
Mebendazole Mediates Proteasomal Degradation of GLI Transcription Factors in Acute Myeloid Leukemia

 , ,
, ,
Abstract
:1. Introduction
2. Results
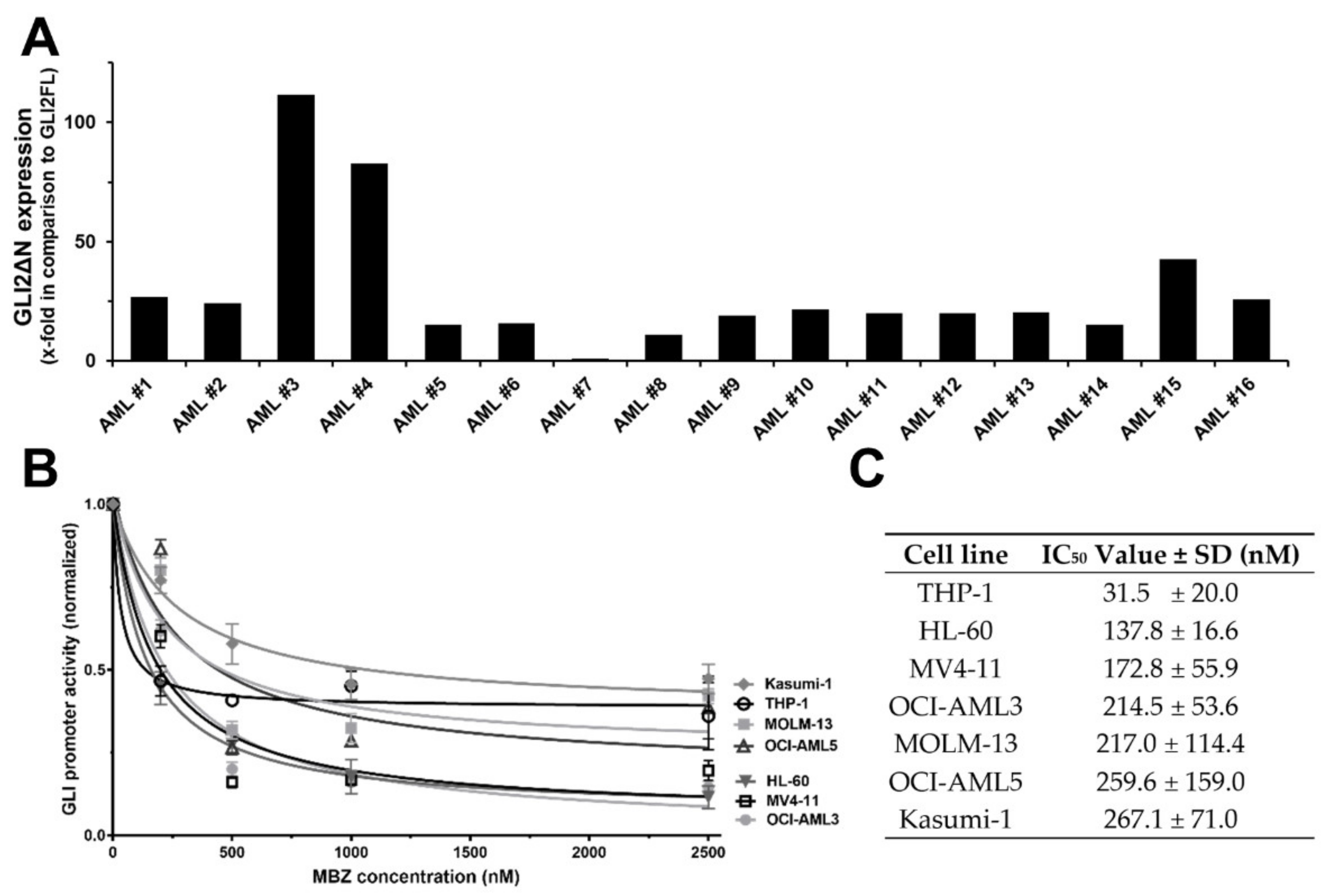
2.1. MBZ Inhibits SMO Independent Non-Canonical GLI Signaling Predominant in AML
2.2. MBZ Promotes Proteasomal Degradation of GLI
2.3. MBZ Promotes Degradation of GLI Transcription Factors via Inhibition of HSP70/90-Chaperone Activity
2.4. In Silico Modeling of MBZ Bound to HSP90
2.5. Mebendazole and the GLI Inhibitor GANT-61 Exhibit Synergistic Anti-Leukemic Effects
2.6. MBZ Sensitizes AML Cells to Chemotherapy
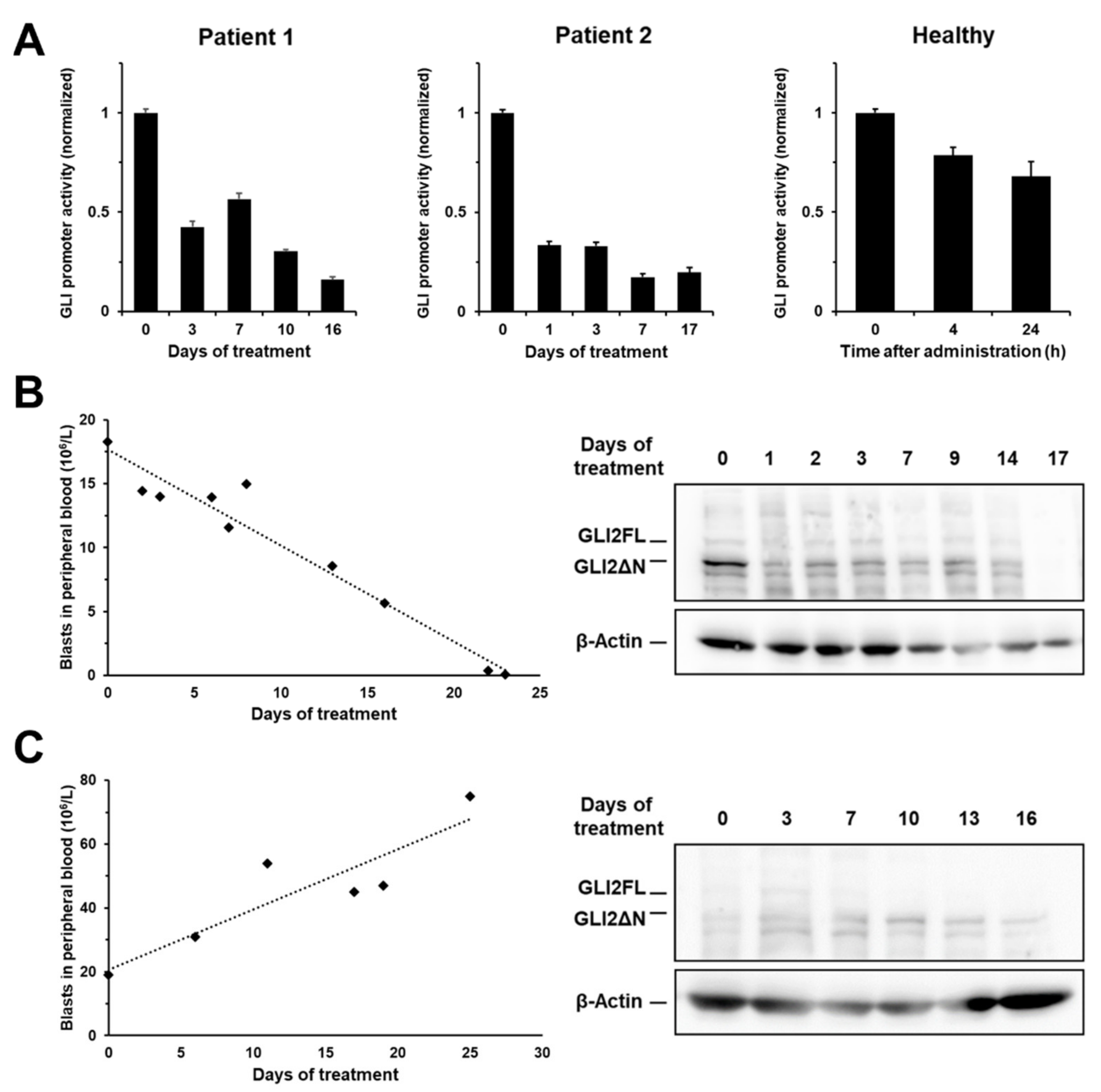
2.7. MBZ Effectively Inhibits GLI Signaling in Clinically Achievable Plasma Levels
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Cell Culture
4.2. Inhibitors and Reagents
4.3. Reverse Transcription and Quantitative PCR
4.4. Protein Isolation and Western Blot
4.5. GLI Reporter Assays
4.6. Plasma Inhibitory Assay
4.7. Refolding Assay
4.8. Proliferation Assay
4.9. Apoptosis Assay
4.10. Colony Formation Assay
4.11. Cloning and Lentiviral Transduction
4.12. In Silico Modeling
4.13. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Sense | Anti-Sense |
|---|---|---|
| GLI1 | CTACATCAACTCCGGCCAAT | CGGCTGACAGTATAGGCAGA |
| GLI2 | CCCCTACCGATTGACATGCG | GAAAGCCGGATCAAGGAGATG |
| GLI2-ALL | AACCCTGTCGCCATTCACAA | CCAGTGGCAGTTGGTCTCAT |
| GLI2-FL | TCAGCCTTTGGACACACACC | TGCACTTGTGGGGCTTCTC |
| GLI2-C-term | GCTGCAACAAAGCCTTCTCC | TTCTCTTTGAGCAGCGGTGT |
| GAPDH | GTCAGTGGTGGACCTGACCT | TGCTGTAGCCAAATTCGTTG |
References
- Jiang, J.; Hui, C. Hedgehog Signaling in Development and Cancer. Dev. Cell 2008, 15, 801–812. [Google Scholar] [CrossRef] [Green Version]
- Aberger, F.; Hutterer, E.; Sternberg, C.; Del Burgo, P.J.; Hartmann, T.N. Acute Myeloid Leukemia-Strategies and Challenges for Targeting Oncogenic Hedgehog/GLI Signaling. Cell Commun. Signal 2017, 15, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latuske, E.M.; Stamm, H.; Klokow, M.; Vohwinkel, G.; Muschhammer, J.; Bokemeyer, C.; Jücker, M.; Kebenko, M.; Fiedler, W.; Wellbrock, J. Combined Inhibition of GLI and FLT3 Signaling Leads to Effective Anti-Leukemic Effects in Human Acute Myeloid Leukemia. Oncotarget 2017, 8, 29187–29201. [Google Scholar] [CrossRef] [Green Version]
- Teglund, S.; Toftgård, R. Hedgehog beyond Medulloblastoma and Basal Cell Carcinoma. Biochim. Biophys. Acta-Rev. Cancer 2010, 1805, 181–208. [Google Scholar] [CrossRef] [PubMed]
- Dierks, C.; Beigi, R.; Guo, G.R.; Zirlik, K.; Stegert, M.R.; Manley, P.; Trussell, C.; Schmitt-Graeff, A.; Landwerlin, K.; Veelken, H.; et al. Expansion of Bcr-Abl-Positive Leukemic Stem Cells Is Dependent on Hedgehog Pathway Activation. Cancer Cell 2008, 14, 238–249. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Gao, J.; Tao, K.; Yu, P. Prognostic Role of Gli1 Expression in Solid Malignancies: A Meta-Analysis. Sci. Rep. 2016, 6, 22184. [Google Scholar] [CrossRef]
- Wellbrock, J.; Latuske, E.; Kohler, J.; Wagner, K.; Stamm, H.; Vettorazzi, E.; Vohwinkel, G.; Klokow, M.; Uibeleisen, R.; Ehm, P.; et al. Expression of Hedgehog Pathway Mediator GLI Represents a Negative Prognostic Marker in Human Acute Myeloid Leukemia and Its Inhibition Exerts Antileukemic Effects. Clin. Cancer Res. 2015, 21, 2388–2398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolska-Washer, A.; Robak, T. Glasdegib in the Treatment of Acute Myeloid Leukemia. Future Oncol. 2019, 15, 3219–3232. [Google Scholar] [CrossRef]
- Guerini, A.E.; Triggiani, L.; Maddalo, M.; Bonù, M.L.; Frassine, F.; Baiguini, A.; Alghisi, A.; Tomasini, D.; Borghetti, P.; Pasinetti, N.; et al. Mebendazole as a Candidate for Drug Repurposing in Oncology: An Extensive Review of Current Literature. Cancers 2019, 11, 1284. [Google Scholar] [CrossRef] [Green Version]
- WHO Informal Working Group on Echinococcosis. Guidelines for Treatment of Cystic and Alveolar Echinococcosis in Humans. Bull. World Health Organ. 1996, 74, 231–242. [Google Scholar]
- Walf-Vorderwülbecke, V.; Pearce, K.; Brooks, T.; Hubank, M.; Van Den Heuvel-Eibrink, M.M.; Zwaan, C.M.; Adams, S.; Edwards, D.; Bartram, J.; Samarasinghe, S.; et al. Targeting Acute Myeloid Leukemia by Drug-Induced c-MYB Degradation. Leukemia 2018, 32, 882–889. [Google Scholar] [CrossRef]
- Kinzler, K.W.; Bigner, S.H.; Bigner, D.D.; Trent, J.M.; Law, M.L.; O’Brien, S.J.; Wong, A.J.; Vogelstein, B. Identification of an Amplified, Highly Expressed Gene in a Human Glioma. Science 1987, 236, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Sadam, H.; Liivas, U.; Kazantseva, A.; Pruunsild, P.; Kazantseva, J.; Timmusk, T.; Neuman, T.; Palm, K. GLI2 Cell-Specific Activity Is Controlled at the Level of Transcription and RNA Processing: Consequences to Cancer Metastasis. Biochim. Biophys. Acta-Mol. Basis Dis. 2016, 1862, 46–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, H.; Nishizaki, Y.; Hui, C.C.; Nakafuku, M.; Kondoh, H. Regulation of Gli2 and Gli3 Activities by an Amino-Terminal Repression Domain: Implication of Gli2 and Gli3 as Primary Mediators of Shh Signaling. Development 1999, 126, 3915–3924. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Ponomaryov, T.; Ornell, K.J.; Zhou, P.; Dabral, S.K.; Pak, E.; Li, W.; Atwood, S.X.; Whitson, R.J.; Chang, A.L.S.; et al. RAS/MAPK Activation Drives Resistance to Smo Inhibition, Metastasis, and Tumor Evolution in Shh Pathway-Dependent Tumors. Cancer Res. 2015, 75, 3623–3635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhry, P.; Singh, M.; Triche, T.J.; Guzman, M.; Merchant, A.A. GLI3 Repressor Determines Hedgehog Pathway Activation and Is Required for Response to SMO Antagonist Glasdegib in AML. Blood 2017, 129, 3465–3475. [Google Scholar] [CrossRef] [PubMed]
- Dayan, A.D. Albendazole, Mebendazole and Praziquantel. Review of Non-Clinical Toxicity and Pharmacokinetics. Acta Trop. 2003, 86, 141–159. [Google Scholar] [CrossRef]
- Fiedler, W.; Freisleben, F.; Wellbrock, J.; Kirschner, K. Mebendazole’s Conformational Space and its Predicted Binding to Human Heat-Shock Protein 90. ChemRxiv 2021, preprint. [Google Scholar]
- Long, B.; Wang, L.-X.; Zheng, F.-M.; Lai, S.-P.; Xu, D.-R.; Hu, Y.; Lin, D.-J.; Zhang, X.-Z.; Dong, L.; Long, Z.-J.; et al. Targeting GLI1 Suppresses Cell Growth and Enhances Chemosensitivity in CD34+ Enriched Acute Myeloid Leukemia Progenitor Cells. Cell. Physiol. Biochem. 2016, 38, 1288–1302. [Google Scholar] [CrossRef]
- Michaelis, M.; Agha, B.; Rothweiler, F.; Löschmann, N.; Voges, Y.; Mittelbronn, M.; Starzetz, T.; Harter, P.N.; Abhari, B.A.; Fulda, S.; et al. Identification of Flubendazole as Potential Anti-Neuroblastoma Compound in a Large Cell Line Screen. Sci. Rep. 2015, 5, 8202. [Google Scholar] [CrossRef]
- Rimkus, T.K.; Carpenter, R.L.; Qasem, S.; Chan, M.; Lo, H.W. Targeting the Sonic Hedgehog Signaling Pathway: Review of Smoothened and GLI Inhibitors. Cancers 2016, 8, 22. [Google Scholar] [CrossRef] [Green Version]
- Larsen, A.R.; Bai, R.Y.; Chung, J.H.; Borodovsky, A.; Rudin, C.M.; Riggins, G.J.; Bunz, F. Repurposing the Antihelmintic Mebendazole as a Hedgehog Inhibitor. Mol. Cancer Ther. 2015, 14, 3–13. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Pak, E.; Ornell, K.J.; Murphy, M.F.P.; Mackenzie, E.L.; Chadwick, E.J.; Ponomaryov, T.; Kelleher, J.F.; Segal, R.A. A Transposon Screen Identifies Loss of Primary Cilia as a Mechanism of Resistance to SMO Inhibitors. Cancer Discov. 2017, 7, 1436–1439. [Google Scholar] [CrossRef] [Green Version]
- Singh, M.; Chaudhry, P.; Merchant, A.A. Primary Cilia Are Present on Human Blood and Bone Marrow Cells and Mediate Hedgehog Signaling. Exp. Hematol. 2016, 44, 1181–1187. [Google Scholar] [CrossRef] [Green Version]
- Rosenzweig, R.; Nillegoda, N.B.; Mayer, M.P.; Bukau, B. The Hsp70 Chaperone Network. Nat. Rev. Mol. Cell Biol. 2019, 20, 665–680. [Google Scholar] [CrossRef]
- Taipale, M.; Jarosz, D.F.; Lindquist, S. HSP90 at the Hub of Protein Homeostasis: Emerging Mechanistic Insights. Nat. Rev. Mol. Cell Biol. 2010, 11, 515–528. [Google Scholar] [CrossRef]
- Butler, L.M.; Ferraldeschi, R.; Armstrong, H.K.; Centenera, M.M.; Workman, P. Maximizing the Therapeutic Potential of HSP90 Inhibitors. Mol. Cancer Res. 2015, 13, 1445–1451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morán Luengo, T.; Kityk, R.; Mayer, M.P.; Rüdiger, S.G.D. Hsp90 Breaks the Deadlock of the Hsp70 Chaperone System. Mol. Cell 2018, 70, 545–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, C.; Sampson, S.B. HSF1: Guardian of Proteostasis in Cancer. Trends Cell Biol. 2016, 26, 17–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Im, C.N.; Yun, H.H.; Lee, J.H. Heat Shock Factor 1 Depletion Sensitizes A172 Glioblastoma Cells to Temozolomide via Suppression of Cancer Stem Cell-like Properties. Int. J. Mol. Sci. 2017, 18, 468. [Google Scholar] [CrossRef]
- Kise, Y.; Takenaka, K.; Tezuka, T.; Yamamoto, T.; Miki, H. Fused Kinase Is Stabilized by Cdc37/Hsp90 and Enhances Gli Protein Levels. Biochem. Biophys. Res. Commun. 2006, 351, 78–84. [Google Scholar] [CrossRef]
- Han, Y.; Wang, B.; Cho, Y.S.; Zhu, J.; Wu, J.; Chen, Y.; Jiang, J. Phosphorylation of Ci/Gli by Fused Family Kinases Promotes Hedgehog Signaling. Dev. Cell 2019, 50, 610–626. [Google Scholar] [CrossRef]
- Nygren, P.; Fryknäs, M.; Ågerup, B.; Larsson, R. Repositioning of the Anthelmintic Drug Mebendazole for the Treatment for Colon Cancer. J. Cancer Res. Clin. Oncol. 2013, 139, 2133–2140. [Google Scholar] [CrossRef] [Green Version]
- Simbulan-Rosenthal, C.M.; Dakshanamurthy, S.; Gaur, A.; Chen, Y.S.; Fang, H.B.; Abdussamad, M.; Zhou, H.; Zapas, J.; Calvert, V.; Petricoin, E.F.; et al. The Repurposed Anthelmintic Mebendazole in Combination with Trametinib Suppresses Refractory NRASQ61K Melanoma. Oncotarget 2017, 8, 12576–12595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, J.I.; Ramesh, R.; Chada, S.; Gomyo, Y.; Roth, J.A.; Mukhopadhyay, T. The Anthelmintic Drug Mebendazole Induces Mitotic Arrest and Apoptosis by Depolymerizing Tubulin in Non-Small Cell Lung Cancer Cells. Mol. Cancer Ther. 2002, 1, 1201–1209. [Google Scholar] [PubMed]
- Bai, R.Y.; Staedtke, V.; Aprhys, C.M.; Gallia, G.L.; Riggins, G.J. Antiparasitic Mebendazole Shows Survival Benefit in 2 Preclinical Models of Glioblastoma Multiforme. Neuro-Oncology 2011, 13, 974–982. [Google Scholar] [CrossRef]
- Jamieson, C.; Martinelli, G.; Papayannidis, C.; Cortes, J.E. Hedgehog Pathway Inhibitors: A New Therapeutic Class for the Treatment of Acute Myeloid Leukemia. Blood Cancer Discov. 2020, 1, 134–145. [Google Scholar] [CrossRef]
- Patil, V.M.; Bhelekar, A.; Menon, N.; Bhattacharjee, A.; Simha, V.; Abhinav, R.; Abhyankar, A.; Sridhar, E.; Mahajan, A.; Puranik, A.D.; et al. Reverse Swing-M, Phase 1 Study of Repurposing Mebendazole in Recurrent High-Grade Glioma. Cancer Med. 2020, 9, 4676–4685. [Google Scholar] [CrossRef]
- Fernandez-Banares, F.; Gonzalez-Huix, F.; Xiol, X.; Catalá, I.; Miró, J.; López, N.; Casais, L. Marrow Aplasia during High Dose Mebendazole Treatment. Am. J. Trop. Med. Hyg. 1986, 35, 350–351. [Google Scholar] [CrossRef] [Green Version]
- DiNardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Döhner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N. Engl. J. Med. 2020, 383, 617–629. [Google Scholar] [CrossRef]
- Braithwaite, P.A.; Roberts, M.S.; Allan, R.J.; Watson, T.R. Clinical Pharmacokinetics of High Dose Mebendazole in Patients Treated for Cystic Hydatid Disease. Eur. J. Clin. Pharmacol. 1982, 22, 161–169. [Google Scholar] [CrossRef]
- Münst, G.J.; Karlaganis, G.; Bircher, J. Plasma Concentrations of Mebendazole during Treatment of Echinococcosis—Preliminary Results. Eur. J. Clin. Pharmacol. 1980, 17, 375–378. [Google Scholar] [CrossRef]
- Pabst, C.; Krosl, J.; Fares, I.; Boucher, G.; Ruel, R.; Marinier, A.; Lemieux, S.; Hébert, J.; Sauvageau, G. Identification of Small Molecules That Support Human Leukemia Stem Cell Activity Ex Vivo. Nat. Methods 2014, 11, 436–442. [Google Scholar] [CrossRef]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A Tool to Design Target-Specific Primers for Polymerase Chain Reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. Available online: https://primer3.ut.ee/ (accessed on 30 September 2021). [CrossRef] [PubMed] [Green Version]
- Pfaffl, M.W. A New Mathematical Model for Relative Quantification in Real-Time RT–PCR. Nucleic Acids Res. 2001, 29, 2002–2007. [Google Scholar] [CrossRef]
- Camacho Londoño, J.; Philipp, S.E. A Reliable Method for Quantification of Splice Variants Using RT-QPCR. BMC Mol. Biol. 2016, 17, 8. [Google Scholar] [CrossRef] [Green Version]
- Freisleben, F.; Behrmann, L.; Thaden, V.; Muschhammer, J.; Bokemeyer, C.; Fiedler, W.; Wellbrock, J. Downregulation of GLI3 Expression Mediates Chemotherapy Resistance in Acute Myeloid Leukemia. Int. J. Mol. Sci. 2020, 21, 5084. [Google Scholar] [CrossRef] [PubMed]
- Weber, K.; Mock, U.; Petrowitz, B.; Bartsch, U.; Fehse, B. Lentiviral Gene Ontology (LeGO) Vectors Equipped with Novel Drug-Selectable Fluorescent Proteins: New Building Blocks for Cell Marking and Multi-Gene Analysis. Gene Ther. 2010, 17, 511–520. Available online: http://www.lentigo-vectors.de/vectors.htm (accessed on 30 September 2021). [CrossRef] [PubMed] [Green Version]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D.; Giambasu, G.; et al. Lessons learned from comparing molecular dynamics engines on the SAMPL5 dataset. J. Comput.-Aided Mol. Des. 2017, 31, 147–161. [Google Scholar]
- Brough, P.A.; Barril, X.; Borgognoni, J.; Chene, P.; Davies, N.G.M.; Davis, B.; Drysdale, M.J.; Dymock, B.; Eccles, S.A.; Garcia-Echeverria, C.; et al. Combining Hit Identification Strategies: Fragment-Based and in Silico Approaches to Orally Active 2-Aminothieno[2,3−d]Pyrimidine Inhibitors of the Hsp90 Molecular Chaperone. J. Med. Chem. 2009, 52, 4794–4809. [Google Scholar] [CrossRef] [PubMed]
- Dymock, B.W.; Barril, X.; Brough, P.A.; Cansfield, J.E.; Massey, A.; McDonald, E.; Hubbard, R.E.; Surgenor, A.; Roughley, S.D.; Webb, P.; et al. Novel, Potent Small-Molecule Inhibitors of the Molecular Chaperone Hsp90 Discovered through Structure-Based Design. J. Med. Chem. 2005, 48, 4212–4215. [Google Scholar] [CrossRef] [PubMed]
- McBride, C.M.; Levine, B.; Xia, Y.; Bellamacina, C.; Machajewski, T.; Gao, Z.; Renhowe, P.; Antonios-McCrea, W.; Barsanti, P.; Brinner, K.; et al. Design, Structure-Activity Relationship, and in Vivo Characterization of the Development Candidate NVP-HSP990. J. Med. Chem. 2014, 57, 9124–9129. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. Ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from Ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.-P.; McKiernan, K.A.; Gomes, J.; Beauchamp, K.A.; Head-Gordon, T.; Rice, J.E.; Swope, W.C.; Martínez, T.J.; Pande, V.S. Building a More Predictive Protein Force Field: A Systematic and Reproducible Route to AMBER-FB15. J. Phys. Chem. B 2017, 121, 4023–4039. [Google Scholar] [CrossRef]
- Alenaizan, A.; Burns, L.A.; Sherrill, C.D. Python Implementation of the Restrained Electrostatic Potential Charge Model. Int. J. Quantum Chem. 2019, 120, e26035. [Google Scholar] [CrossRef]
- Chou, T.C. Drug Combination Studies and Their Synergy Quantification Using the Chou-Talalay Method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Freisleben, F.; Modemann, F.; Muschhammer, J.; Stamm, H.; Brauneck, F.; Krispien, A.; Bokemeyer, C.; Kirschner, K.N.; Wellbrock, J.; Fiedler, W. Mebendazole Mediates Proteasomal Degradation of GLI Transcription Factors in Acute Myeloid Leukemia. Int. J. Mol. Sci. 2021, 22, 10670. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910670
Freisleben F, Modemann F, Muschhammer J, Stamm H, Brauneck F, Krispien A, Bokemeyer C, Kirschner KN, Wellbrock J, Fiedler W. Mebendazole Mediates Proteasomal Degradation of GLI Transcription Factors in Acute Myeloid Leukemia. International Journal of Molecular Sciences. 2021; 22(19):10670. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910670
Chicago/Turabian StyleFreisleben, Fabian, Franziska Modemann, Jana Muschhammer, Hauke Stamm, Franziska Brauneck, Alexander Krispien, Carsten Bokemeyer, Karl N. Kirschner, Jasmin Wellbrock, and Walter Fiedler. 2021. "Mebendazole Mediates Proteasomal Degradation of GLI Transcription Factors in Acute Myeloid Leukemia" International Journal of Molecular Sciences 22, no. 19: 10670. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms221910670