1. Introduction
Recent advances in CRISPR/Cas-based gene editing have led to a renewed interest in optimizing plant transformation and regeneration techniques to enable more rapid development of gene edited lines for crop improvement.
Agrobacterium tumefaciens-mediated transformation and biolistic/particle bombardment-based delivery have been used for plant transformation for over 30 years [
1,
2], with advantages and disadvantages to each method. Transformation using binary vectors for
Agrobacterium-mediated delivery and integration of the T-DNA often results in single copy insertions, but has species and genotype-specific differences in protocols and efficiencies. At the same time, biolistic-based delivery has fewer constraints on the target species, but often results in multiple insertions or insertion of partial DNA fragments. Both delivery methods require slow, labor-intensive, and often complex sets of species- and genotype-specific protocols for in vitro tissue culture and regeneration from transformed explants.
The past few decades have seen significant improvements in tissue culture techniques which have drastically increased the number of species capable of successfully passing through the lengthy tissue culture process. In addition to the components of the culture media, the explant origin has long been known to play an important role in tissue culture success [
3]. Even problems in otherwise simple steps such as embryogenic calli formation can be difficult to overcome, especially in recalcitrant crops. Often, the plant’s regeneration step is a greater bottleneck than the gene integration itself. Most crop gene editing studies to date have employed model genotypes that are relatively easy to transform; however, gene editing will need to be performed in high-yielding modern cultivars to make an immediate impact in plant breeding. Unfortunately, for many crops, modern cultivars and elite breeding lines, unlike the model genotypes, tend to be tissue culture recalcitrant, and attempting to directly apply novel gene editing techniques to manipulate desirable agronomic traits in high-performing breeding lines is not an easy task. Thus, although in vitro plant transformation, tissue culture, and regeneration techniques have made tremendous progress over the past few decades, there are still several bottlenecks that hamper faster progress, including low frequency of transformed events, long periods of time needed for tissue culture and regeneration, and low precision of biolistic-mediated transformation [
4]. Therefore, to benefit from the advantages of gene editing technology, callus induction,
Agrobacterium-mediated transformation (preferred for its precision and single copy insertions), and plant regeneration must be optimized for routine transformation and delivery of CRISPR reagents.
As a model monocot species, rice (
Oryza sativa) has a long history of research to optimize
Agrobacterium-mediated transformation and regeneration; however, this has largely focused on relatively easy-to-transform
temperate japonica varieties such as Nipponbare and Kitaake [
5,
6,
7,
8,
9,
10]. These varieties have efficient callus induction from seeds, high efficiency of transformation by
Agrobacterium, and good regeneration capacity from callus tissue [
11]. However, previous research has indicated that it is much more difficult to achieve successful transformation and tissue culture in other rice subgroups. For example, an early study examined rates of callus induction and regeneration across 60 rice genotypes using mature seeds and found much lower rates of callus induction and regeneration in
indica and
tropical japonica (previously referred to as “
javanica”), in comparison to
temperate japonica [
12]. For recalcitrant rice genotypes that have difficulty in callus induction from mature seeds, greater success has been made starting from immature embryos, especially for
indica varieties, although with the added downside of having to grow plants to the flowering stage and harvest and extract the embryos from the immature seeds [
9,
13,
14,
15,
16,
17,
18,
19,
20].
In contrast with the large number of studies working to improve
indica transformation efficiency, just a handful of studies have demonstrated successful transformation of
tropical japonica rice varieties. One early study showed a significantly lower transformation efficiency for the
tropical japonica variety “Lemont” in comparison to the
indica “IR64” using seedling shoots as initial explants [
21]. Subsequently,
Agrobacterium-mediated transformation using immature embryos was optimized for two U.S.
tropical japonica varieties, and transformation efficiencies ranged from 4–15% for Jefferson and 9–20% for Gulfmont [
22]. Another study optimized the transformation frequency of the Indonesian
tropical japonica variety “Rojolele” to 23% of the efficiency of Nipponbare [
23]; while the same group found a lower transformation efficiency for four other
tropical japonica varieties, ranging from 3.75 to 10% using callus induction from mature seeds [
24]. A more recent study optimized transformation and regeneration for the Indonesian
tropical japonica black rice variety “Cempo Ireng,” but ended up with a transformation efficiency of 1.5% using callus induction from mature seeds [
25]. These studies suggest that many
tropical japonica genotypes are even more difficult to transform and regenerate than
indica varieties.
Phytoene desaturase (PDS) is a key enzyme in the carotenoid biosynthesis pathway and plays a critical role in the production of carotenoids, xanthophylls, alpha-carotene, and beta-carotene [
26]. Knocking out the function of the
PDS gene leads to albinism and slow plant growth [
27,
28]. As such,
PDS has been used as an easy visual phenotypic marker for CRISPR/Cas9-mediated gene knockouts in various plant species, including rice, tobacco, apple, tomato, cassava, banana, wheat, and melon [
29,
30,
31,
32,
33,
34,
35,
36,
37]. Another gene that may provide a visual phenotypic marker for gene knockouts in rice is the
young seedling albino (
YSA) mutant, which affects the levels of total chlorophyll content in young rice seedlings [
38]. YSA is a member of a large family of pentatricopeptide repeat (PPR) proteins and plays a role in chloroplast development. A gamma-ray induced
YSA mutant was characterized as having an albino phenotype before the three-leaf stage, but then turned green again by the six-leaf stage after recovering all pigment levels, including total chlorophyll and carotenoids [
38]. Subsequently, knocking out the
YSA gene in rice with CRISPR/Cas9 was confirmed to result in a young seedling albino phenotype [
39].
Although the majority of global rice production is
indica rice, most rice varieties grown in the Southern U.S. and in South America are
tropical japonica. For example, Presidio, a long-grain
tropical japonica cultivar, is a high-yielding rice variety with excellent milling quality, strong resistance to blast and sheath blight diseases, and good ratooning potential for rice-growing areas in Texas and Louisiana [
40]. Even though it was released over 15 years ago, it is still the second most popular inbred long-grain cultivar grown in Texas (after the Clearfield variety CL153) and has been grown in Texas on average over 9000 hectares per year the past four years [
41]. Unfortunately, transformation of Presidio using the established Nipponbare protocol has been unsuccessful in our hands, indicating that this cultivar needs optimization for tissue culture and regeneration (unpublished data). In this study, we report the optimization of the rice transformation and regeneration protocol of Presidio with mature seeds and confirmation of efficient CRISPR/Cas9 gene editing by creating knockout mutants of
phytoene desaturase (
PDS) and
young seedling albino (
YSA) using both
Agrobacterium-mediated transformation and particle bombardment. Optimization of an efficient mature-seed transformation protocol for this
tropical japonica variety provides rapid and efficient CRISPR-based gene editing in the background of a popular, high yielding, long-grain rice cultivar, paving the way for more efficient gene editing across similar
tropical japonica varieties for rice production in the U.S. and South America.
3. Discussion
The current study has optimized an efficient transformation and gene editing system for the popular U.S. long grain
tropical japonica variety Presidio. As Presidio could not be transformed using the previous best practice protocol for rice transformation [
9], a number of modifications were tested and found necessary to proceed to the next step of the tissue culture process. First, during the callus induction phase, 10 days were not adequate for the induction; only after increasing to 21 days was it possible to achieve actively proliferating calli to be used as initial explants prior to the infection/bombardment step. A similar result was observed by another group working on elite
japonica varieties [
11]. Next, on the
Agrobacterium-mediated transformation pipeline, co-cultivation can be challenging in an in vitro environment as bacteria overgrowth can lead to irreversible damage to the tissue, jeopardizing further steps. In our hands, the three days of co-cultivation had to be reduced to one day to avoid bacteria overgrowth. Another major change implemented during the calli selection phase was to keep the samples in the dark. During optimization, we noticed that light conditions for Presidio accelerated lethal tissue necrosis on the explants when compared to dark conditions; however, the putative transformed tissue was able to survive and proliferate for both selection stages when kept in the dark. Lastly, hormones play a very important role during the critical regeneration stage and are known to have varying levels of effectiveness across different ranges of concentration, hormone combinations, and length of exposure. After negative regeneration results were obtained using 0.5mg/L kinetin concentration for 14 days as proposed by the literature [
9], modifications were implemented to stimulate regeneration responses from the embryogenic tissue. As described in several
indica rice transformation studies [
11,
43], often higher concentrations of cytokine and a longer regeneration phase are required. Presidio regeneration was finally successfully obtained using 40 days under 3mg/L of kinetin. Only after identifying the ideal combination of modifications described above were we able to develop a robust transformation protocol for Presidio ready to support a successful gene editing approach.
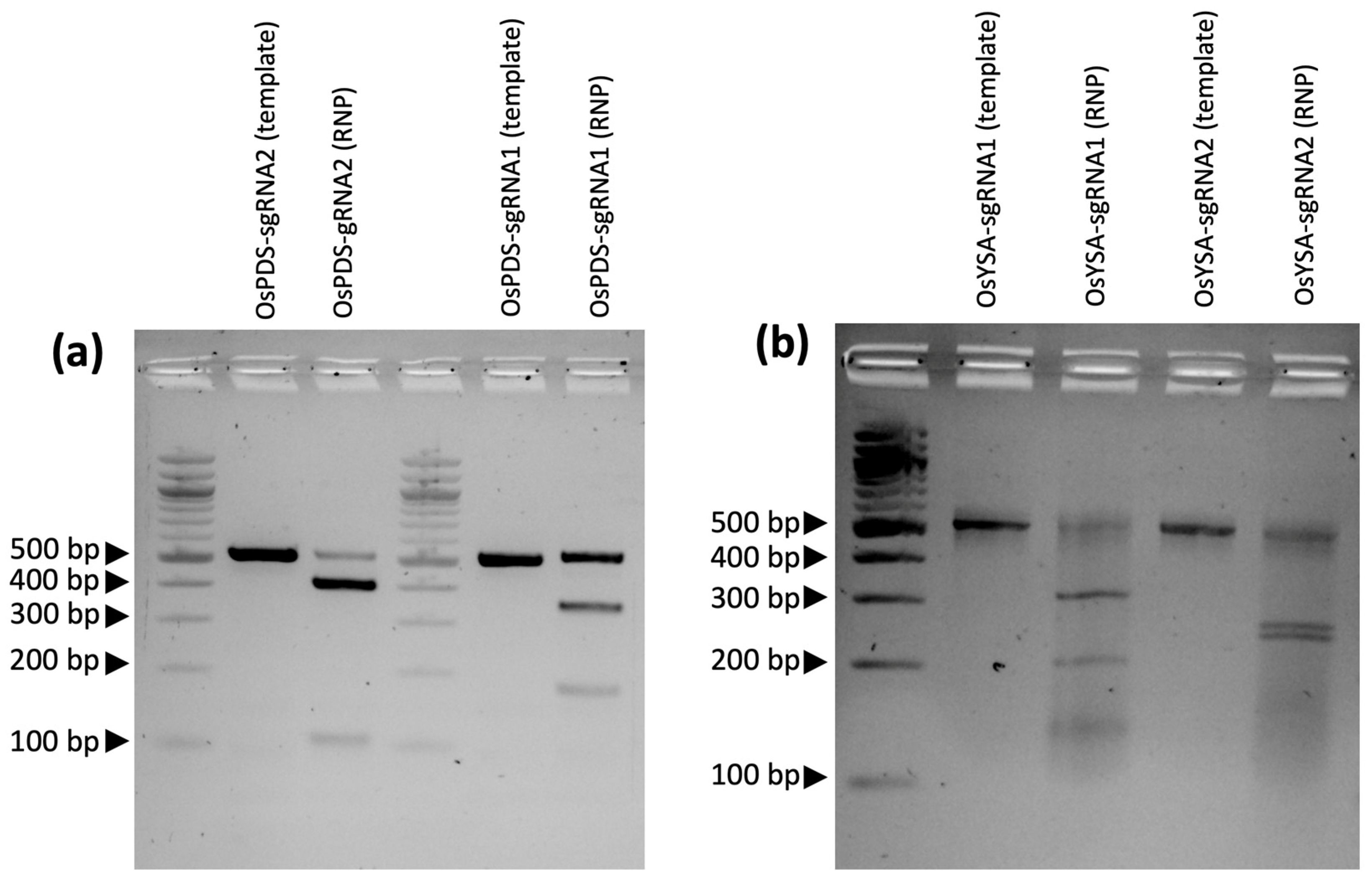
The first step to ensure a successful gene knockout project is robust gRNA design targeting coding sequences near the start of the gene to increase the chance that a frameshift mutation will lead to a premature stop codon that will knock out the gene function. Best practices call for sequencing the target site in the genotype of interest to detect any polymorphisms that differ from the reference genome that might interfere with the gRNA or PAM sequences. In the current study, Presidio was sequenced at the target regions to avoid any polymorphic regions when designing the gRNAs. Once the target regions were identified, an online gRNA design tool, in this case CRISPRdirect, was used to ensure proper design of the gRNA next to a protospacer adjacent motif (PAM) sequence, while at the same time avoiding potential off-target sites across the genome that may share similar sequences. Another validation step is running the guide RNAs through an in vitro ribonucleoprotein (RNP) assay to ensure that the gRNAs are functional in cutting the target sequence. Interestingly, in the current study, the predicted efficiency of each guide RNA based on how well each gRNA digested the target DNA fragment in the in vitro RNP assays (
Figure 1) matched the relative mutation rates from the transformation experiment: for the PDS target gene, gRNA2 had more mutations than gRNA1, while for YSA, gRNA1 had a higher number of mutations than gRNA2 (
Supplementary Table S2).
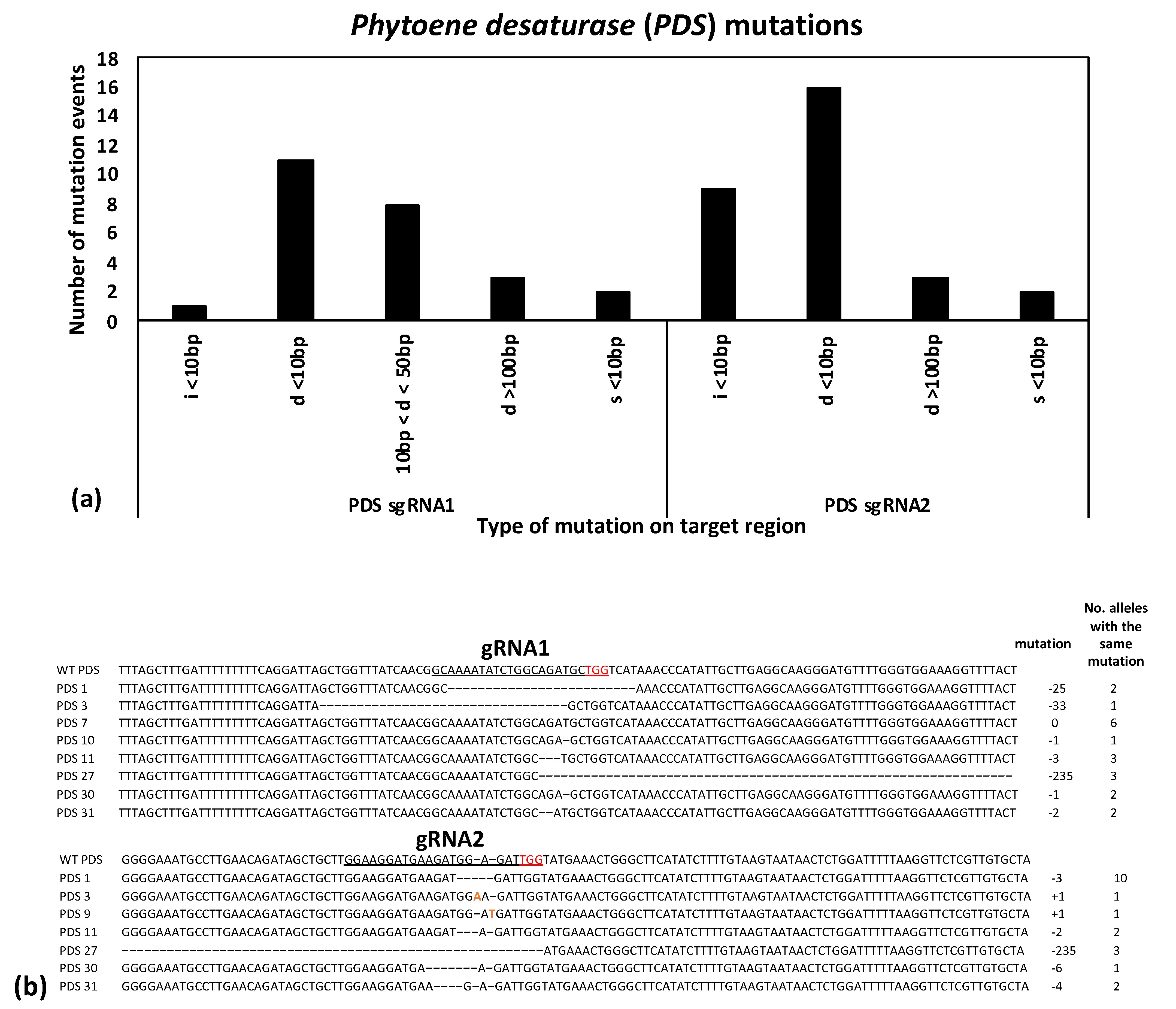
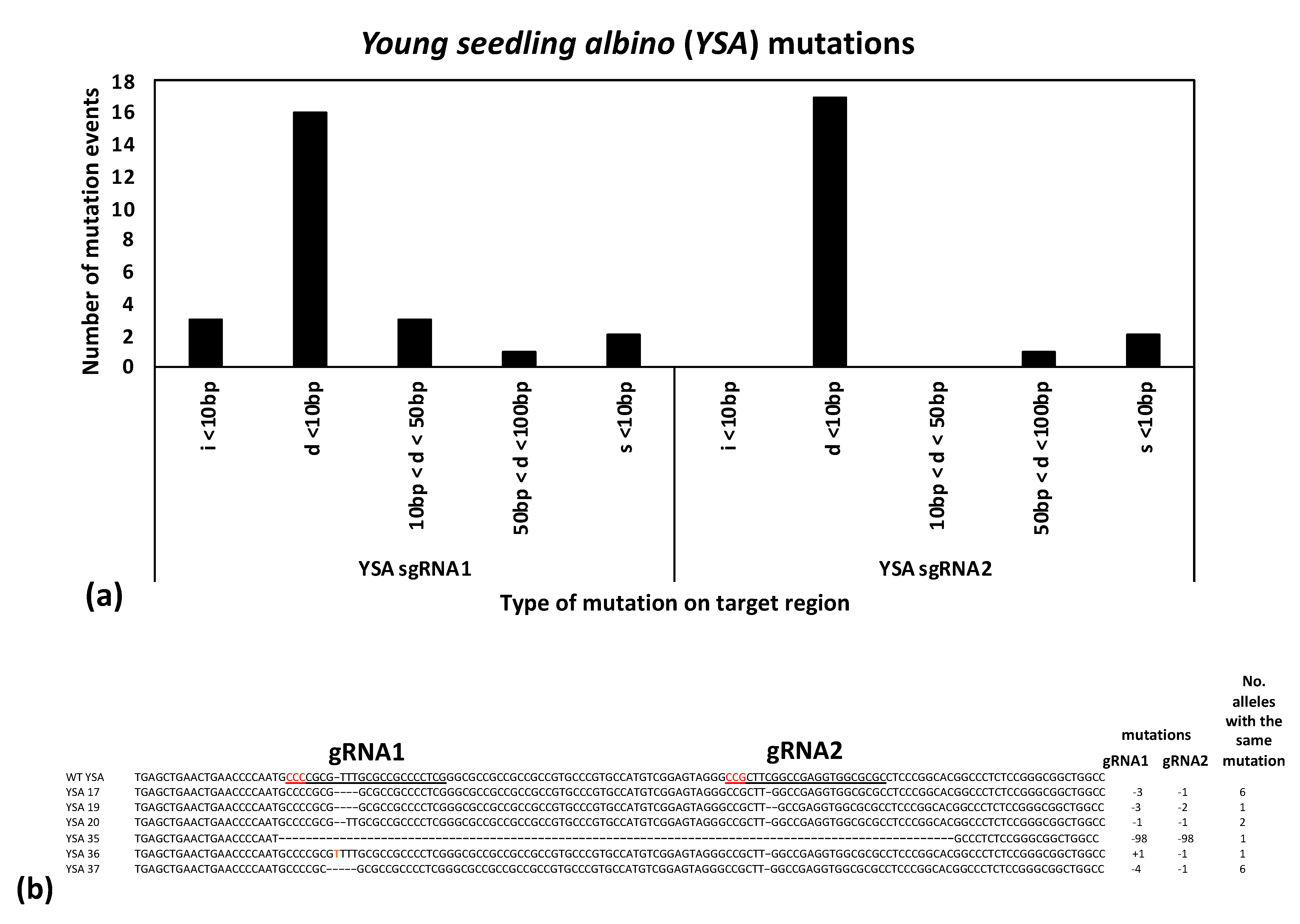
To further ensure successful gene editing, two gRNAs were designed in the coding sequence for each gene target spaced between 40–250 bp apart. If both are successful in creating mutations, they can either create a deletion spanning the two sites or induce two smaller mutations at each target site. Likewise, if one gRNA unexpectantly fails or induces mutations at a lower rate, the second gRNA serves as a backup, providing a high overall chance of induced mutations knocking out the gene function. All combinations were observed in the present study in the edited progeny: deletions spanning both gRNA sites, small mutations at both gRNA sites, and small mutations at one of the gRNA sites (
Figure 3 and
Figure 4). Notably, most of the mutations observed in the current study were predicted to lead to frameshift mutations (such as the 1, 2, or 4 bp deletions or insertions). Moreover, in all cases where an in-frame mutation was expected at one gRNA site (i.e., four observed 3 bp deletions and one observed 6 bp deletion), the second gRNA site in the same plant had a predicted frameshift mutation (
Figure 3 and
Figure 4). Frameshift mutations are predicted to have a high chance of gene knockouts by leading to premature stop codons and truncated proteins.
Another important component of a successful gene editing project is robust gRNA expression. Although both gRNAs can each be expressed with their own small nuclear RNA promoters, such as U3 or U6, greater levels of expression have previously been demonstrated by taking advantage of the endogenous tRNA processing system [
44]. By inserting a synthetic polycistronic tRNA–gRNA (PTG) module into the plant, multiple gRNAs can be expressed at a high level and precisely excised in planta to produce functional gRNAs [
44]. The current study deployed this system to express the two gRNAs for each target gene by ordering a synthetic DNA fragment specific for each gene target, which was then expressed with a U3 promoter, along with the Cas9 gene expressed with a rice ubiquitin promoter (
Supplementary Figure S4). These vectors were tested using both
Agrobacterium-mediated transformation and particle bombardment: calli were induced from mature seeds, transformed, regenerated, and grown until the desired visible phenotype was observed (
Figure 1). As the
Agrobacterium-mediated transformation and particle bombardment provided similar results, we chose to move forward with characterizing a subset of the
Agrobacterium-transformed plants to avoid complications from multiple copies and/or fragmented insertions from bombardment. These results showed that stable integration by
Agrobacterium-mediated transformation of a T-DNA containing the U3-PTG and UBI-Cas9 led to expression of both gRNAs and successful editing of each target gene (
Supplementary Table S2).
The PDS protein plays a critical role in the carotenoid biosynthesis pathway and is involved in the synthesis of key photosynthetic pigments; notably, disruption of the
PDS gene affects chloroplast development and leads to an albino phenotype [
27]. Likewise, the YSA protein is important for the development of chloroplasts during early seedling development; disrupting the
YSA gene causes an albino phenotype at the third-leaf stage; however, plants recover to a normal green by the six-leaf stage [
38]. One objective of the current study was to compare the usefulness of knocking out the
PDS and
YSA genes to provide a visible phenotype when optimizing plant transformation and gene editing protocols in rice. Using CRISPR/Cas9 to edit the PDS gene produced both variegated and full albino plants (
Supplementary Figure S1), while mutating the YSA gene produced a mix of green and more subtle variegated phenotypes (
Supplementary Figure S2). Sanger sequencing was performed to confirm the mutations underlying the two gRNAs for each of the
PDS and
YSA target sites. For the
PDS target, the sequence results largely matched the phenotypes—all of the albino seedlings had either a homogyous (i.e., two identical mutant alleles) or biallelic (i.e., two different mutant alleles) at one or both of the gRNA sites (
Supplementary Table S2). Likewise, one of the green seedlings (PDS-4) had a heterozygous mutation at gRNA1 and was wild type of gRNA2, suggesting that the heterozygous mutation was not enough to cause an albino phenotype. At the same time, four other green PDS-edited seedlings (PDS-5, 6, 8, and 9) showed heterozygous mutations at gRNA1, but were homozygous or biallelic at gRNA2. A closer examination of the sequences from the five cloned PCR products for each transformed sample revealed that all four samples were heterozygous at gRNA1 (i.e., one wild type and one mutated allele) and that the homozygous or biallelic mutations at gRNA2 were largely 1 bp substitutions or 3 bp deletions, which presumably are not enough to disrupt the protein function (data not shown). On the other hand, mutations in the
YSA gene did not lead to clear albino phenotypes. While all of the sequenced progenies had homozygous or biallelic mutations at both gRNA1 and gRNA2, only four plants showed either young white or variegated leaves (
Supplementary Table S2). Although the published young seedling albino mutations had obvious albino phenotypes at the three-leaf stage, that was in the
indica genetic background of Pei’ai64S [
38]. It may be possible that the
tropical japonica background (in this case, Presidio) has other related genes that compensate for the
YSA mutations, leading to a reduced phenotypic effect. Therefore, the results of our study suggest that
PDS is a more reliable phenotypic indicator than
YSA for testing CRISPR-based knockouts in a
tropical japonica genetic background.
4. Materials and Methods
4.1. Amplification of the PDS and YSA Genes in Nipponbare and Presidio
About 30–50 mg of the tips of rice leaves were collected and placed in 2.0 mL screw cap tubes (Cat #1420-9600, USA Scientific, Ocala, FL, USA) for DNA isolation. Genomic DNA extractions of rice leaf tissues were carried out following a modified CTAB protocol [
45]. Briefly, leaf tissues were homogenized in 600 μL of CTAB buffer, 40 mg of polyvinylpyrrolidone (PVP-40) and 4 μL of 2-mercaptoethanol. Each sample was topped with 10 μL of RNase A (10 mg/mL), and then incubated at 60 °C for 15 min. Afterward, samples were extracted with chloroform: isoamyl alcohol (24:1), and genomic DNA was precipitated with 500 μL of isopropanol and resuspended in 100 μL of water.
To compare the similarity of both PDS and YSA between the Nipponbare and Presidio, primers were designed to the Nipponbare reference genome to amplify a portion of the PDS (Gramene ID: Os03g0184000) and YSA (Gramene ID: Os03g0597200). A portion of the PDS gene, which included exon 3 and exon 4, was amplified using the following primers: OsPDS forward primer (5’ CCATTACAGGTCGTGATTGCT) and OsPDS reverse primer (5’ ATCTATCAGTGCTGGCGGTAA). The following primers were used to amplify a part of the first exon of the YSA gene: OsYSA forward primer (5’ CCTGTGCATGCGCTCTCTTC) and OsYSA reverse primer (5’ AGGGGCACCAGGTGAAATTG). The PCR protocol for PDS is as follows: 98 °C for 30 s, followed by 30 cycles of 98 °C for 10 s, 60 °C for 30 s, and 72 °C for 30 s and a final extension of 72 °C for 5 min. The same PCR protocol was followed for YSA, with the exception that the annealing temperature was 58 °C. The fragments of the PDS and YSA genes were amplified in Nipponbare and Presidio cultivars using PhusionTM High-Fidelity DNA polymerase (Cat #F530L, ThermoFisher Scientific, Waltham, MA, USA) and viewed in a 1.2% agarose gel. The PCR products were extracted and purified using the QIAquick Gel Extraction Kit (Cat #28704, Qiagen, Germantown, MD, USA) and sequenced by Sanger sequencing (Texas A&M Laboraotry for Genome Technology, College Station, TX, USA). Conserved sequences between Presidio and Nipponbare were used for gRNA design.
4.2. Design of gRNAs and Testing by In Vitro Digestion with the RNP Assay
Two gRNAs were designed for the coding regions of both
PDS and
YSA. All gRNAs were designed using the online CRISPRdirect design tool [
46], and gRNAs with the NGG PAM sequence were selected. The
PDS gene is located on chromosome 3 and has 14 exons. Two gRNAs targeting the third and fourth exon were designed (
Supplementary Table S1). Coincidently, the gRNA targeting the fourth exon has been shown to work previously [
47]. Both gRNAs are 204 bp apart from each other. The
YSA gene, also located on chromosome 3, has one exon; therefore, both gRNAs were designed for this exon. The gRNAs targeting the
YSA exon are 40 bp apart.
Synthetic gRNAs targeting
PDS and
YSA were ordered from Synthego and their functionality was tested by in vitro digestion of target PCR amplicons using a ribonucleoprotein (RNP) assay. The PCR-amplified fragments of the
PDS and
YSA gene targets from wild-type Presidio were gel purified using a QIAquick gel extraction kit. The RNP complex was developed following a protocol from Integrated DNA Technologies (IDT) as follows: 11 μL of gRNAs with a concentration of 110 μM, 1.6 μL of 62 μM Cas9 protein from
Streptococcus pyogenes (Cat #1081059, IDT, Coralville, IA, USA), and 87.4 μL of PBS buffer were combined and incubated at room temperature for 10 min. Then, 1 μL of RNP complex (1 μM) was mixed with 1 μL of 10x Cas9 nuclease reaction buffer (200 mM HEPES, 100 mM MgCl2, 5 mM DTT, and 167 mM KCl), 50 nM of DNA substrate, and 7.5 μL of nuclease-free water. Afterward, the mixture was incubated at 37 °C for 60 min, followed by the addition of 1 μL of 20 mg/mL proteinase K, which was then incubated at 56 °C for 10 min. Results were then visualized by electrophoresis on a 1.2% agarose gel. The cleavage efficiency was determined by densitometry using ImageJ 1.52v (
https://imagej.nih.gov/ij/ accessed on 20 April 2020). The control contained everything but the gRNAs, which resulted in an uncut PCR amplicon of either
PDS or
YSA, and the controls were compared to the products that were cut by the gRNA.
4.3. Construction of the tRNA–gRNA Vector and Agrobacterium Cloning
To obtain efficient expression of both gRNAs targeting the
PDS and
YSA genes, we used an endogenous tRNA-processing system, which has previously been shown to work well in rice [
44]. BsaI restriction sites flanking both ends of the tRNA–gRNA construct were used for the ligation to the pRGEB32 vector (Addgene plasmid #63142;
http://www.addgene.org/63142/ accessed on 20 April 2020). The tRNA–gRNA construct was synthesized by GenScript and cloned into a pUC57 vector by GenScript. This vector was then digested with BsaI to extract the tRNA–gRNA construct and ligated to the pRGEB32 vector. The end result was the pRGEB32 binary vector with either the
PDS tRNA–gRNA or
YSA tRNA–gRNA construct (
Supplementary Figure S4), which was then transformed into 5-alpha
E. coli competent cells (Cat #C2988J, NEB, Ipswich, MA, USA). The
E. coli transformation protocol was performed as suggested by NEB. The tRNA–gRNA and pRGEB32 plasmid were isolated from
E. coli using the QIAprep Spin Miniprep Kit (Cat #27104, Qiagen, Germantown, MD, USA), validated by both restriction digestion and Sanger sequencing. Validated plasmids were then transformed into
Agrobacterium tumefaciens EHA105 cells.
EHA105 transformations with the tRNA–gRNA fragment in pRGEB32 were carried out using the freeze and thaw method [
48]. Briefly, 50 μL of EHA105
Agrobacterium cells were thawed on ice, and 1 μL (100–1000 ng) of plasmid DNA was added. The tube was gently flicked to mix, and the mixture was kept on ice for 2 min. The mixture was then transferred into liquid nitrogen for 5 min and then incubated in a 37 °C water bath for 5 min. After the incubation, 1 mL of liquid LB media was added to the mixture and incubated for 4 h on a shaking incubator at 28 °C. The tubes were then placed in a centrifuge for 2 min at 6000 rpm to pellet the cells. The excess supernatant was removed and left with 50 μL of liquid LB media. All 50 μL were then suspended on Petri dishes containing solid LB media with kanamycin antibiotics and incubated for 2 days at 28 °C. Colonies were picked and placed in 5 mL of liquid LB media on a shaking incubator at 28 °C for three days to allow the cells to grow. Approximately 4 mL of the plasmid was isolated for validation by restriction digestion and Sanger sequencing. The other 1 mL was stored to be used for the plant transformations.
4.4. Agrobacterium-Mediated Transformation
The tissue culture transformation approach using the callus method for mature seeds was adapted from a previously published protocol [
9] with some modifications.
Agrobacterium tumefaciens EHA105 was cultured on an AB plate that contains appropriate antibiotics in the dark at 28 °C for 3 days. The bacteria were then collected with a loop, suspended in 1 mL of AAM medium at a final density OD600 = 0.3. Well-maintained mature Presidio seeds were dehusked and collected in a 50 mL sterile tube. Next, the seeds were surface sterilized in 20 mL of 70% ethanol for 10 sonds, followed by 25 mL of 2% sodium hypochlorite that contains one drop of Tween-20 under stirring for 30 min. The seeds were then washed in sterile water three to five times, placed them on 2N6 callus induction medium and incubated at 30 °C in the dark for 7 days. Scutella from cultured seeds were then cut out, transferred to fresh 2NBK medium with the scutella facing up, and incubated at 30 °C in the dark for 21 days for calli formation. The actively proliferating calli were cut/isolated into small pieces of 0.5–1 mm in diameter using a scalpel and were transferred with forceps to the 2N6 medium. Next, the plate was sealed with surgical tape, and 200–250 calli can be placed on a single plate. Following this, the plates were incubated under continuous illumination (5000 lux) at 32 °C for 3 days.
The new actively proliferating calli were soaked in 5 mL sterile water in a 90-mm Petri dish. After that, the calli were separated and the water was removed. A total of 1.0 mL of bacterial suspension was added and mixed with the calli by pipetting; the mixture was then incubated for 2 min at 25 °C. The mixing by pipetting was repeated again and the bacterial suspension was then removed. Following this, approximately 150 calli were transferred with forceps to a co-cultivation medium, and the plate was sealed with Parafilm and incubated in the dark at 25 °C for 1 day. After co-cultivation, the calli were transferred to selective medium 2NBKCH40,30 to 60 calli can be placed on a single plate. Next, the plates were incubated under dark conditions at 32 °C for 2 weeks. Subsequently, the proliferated, yellowish white calli were transferred to the second selective medium NBKCH40 (modified) and the plate was sealed with surgical tape. The plates were then incubated under dark conditions at 32 °C for 7 days. Again, the proliferated calli were transferred to the regeneration medium NBKRH40, and the plate was sealed with surgical tape. The plates were then incubated under continuous illumination (5000 lux) at 32 °C. Thereafter, the subculture was performed into fresh media every three weeks until plant regeneration (40–60 days). Regenerated plantlets advanced to rooting induction media NBKFH40 under continuous illumination at 32 °C.
4.5. Particle Bombardment-Mediated Transformation
Preparation of gold particle stocks was performed as follows: a total of 20 mg of 0.6 μm gold microcarriers (Cat #165-2262, Bio-Rad, Hercules, CA, USA) were weighed in a sterile 1.5 mL Eppendorf tube followed by the addition of 1mL of 100% ethanol. The mixture containing gold and ethanol underwent water bath sonication (Cat #CPX-952-218R, Branson, Brookfield, CT, USA) for 2 min and was then microcentrifuged for 3 s at top speed. The supernatant was removed, and the ethanol wash was repeated once more. The wash with ethanol was performed twice. Afterward, 1 mL of sterile distilled water was added, and water bath was sonicated for 2 min. The gold microcarriers were then centrifuged for 3 s at top speed. The wash with sterile distilled water was repeated once more. The washes with distilled water were performed twice. Thereafter, the gold microcarriers were resuspended in 1 mL of sterile distilled water. The resuspension was aliquoted into 50 μL amongst 1.5 mL Eppendorf tubes and stored at −20°C for future use.
4.6. Coating the Gold Microcarriers with Plasmid DNA
An aliquot of the gold microcarriers was defrosted and water-bath sonicated for 1 min. Plasmid DNA isolated with the Plasmid Maxi Kit (Cat #12162, Qiagen, Germantown, MD, USA) was used for particle bombardment in order to obtain a high concentration, which decreases the volume used during the experiment. Our experiments seemed to work best when the volume of DNA was 5 μL or less. At least 1 mg/mL of plasmid DNA was added to the gold microcarriers, and the Eppendorf tube was vortexed for 1 min. After mixing the components together, 50 μL of 2.5 M calcium chloride and 20 μL of 0.1 M spermidine was added onto the inside of the cap of the Eppendorf tube. The calcium chloride and spermidine were mixed together on the cap by pipetting up and down 2–3 times. The cap of the Eppendorf tube was closed, and the tube was tapped down to let all the components mix together at the bottom of the Eppendorf tube. The mixture was microcentrifuged for 5 s at top speed, and the supernatant was removed. The pellet was resuspended in 150 μL of 100% ethanol and vortexed for 1 min. The components were microcentrifuged for 5 s at top speed, and the supernatant was removed. The pellet was then resuspended in 40 μL of 100%.
4.7. Preparation of Plasmid DNA Delivery
The macrocarrier holders (Cat #165-2322, Bio-Rad, Hercules, CA, USA) and stopping screens (Cat #165-2336, Bio-Rad, Hercules, CA, USA) were sterilized by autoclave. The macrocarriers (Cat #165-2335, Bio-Rad, Hercules, CA, USA) and 900 psi rupture disks (Cat #165-2328, Bio-Rad, Hercules, CA, USA) were sterilized with 100% ethanol on the day of use. The Biolistic® PDS-1000/He particle delivery system (Cat #165-2257, Bio-Rad, Hercules, CA, USA) was cleaned and sterilized with 75% ethanol before and after use. Once cleaned with 100% ethanol, macrocarriers and rupture disks are placed in sterile Petri dishes and allowed to air dry. Once airdried, the macrocarriers are loaded on the macrocarrier holders. The gold-plasmid complex in the 100% ethanol solution was slowly loaded onto the center of the macrocarrier, which is within the macrocarrier holder. The gold-plasmid DNA complex was then allowed to air dry on the macrocarriers for 5–10 min.
Once the gold-plasmid DNA has dried, the macrocarrier holder is loaded onto the delivery system. The set-up for the Biolistic® PDS-1000/He particle delivery system was performed following the provided manual. The settings used for the Biolistic® PDS-1000/He particle delivery system were as follows: 2.5 cm distance gap between the rupture disk and macrocarrier, 9 cm target distance between the stopping screen and the target plate, 0.8 cm distance between the macrocarrier and the stopping screen, 28–29” Hg vacuum, 5.0 vacuum flow rate, and 4.5 vacuum vent rate. Presidio rice calli on osmotic media where bombarded, and the plates were removed from the delivery system and wrapped with 3M micropore tape. The osmotic media was prepared as follows: 4.4 g of Murashige and Skoog (MS) media (Cat #M5524, Sigma-Aldrich, St. Louis, MO, USA), 5 mL of 2,4-dichlorophenoxyacetic acid (1mg/mL) (Cat #D72724, Sigma-Aldrich, St. Louis, MO, USA), 72.867g of mannitol (Cat #M1902, Sigma-Aldrich, St. Louis, MO, USA), and 3.2 g of phytagel (Cat #P8169, Sigma-Aldrich, St. Louis, MO, USA), and the pH was adjusted to 5.8 (Liang et al. 2018). The plates were placed in the dark at 25 °C overnight. After the overnight incubation in the dark, plates were kept under the same conditions as calli treated with Agrobacterium.
4.8. Validation of Edited Plants
Leaf samples from the regenerated plants, transformed with either a
PDS tRNA–gRNA or
YSA tRNA–gRNA plasmid, were collected. Genomic DNA was extracted as described above, and the regions of
PDS and
YSA targeted by the sgRNAs were amplified by PCR. The purified PCR samples were gel purified and ligated to pCR–Blunt vector (Cat #K270020, ThermoFisher Scientific, Waltham, MA, USA). The ligated vector and PCR product were then transformed into DH5-alpa competent
E. coli cells. For each sample, five colonies were randomly selected, and DNA plasmids were then isolated using QIAprep Spin Miniprep Kit and were sent for Sanger sequencing at the Laboratory for Genome Technology at Texas A&M University using an ABI 3130 Genetic Analyzer. Sequencing data were then analyzed using the MAFFT Multiple Sequence Alignment Software Version 7 from Benchling (
https://www.benchling.com/ accessed on 20 April 2020).


 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}