.; Fersht, A.R. Ultra-fast barrier-limited folding in the peripheral subunit-binding domain family. J. Mol. Biol. 2005, 353, 427–446. [Google Scholar] [CrossRef] [PubMed]
- Matoba, Y.; Kumagai, T.; Yamamoto, A.; Yoshitsu, H.; Sugiyama, M. Crystallographic evidence that the dinuclear copper center of tyrosinase is flexible during catalysis. J. Biol. Chem. 2006, 281, 8981–8990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Improta, S.; Politou, A.S.; Pastore, A. Immunoglobulin-like modules from titin I-band: Extensible components of muscle elasticity. Structure 1996, 4, 323–337. [Google Scholar] [CrossRef] [Green Version]
- Artymiuk, P.J.; Blake, C.C. Refinement of human lysozyme at 1.5 A resolution analysis of non-bonded and hydrogen-bond interactions. J. Mol. Biol. 1981, 152, 737–762. [Google Scholar] [CrossRef]
- Rafferty, J.B.; Somers, W.S.; Saint-Girons, I.; Phillips, S.E. Three-dimensional crystal structures of Escherichia coli met repressor with and without corepressor. Nature 1989, 341, 705–710. [Google Scholar] [CrossRef]
- Pebay-Peyroula, E.; Rummel, G.; Rosenbusch, J.P.; Landau, E.M. X-ray structure of bacteriorhodopsin at 2.5 angstroms from microcrystals grown in lipidic cubic phases. Science 1997, 277, 1676–1681. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.P.; Stimson, E.R.; Ghilardi, J.R.; Mantyh, P.W.; Lu, Y.A.; Felix, A.M.; Llanos, W.; Behbin, A.; Cummings, M.; Van Criekinge, M. 1H NMR of A beta amyloid peptide congeners in water solution. Conformational changes correlate with plaque competence. Biochemistry 1995, 34, 5191–5200. [Google Scholar] [CrossRef]
- Jayawickrama, D.; Zink, S.; Velde, D.V.; Effiong, R.I.; Larive, C.K. Conformational Analysis of the β-amyloid Peptide Fragment, β(12–28). J. Biomol. Struct. Dyn. 1995, 13, 229–244. [Google Scholar] [CrossRef]
- Kohno, T.; Kobayashi, K.; Maeda, T.; Sato, K.; Takashima, A. Three-dimensional structures of the amyloid beta peptide (25–35) in membrane-mimicking environment. Biochemistry 1996, 35, 16094–16104. [Google Scholar] [CrossRef]
- Banach, M.; Konieczny, L.; Roterman, I. The fuzzy oil drop model, based on hydrophobicity density distribution, generalizes the influence of water environment on protein structure and function. J. Theor. Biol. 2014, 359, 6–17. [Google Scholar] [CrossRef] [Green Version]
- Roterman, I.; Banach, M.; Kalinowska, B.; Konieczny, L. Influence of the Aqueous Environment on Protein Structure—A Plausible Hypothesis Concerning the Mechanism of Amyloidogenesis. Entropy 2016, 18, 351. [Google Scholar] [CrossRef] [Green Version]
- Ptak-Kaczor, M.; Banach, M.; Stapor, K.; Fabian, P.; Konieczny, L.; Roterman, I. Solubility and Aggregation of Selected Proteins Interpreted on the Basis of Hydrophobicity Distribution. Int. J. Mol. Sci. 2021, 22, 5002. [Google Scholar] [CrossRef] [PubMed]
- Banach, M.; Kalinowska, B.; Konieczny, L.; Roterman, I. Role of Disulfide Bonds in Stabilizing the Conformation of Selected Enzymes—An Approach Based on Divergence Entropy Applied to the Structure of Hydrophobic Core in Proteins. Entropy 2016, 18, 67. [Google Scholar] [CrossRef] [Green Version]
- Banach, M.; Konieczny, L.; Roterman, I. The Amyloid as a Ribbon-Like Micelle in Contrast to Spherical Micelles Represented by Globular Proteins. Molecules 2019, 24, 4395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fändrich, M.; Dobson, C.M. The behaviour of polyamino acids reveals an inverse side chain effect in amyloid structure formation. EMBO J. 2002, 21, 5682–5690. [Google Scholar] [CrossRef] [Green Version]
- Cordy, J.M.; Hooper, N.M.; Turner, A.J. The involvement of lipid rafts in Alzheimer’s disease. Mol. Membr. Biol. 2006, 23, 111–122. [Google Scholar] [CrossRef]
- Konieczny, L.; Roterman, I. Globular or ribbon-like micelle. In From Globular Proteins to Amyloids; Roterman-Konieczna, I., Ed.; Elsevier: Amsterdam, The Netherlands; Oxford, UK; Cambridge, MA, USA, 2020; pp. 41–54. [Google Scholar]
- Blancas-Mejia, L.M.; Misra, P.; Dick, C.J.; Cooper, S.A.; Redhage, K.R.; Bergman, M.R.; Jordan, T.L.; Maar, K.; Ramirez-Alvarado, M. Immunoglobulin light chain amyloid aggregation. Chem. Commun. 2018, 54, 10664–10674. [Google Scholar] [CrossRef]
- Eneqvist, T.; Andersson, K.; Olofsson, A.; Lundgren, E.; Sauer-Eriksson, A.E. The beta-slip: A novel concept in transthyretin amyloidosis. Mol. Cell 2000, 6, 1207–1218. [Google Scholar] [CrossRef]
- Banach, M.; Stapor, K.; Fabian, P.; Konieczny, L.; Roterman, I. Divergence Entropy-Based Evaluation of Hydrophobic Core in Aggressive and Resistant Forms of Transthyretin. Entropy 2021, 23, 458. [Google Scholar] [CrossRef]
- Manganelli, F.; Fabrizi, G.M.; Luigetti, M.; Mandich, P.; Mazzeo, A.; Pareyson, D. Hereditary transthyretin amyloidosis overview. Neurol. Sci. 2020, 1–10, Online ahead of print. [Google Scholar] [CrossRef]
- Weingarten, M.D.; Lockwood, A.H.; Hwo, S.Y.; Kirschner, M.W. A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. USA 1975, 72, 1858–1862. [Google Scholar] [CrossRef] [Green Version]
- Barinova, K.V.; Kuravsky, M.L.; Arutyunyan, A.M.; Serebryakova, M.V.; Schmalhausen, E.V.; Muronetz, V.I. Dimerization of Tyr136Cys alpha-synuclein prevents amyloid transformation of wild type alpha-synuclein. Int. J. Biol. Macromol. 2017, 96, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Sluzky, V.; Klibanov, A.M.; Langer, R. Mechanism of insulin aggregation and stabilization in agitated aqueous solutions. Biotechnol. Bioeng. 1992, 40, 895–903. [Google Scholar] [CrossRef] [PubMed]
- Buell, A.K.; Dobson, C.M.; Knowles, T.P. The physical chemistry of the amyloid phenomenon: Thermodynamics and kinetics of filamentous protein aggregation. Essays Biochem. 2014, 56, 11–39. [Google Scholar] [CrossRef] [PubMed]
- Dygut, J.; Kalinowska, B.; Banach, M.; Piwowar, M.; Konieczny, L.; Roterman, I. Structural Interface Forms and Their Involvement in Stabilization of Multidomain Proteins or Protein Complexes. Int. J. Mol. Sci. 2016, 17, 1741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schubeis, T.; Le Marchand, T.; Daday, C.; Kopec, W.; Movellan, K.T.; Stanek, J.; Schwarzer, T.S.; Castiglione, K.; De Groot, B.L.; Pintacuda, G.; et al. A β-barrel for oil transport through lipid membranes: Dynamic NMR structures of AlkL. Proc. Natl. Acad. Sci. USA 2020, 117, 21014–21021. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Boyer, D.R.; Sawaya, M.R.; Ge, P.; Eisenberg, D.S. Cryo-EM structures of four polymorphic TDP-43 amyloid cores. Nat. Struct. Mol. Biol. 2019, 26, 619–627. [Google Scholar] [CrossRef]
- Li, B.; Ge, P.; Murray, K.A.; Sheth, P.; Zhang, M.; Nair, G.; Sawaya, M.R.; Shin, W.S.; Boyer, D.R.; Ye, S.; et al. Cryo-EM of full-length α-synuclein reveals fibril polymorphs with a common structural kernel. Nat. Commun. 2018, 9, 1–10. [Google Scholar] [CrossRef]
- Konieczny, L.; Roterman, I. Description of the fuzzy oil drop model. In From Globular Proteins to Amyloids; Roterman-Konieczna, I., Ed.; Elsevier: Amsterdam, The Netherlands; Oxford, UK; Cambridge, MA, USA, 2020; pp. 1–12. [Google Scholar]
- Banach, M.; Konieczny, L.; Roterman, I. The late-stage intermediate. In Protein Folding in Silico; Roterman-Konieczna, I., Ed.; Woodhead Publishing Limited: Oxford, UK; Cambridge, MA, USA; Philadelphia, PA, USA; New Delhi, India, 2012; pp. 21–38. [Google Scholar]
- Levitt, M. A simplified representation of protein conformations for rapid simulation of protein folding. J. Mol. Biol. 1976, 104, 59–107. [Google Scholar] [CrossRef]
- Kullback, S.; Leibler, R.A. On Information and Sufficiency. Ann. Math. Stat. 1951, 22, 79–86. [Google Scholar] [CrossRef]
- Available online:https://www.ks.uiuc.edu/Research/vmd/ (accessed on 31 May 2021).
- Clark, P.L.; Plaxco, K.W.; Sosnick, T.R. Water as a Good Solvent for Unfolded Proteins: Folding and Collapse are Fundamentally Different. J. Mol. Biol. 2020, 432, 2882–2889. [Google Scholar] [CrossRef]
- Riback, J.A.; Bowman, M.A.; Zmyslowski, A.M.; Knoverek, C.R.; Jumper, J.M.; Hinshaw, J.R.; Kaye, E.B.; Freed, K.F.; Clark, P.L.; Sosnick, T.R. Innovative scattering analysis shows that hydrophobic disordered proteins are expanded in water. Science 2017, 358, 238–241. [Google Scholar] [CrossRef] [Green Version]
- Nagibina, G.S.; Glukhova, K.A.; Uversky, V.N.; Melnik, V.N.; Melnik, T.N. Intrinsic Disorder-Based Design of Stable Globular Proteins. Biomolecules 2019, 10, 64. [Google Scholar] [CrossRef] [Green Version]
- Uversky, V.U. Protein intrinsic disorder and structure-function continuum. Prog Mol. Biol. Transl. Sci. 2019, 166, 1–17. [Google Scholar] [CrossRef]
- Kalinowska, B.; Banach, M.; Konieczny, L.; Marchewka, D.; Roterman, I. Intrinsically disordered proteins—Relation to general model expressing the active role of the water environment. Adv. Protein Chem. Struct. Biol. 2014, 94, 315–346. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://disprot.org/ (accessed on 6 July 2021).
- Hatos, A.; Hajdu-Soltész, B.; Monzon, A.M.; Palopoli, N.; Álvarez, L.; Aykac-Fas, B.; Bassot, C.; Benítez, G.I.; Bevilacqua, M.; Chasapi, A.; et al. DisProt: Intrinsic protein disorder annotation in 2020. Nucleic Acids Res. 2020, 48, D269–D276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vucetic, S.; Obradovic, Z.; Vacic, V.; Radivojac, P.; Peng, K.; Iakoucheva, L.M.; Cortese, M.S.; Lawson, J.D.; Brown, C.J.; Sikes, J.G. DisProt: A database of protein disorder. Bioinformatics 2005, 21, 137–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz, V. Conformational Dynamics and Ensembles in Protein Folding. Annu. Rev. Biophys. Biomol. Struct. 2007, 36, 395–412. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, V.; Thompson, P.A.; Hofrichter, J.; Eaton, W.A. Folding dynamics and mechanism of β-hairpin formation. Nat. Cell Biol. 1997, 390, 196–199. [Google Scholar] [CrossRef]
- Muñoz, V.; Henry, E.; Hofrichter, J.; Eaton, W.A. A statistical mechanical model for -hairpin kinetics. Proc. Natl. Acad. Sci. USA 1998, 95, 5872–5879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz, V.; Cerminara, M. When fast is better: Protein folding fundamentals and mechanisms from ultrafast approaches. Biochem. J. 2016, 473, 2545–2559. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, V.; Campos, L.A.; Sadqi, M. Limited cooperativity in protein folding. Curr. Opin. Struct. Biol. 2016, 36, 58–66. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PROTEIN | WT | AMYLOID | ||||||
|---|---|---|---|---|---|---|---|---|
| PDB-ID | RD T-O-R | RD M-O-T | K | K | RD M-O-T | RD T-O-R | PDB-ID | |
| Transthyretin—A | 1DVQ | 0.562 | 0.342 | 0.5 | ||||
| (11–35) + (57–123) | 1DVQ | 0.584 | 0.331 | 0.5 | 1.8 | 0.242 | 0.757 | 6SDZ |
| IgG – VL | 4BJL | 0.547 | 0.390 | 0.5 | ||||
| (1–37) + (66–105) | 4BJL | 0.516 | 0.405 | 0.4 | 1.1 | 0.229 | 0.755 | 6HUD |
| TAU | 1.2 | 0.323 | 0.674 | 5O3L | ||||
| α-Synuclein | 1XQ8 | 0.643 | 0.340 | 1.3 | ||||
| (30–100) | 1XQ8 | 0.672 | 0.319 | 1.3 | 0.5 | 0.377 | 0.568 | 2N0A |
| Aβ(1–42) (15–40) D23N | 0.5 | 0.349 | 0.626 | 2MPZ | ||||
| Aβ(1–42) (1–40) E22Δ | 0.7 | 0.334 | 0.629 | 2MVX | ||||
| Aβ(1–42) (11–42) | 0.5 | 0.385 | 0.538 | 2MXU | ||||
| PROTEIN | WT | AMYLOID | ||||||
|---|---|---|---|---|---|---|---|---|
| PDB-ID | RD T-O-R | RD M-O-T | K | K | RD M-O-T | RD T-O-R | PDB-ID | |
| Tranthyretin - A | 1DVQ | 0.562 | 0.342 | 0.5 | ||||
| (11–35) + (57–123) | 1DVQ | 0.584 | 0.331 | 0.5 | 1.1 | 0.305 | 0.694 | 6SDZ(F) |
| IgG – VL | 4BJL | 0.547 | 0.390 | 0.5 | ||||
| (1–37) + (66–105) | 4BJL | 0.516 | 0.405 | 0.4 | 1.0 | 0.193 | 0.793 | 6HUD(C) |
| TAU | 1.2 1.2 | 0.334 0.259 | 0.664 0.728 | 5O3L superfibril | ||||
| α-Synuclein | 1XQ8 | 0.643 | 0.340 | 1.3 | ||||
| (30–100) | 1XQ8 | 0.672 | 0.319 | 1.3 | 0.2 | 0.430 | 0.506 | 2N0A(E) |
| Aβ(1–42)(15–40)D23N | 0.4 0.2 | 0.496 0.457 | 0.554 0.491 | 2MPZ(M) superfibril | ||||
| Aβ(1–42) (1–40) E22Δ | 0.8 0.5 | 0.326 0.315 | 0.649 0.567 | 2MVX(E) superfibril | ||||
| Aβ(1–42) (11–42) | 0.3 | 0.436 | 0.515 | 2MXU(F) | ||||
| PROTEIN | PDB-ID | Fragment in Amyloid | Refs. | |
|---|---|---|---|---|
| WT | Amyloid | |||
| Transthyretin | 1DVQ – A | 6SDZ | (11–35) + (57–123) | [38,39] |
| IgG – VL | 4BJL –VL | 6HUD | (1–37) + (66–105) | [40,41] |
| Tau | 5O3L | [42] | ||
| α-synuclein | 1XQ8 | 2N0A | (30–100) | [43,44] |
| Aβ (1–42) | 2MXU | (11–42) | [45] | |
| Aβ (1–42) | 2MPZ | (15–40) D23N | [46] | |
| Aβ (1–42) | 2MVX | (1–40) E22Δ | [47] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roterman, I.; Stapor, K.; Fabian, P.; Konieczny, L. In Silico Modeling of the Influence of Environment on Amyloid Folding Using FOD-M Model. Int. J. Mol. Sci. 2021, 22, 10587. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910587
Roterman I, Stapor K, Fabian P, Konieczny L. In Silico Modeling of the Influence of Environment on Amyloid Folding Using FOD-M Model. International Journal of Molecular Sciences. 2021; 22(19):10587. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910587
Chicago/Turabian StyleRoterman, Irena, Katarzyna Stapor, Piotr Fabian, and Leszek Konieczny. 2021. "In Silico Modeling of the Influence of Environment on Amyloid Folding Using FOD-M Model" International Journal of Molecular Sciences 22, no. 19: 10587. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910587