
α-Galactosidase a Deficiency in Fabry Disease Leads to Extensive Dysregulated Cellular Signaling Pathways in Human Podocytes

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
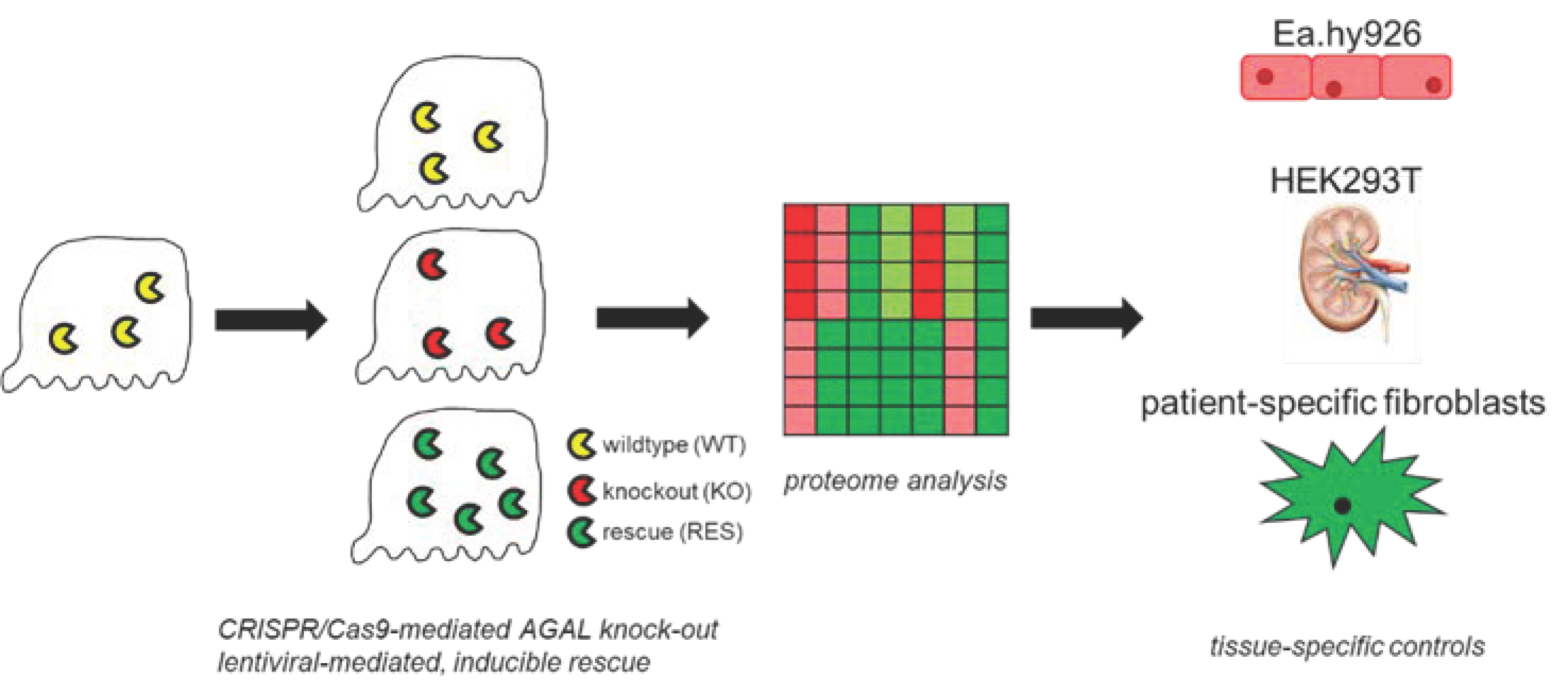
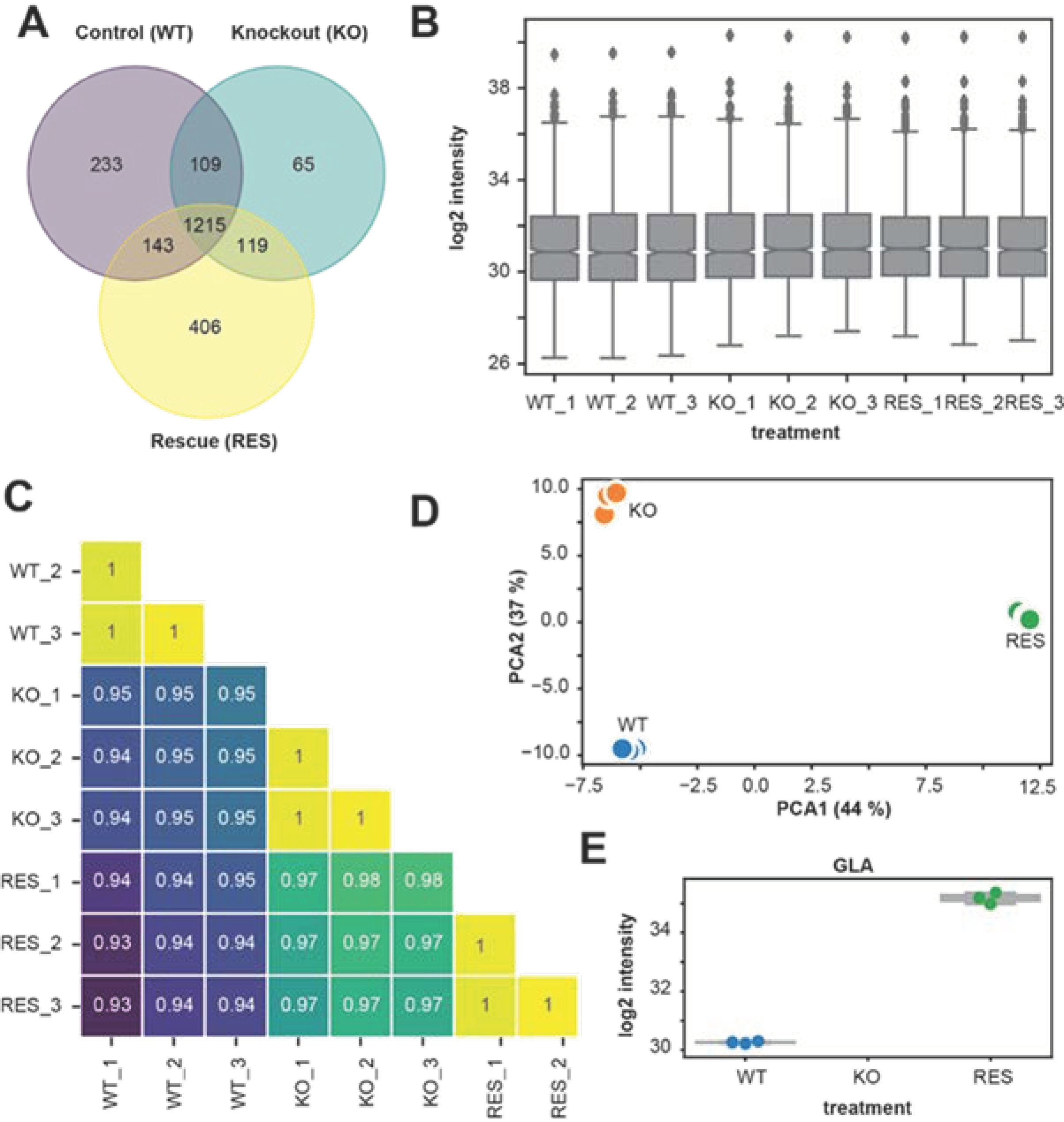
2.1. Proteome Analyses
2.1.1. Comparison of Replicates and Groups for LC-MS/MS Reproducibility and Quality
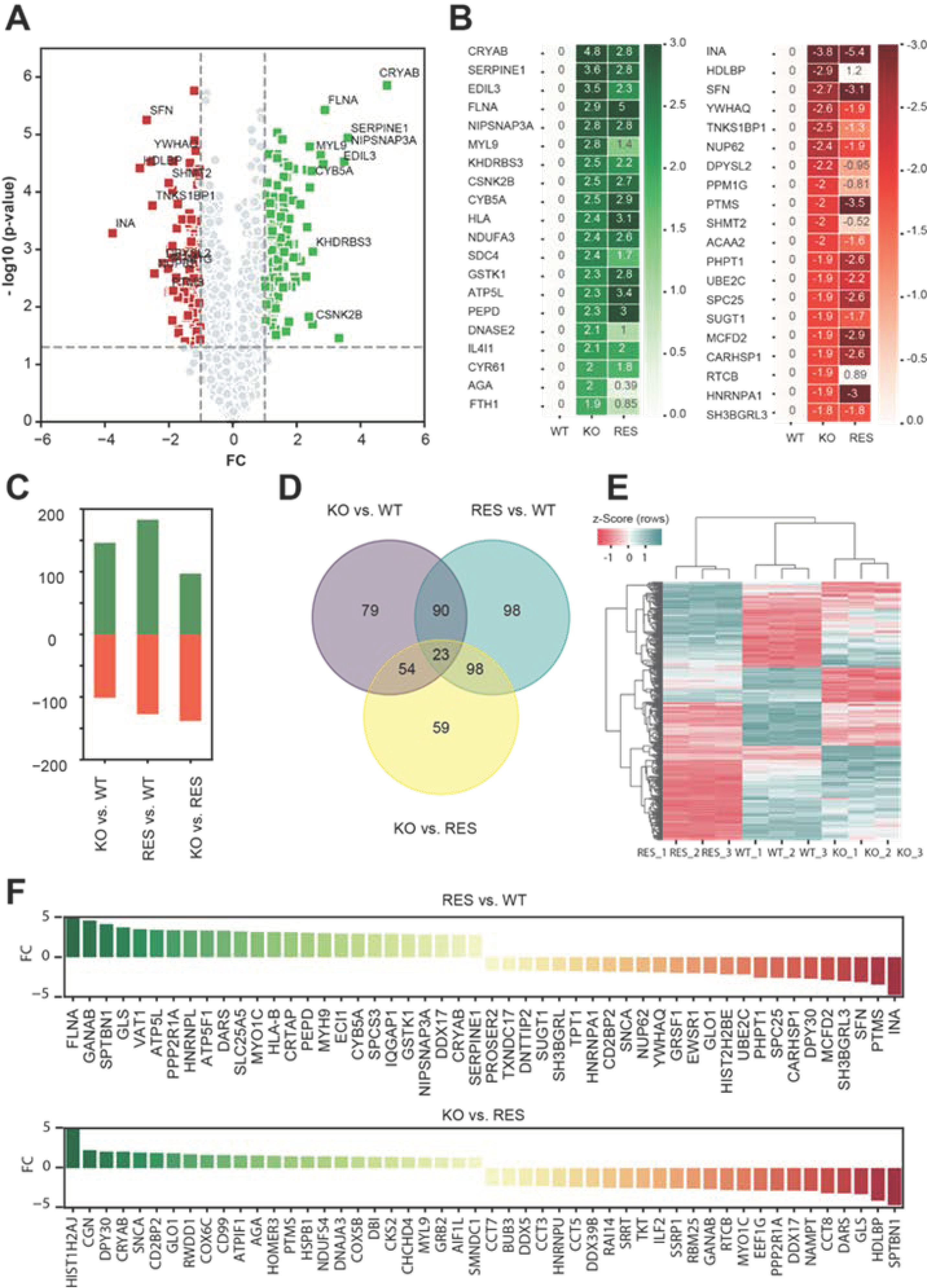
2.1.2. Regulated Proteins in Pairwise Comparison
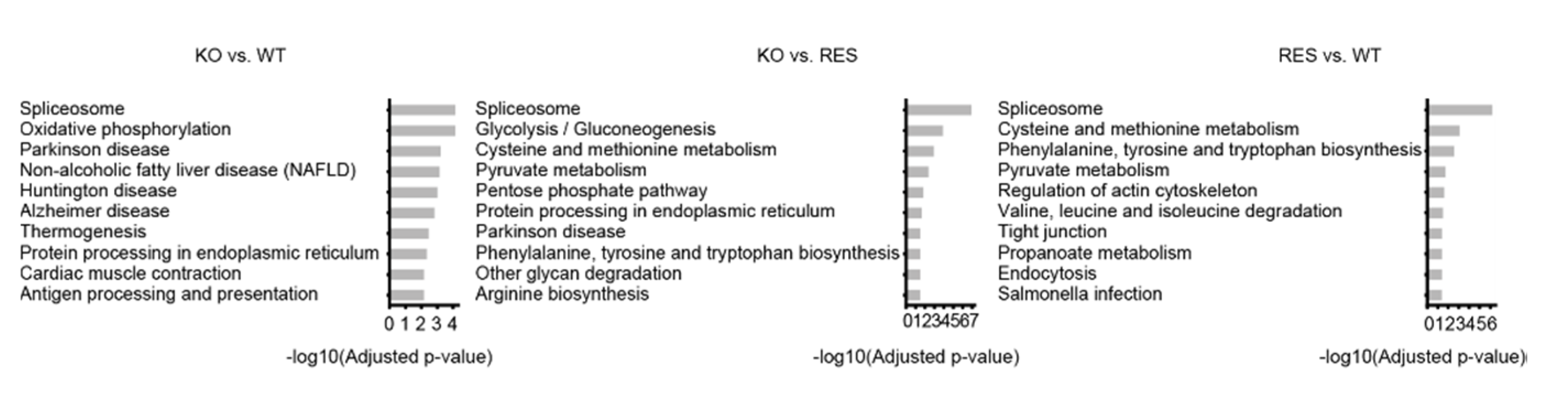
2.1.3. Gene-Set Enrichment Analysis
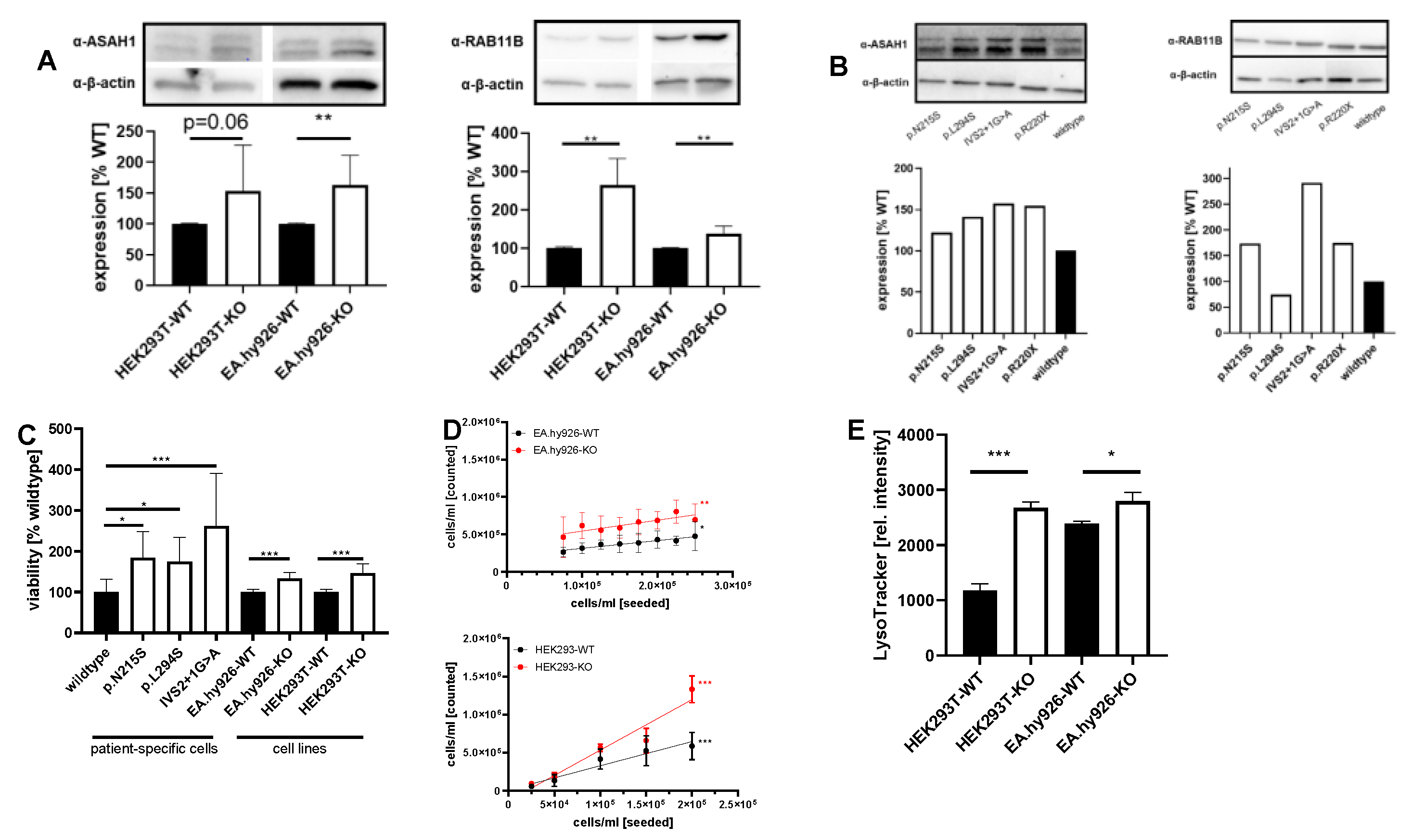
2.2. Expression of GOIs and Functional Analyses of Involved Pathways in Other FD Cell Lines and Patient-Specific Fibroblasts
3. Discussion
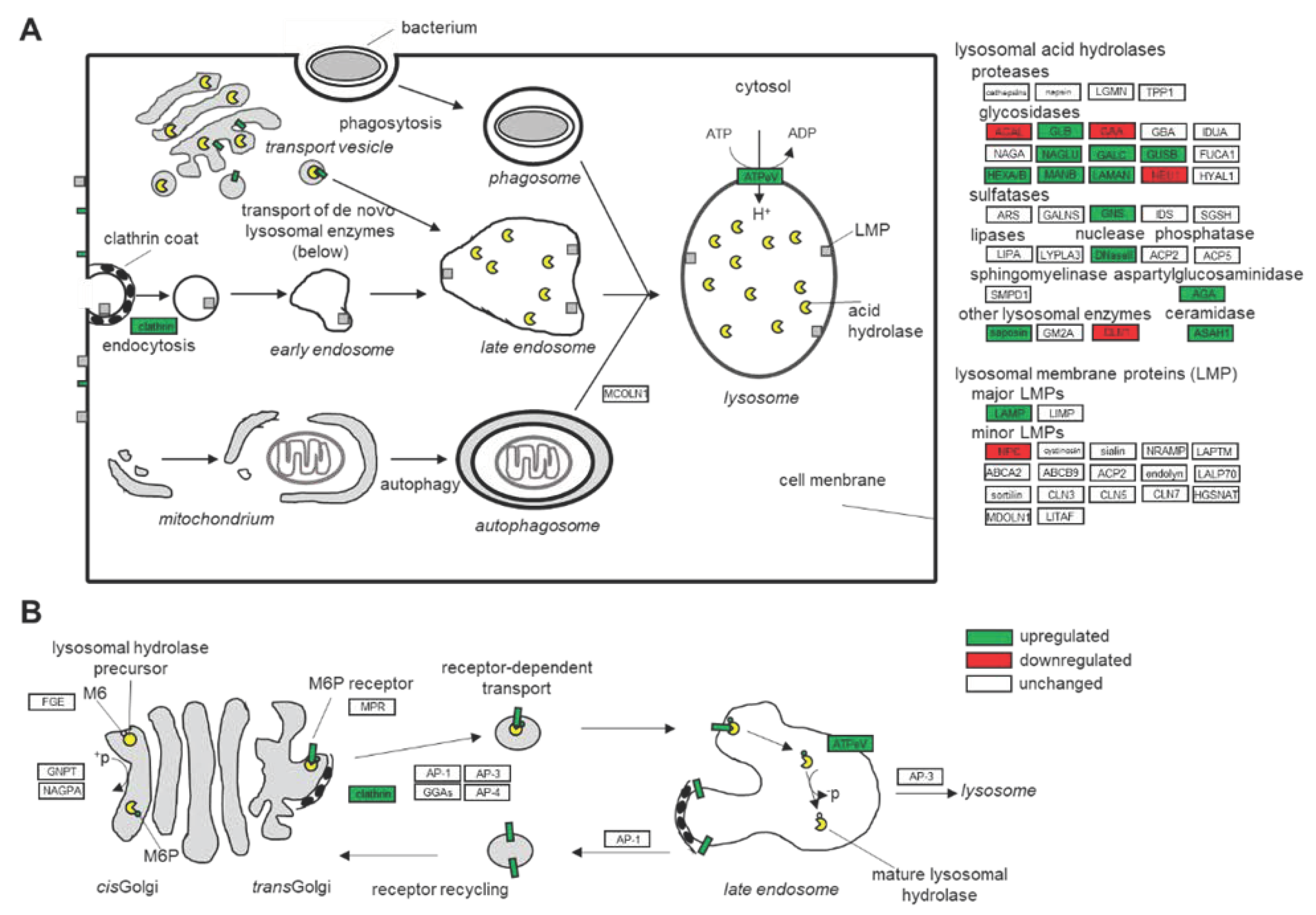
3.1. Dysfunction of the Lysosomal Pathways
3.2. Comparison of the Results to Other Chronic Kidney Diseases and to Other FD-Typical Manifestations
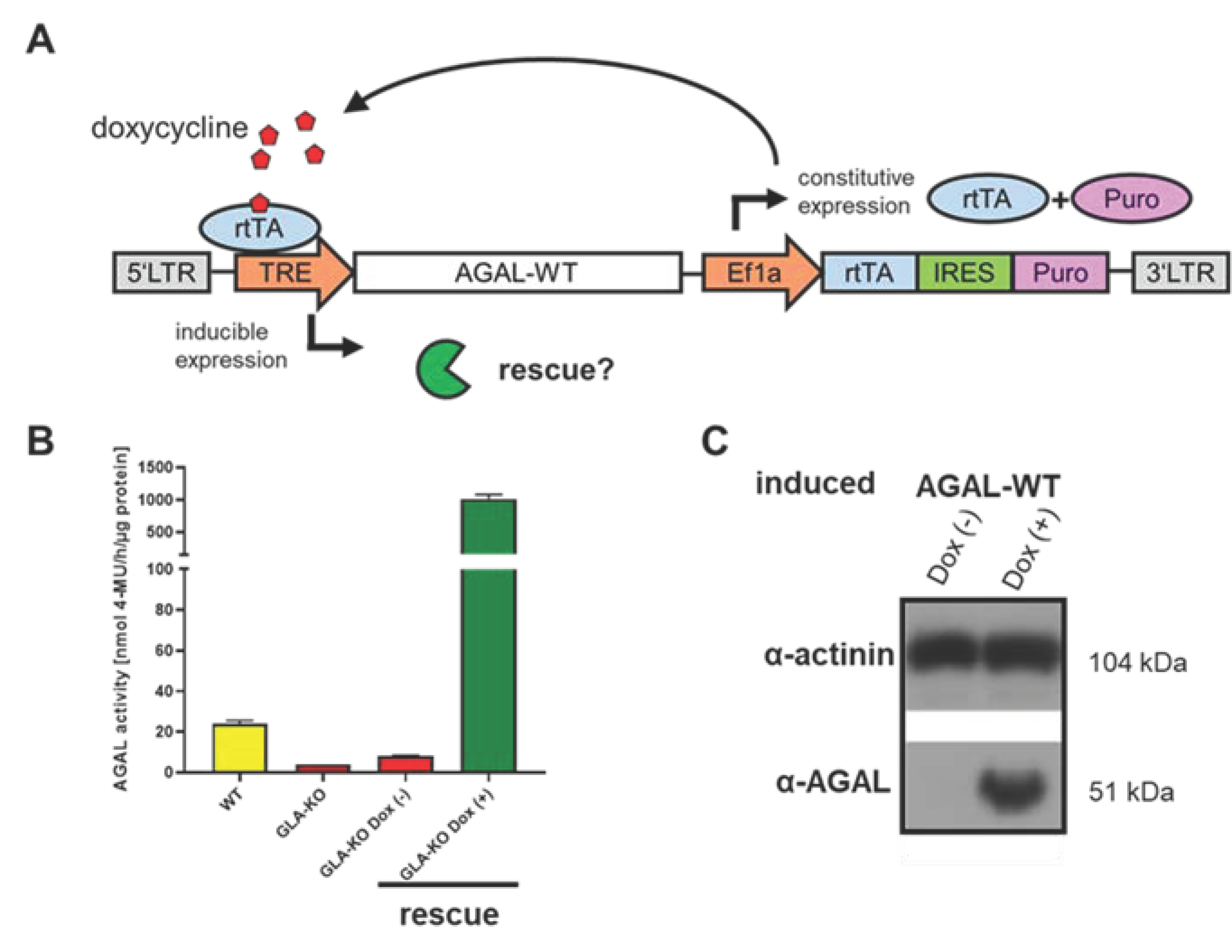
3.3. Rescue and Normalization of Protein Expression
3.4. Validation of Dysregulated Pathways and Functions in Other AGAL-Deficient Cell Lines
3.5. Limitations
4. Materials and Methods
4.1. Cell Culture Maintenance
4.2. Proteome Analysis and Statistics
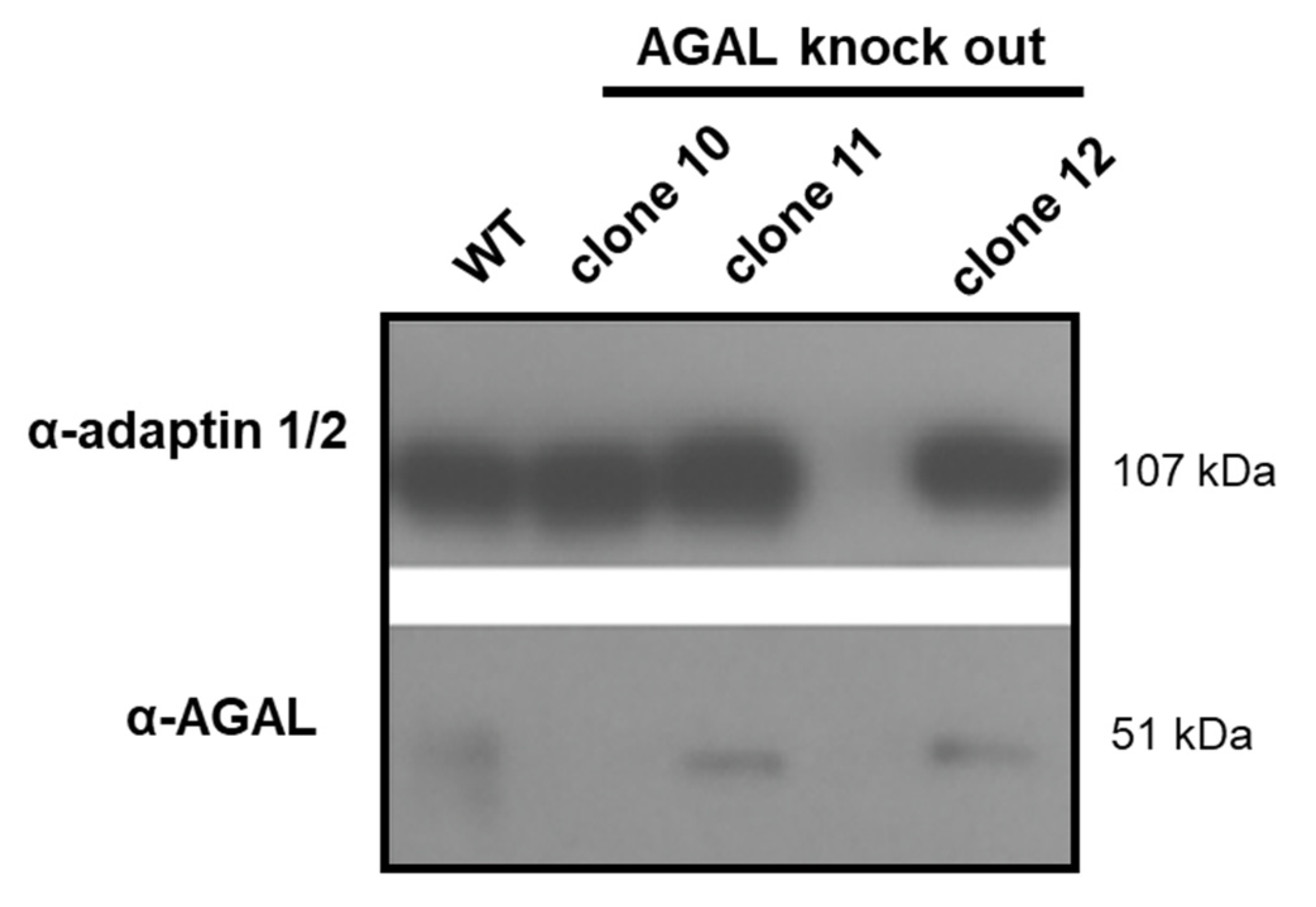
4.3. CRISPR/Cas9-Mediated GLA Knockout in Podocytes
4.4. AGAL Expression and Sample Preparation for Proteome Analyses
4.5. Western Blot Analyses
4.6. AGAL Enzyme Activity Assay
4.7. Proliferation Rate and Lysosome Staining
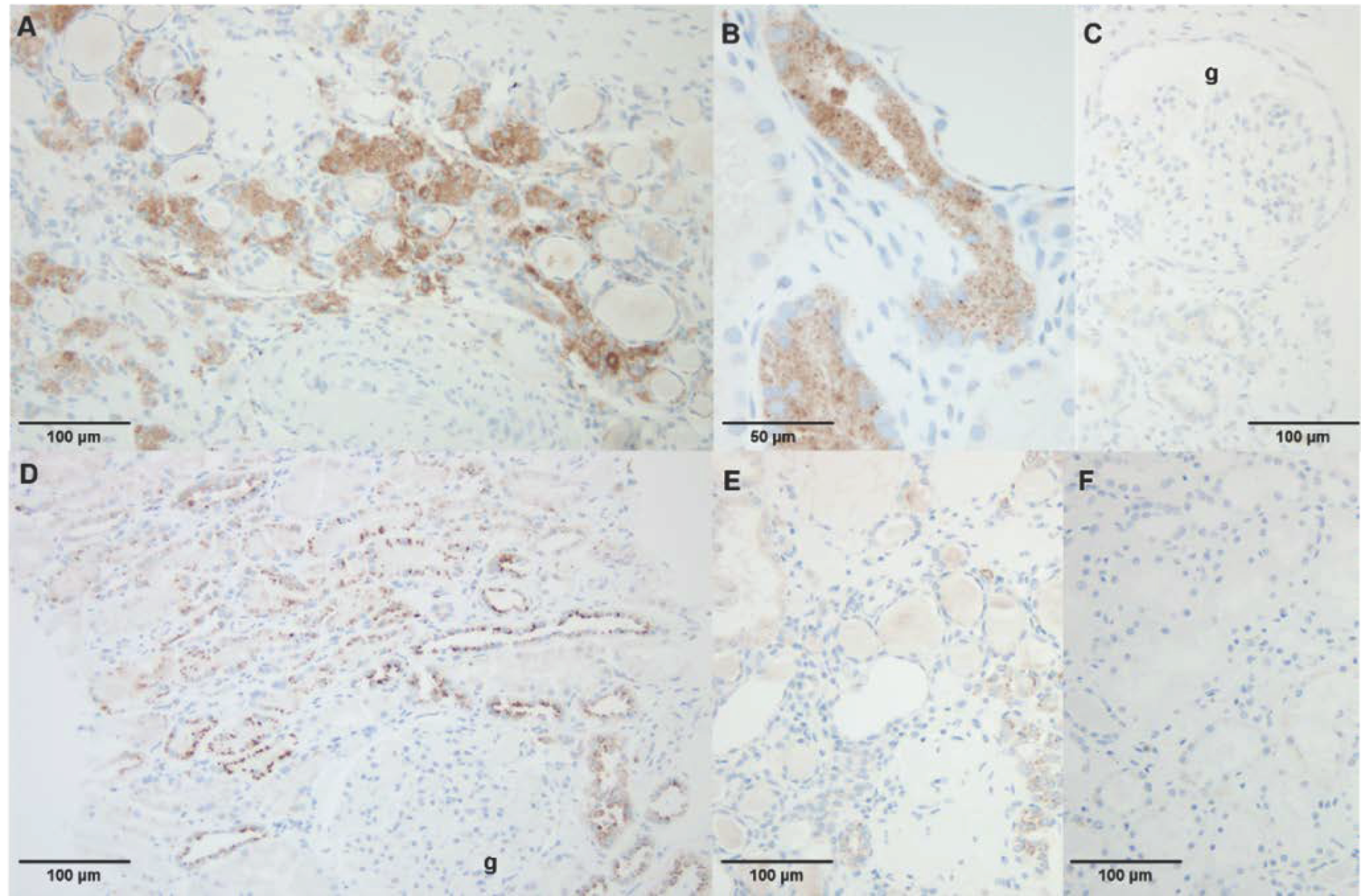
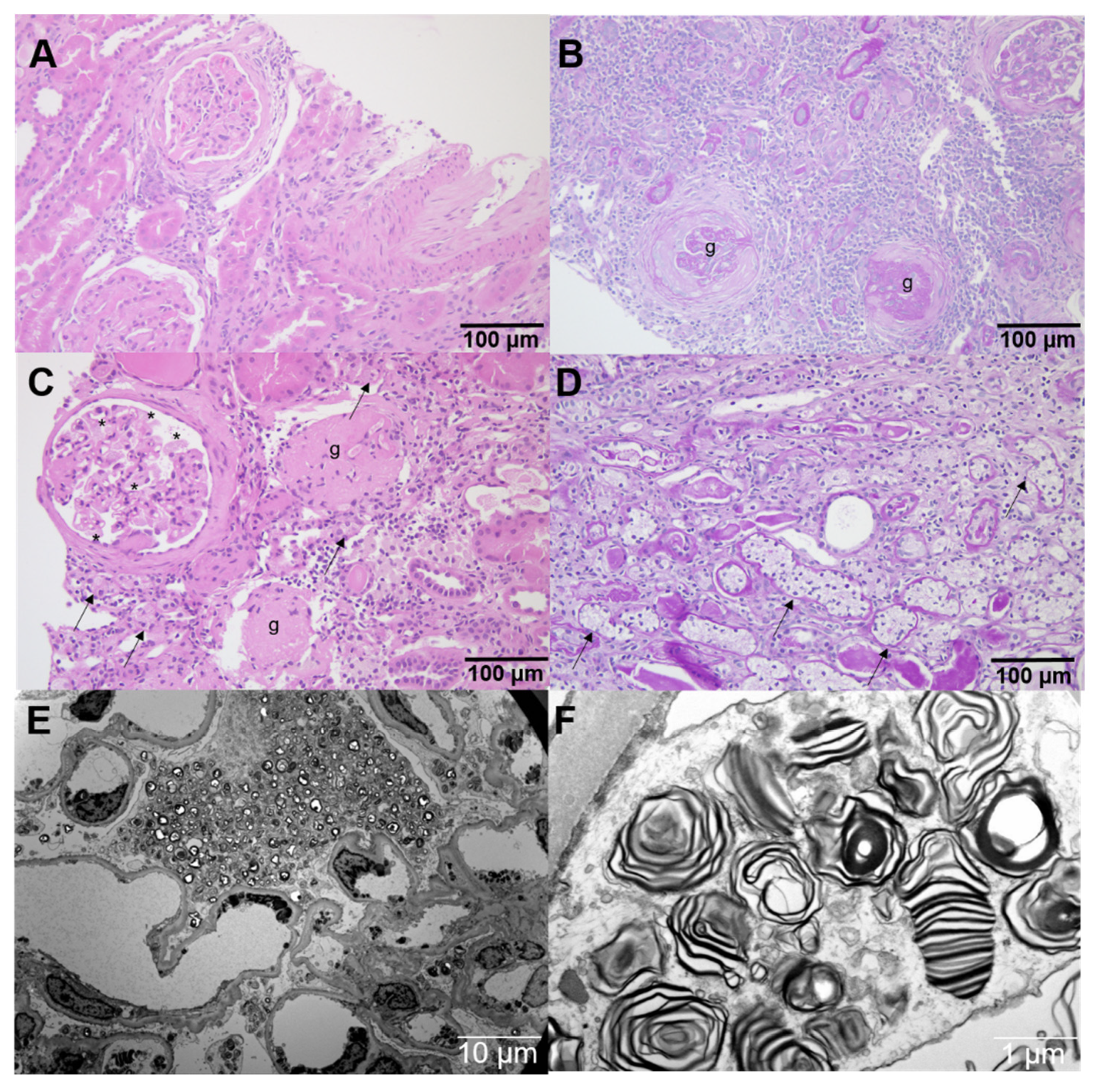
4.8. Immunohistochemistry in Kidney Biopsy Specimen
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A



References
- Zarate, Y.A.; Hopkin, R.J. Fabry’s disease. Lancet 2008, 372, 1427–1435. [Google Scholar] [CrossRef]
- Najafian, B.; Tøndel, C.; Svarstad, E.; Gubler, M.C.; Oliveira, J.P.; Mauer, M. Accumulation of globotriaosylceramide in podocytes in Fabry nephropathy is associated with progressive podocyte loss. J. Am. Soc. Nephrol. 2020, 31, 865–875. [Google Scholar] [CrossRef] [PubMed]
- Fall, B.; Scott, C.R.; Mauer, M.; Shankland, S.; Pippin, J.; Jefferson, J.A.; Wallace, E.; Warnock, D.; Najafian, B. Urinary podocyte loss is increased in patients with Fabry disease and correlates with clinical severity of Fabry nephropathy. PLoS ONE 2016, 11, e0168346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trimarchi, H.; Canzonieri, R.; Schiel, A.; Politei, J.; Stern, A.; Andrews, J.; Paulero, M.; Rengel, T.; Aráoz, A.; Forrester, M.; et al. Podocyturia is significantly elevated in untreated vs treated Fabry adult patients. J. Nephrol. 2016, 29, 791–797. [Google Scholar] [CrossRef]
- Trimarchi, H.; Canzonieri, R.; Schiel, A.; Costales-Collaguazo, C.; Politei, J.; Stern, A.; Paulero, M.; Rengel, T.; Andrews, J.; Forrester, M.; et al. Increased urinary CD80 excretion and podocyturia in Fabry disease. J. Transl. Med. 2016, 14, 289. [Google Scholar] [CrossRef] [Green Version]
- Slaats, G.G.; Braun, F.; Hoehne, M.; Frech, L.E.; Blomberg, L.; Benzing, T.; Schermer, B.; Rinschen, M.M.; Kurschat, C.E. Urine-derived cells: A promising diagnostic tool in Fabry disease patients. Sci. Rep. 2018, 8, 11042. [Google Scholar] [CrossRef]
- Sanchez-Niño, M.D.; Carpio, D.; Sanz, A.B.; Ruiz-Ortega, M.; Mezzano, S.; Ortiz, A. Lyso-Gb3 activates Notch1 in human podocytes. Hum. Mol. Genet. 2015, 24, 5720–5732. [Google Scholar] [CrossRef] [Green Version]
- Braun, F.; Blomberg, L.; Brodesser, S.; Liebau, M.C.; Schermer, B.; Benzing, T.; Kurschat, C.E. Enzyme replacement therapy clears Gb3 deposits from a podocyte cell culture model of Fabry disease but fails to restore altered cellular signaling. Cell Physiol. Biochem. 2019, 52, 1139–1150. [Google Scholar] [PubMed]
- Skrunes, R.; Tøndel, C.; Leh, S.; Larsen, K.K.; Houge, G.; Davidsen, E.S.; Hollak, C.; van Kuilenburg, A.B.P.; Vaz, F.M.; Svarstad, E. Long-term dose-dependent agalsidase effects on kidney histology in Fabry disease. Clin. J. Am. Soc. Nephrol. 2017, 2, 1470–1479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenders, M.; Stappers, F.; Niemietz, C.; Schmitz, B.; Boutin, M.; Ballmaier, P.J.; Zibert, A.; Schmidt, H.; Brand, S.M.; Auray-Blais, C.; et al. Mutation-specific Fabry disease patient-derived cell model to evaluate the amenability to chaperone therapy. J. Med. Genet. 2019, 56, 548–556. [Google Scholar] [CrossRef] [PubMed]
- Lenders, M.; Boutin, M.; Auray-Blais, C.; Brand, E. Effects of orally delivered alpha-Galactosidase A on gastrointestinal symptoms in patients with Fabry disease. Gastroenterology 2020, 159, 1602–1604. [Google Scholar] [CrossRef] [PubMed]
- Politei, J.; Alberton, V.; Amoreo, O.; Antongiovanni, N.; Arán, M.N.; Barán, M.; Cabrera, G.; Di Pietrantonio, S.; Durand, C.; Fainboim, A.; et al. Clinical parameters, LysoGb3, podocyturia, and kidney biopsy in children with Fabry disease: Is a correlation possible? Pediatr. Nephrol. 2018, 33, 2095–2101. [Google Scholar] [CrossRef] [PubMed]
- Liern, M.; Collazo, A.; Valencia, M.; Fainboin, A.; Isse, L.; Costales-Collaguazo, C.; Ochoa, F.; Vallejo, G.; Zotta, E. Podocyturia in paediatric patients with Fabry disease. Nefrologia 2019, 39, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Martineau, T.; Boutin, M.; Côté, A.M.; Maranda, B.; Bichet, D.G.; Auray-Blais, C. Tandem mass spectrometry analysis of urinary podocalyxin and podocin in the investigation of podocyturia in women with preeclampsia and Fabry disease patients. Clin. Chim. Acta 2019, 495, 67–75. [Google Scholar] [CrossRef]
- Schumann, A.; Schaller, K.; Belche, V.; Cybulla, M.; Grünert, S.C.; Moers, N.; Sass, J.O.; Kaech, A.; Hannibal, L.; Spiekerkoetter, U. Defective lysosomal storage in Fabry disease modifies mitochondrial structure, metabolism and turnover in renal epithelial cells. J. Inherit. Metab. Dis. 2021, 44, 1039–1050. [Google Scholar] [CrossRef]
- Rozenfeld, P.A.; de Los Angeles Bolla, M.; Quieto, P.; Pisani, A.; Feriozzi, S.; Neuman, P.; Bondar, C. Pathogenesis of Fabry nephropathy: The pathways leading to fibrosis. Mol. Genet. Metab. 2020, 129, 132–141. [Google Scholar] [CrossRef]
- Koehler, S.; Kuczkowski, A.; Kuehne, L.; Jüngst, C.; Hoehne, M.; Grahammer, F.; Eddy, S.; Kretzler, M.; Beck, B.B.; Höhfeld, J.; et al. Proteome analysis of isolated podocytes reveals stress responses in glomerular sclerosis. J. Am. Soc. Nephrol. 2020, 31, 544–559. [Google Scholar] [CrossRef]
- Imasawa, T.; Rossignol, R. Podocyte energy metabolism and glomerular diseases. Int. J. Biochem. Cell Biol. 2013, 45, 2109–2118. [Google Scholar] [CrossRef]
- Saoud, R.; Jaffa, M.A.; Habib, A.; Zhao, J.; Al Hariri, M.; Zhu, R.; Hasan, A.; Ziyadeh, F.N.; Kobeissy, F.; Mechref, Y.; et al. Modulation of proteomic and inflammatory signals by Bradykinin in podocytes. J. Adv. Res. 2020, 24, 409–422. [Google Scholar] [CrossRef]
- Bird, S.; Hadjimichael, E.; Mehta, A.; Ramaswami, U.; Hughes, D. Fabry disease and incidence of cancer. Orphanet J. Rare Dis. 2017, 12, 150. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Shen, W.; Peng, H.; Li, Y.; Chen, F.; Zheng, L.; Xu, J.; Jia, L. Fibronectin 1 promotes melanoma proliferation and metastasis by inhibiting apoptosis and regulating EMT. Onco Targets Ther. 2019, 12, 3207–3221. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Ou, D.H.; Zhuang, D.W.; Zheng, Z.F.; Lin, M.E. In silico analysis of the immune microenvironment in bladder cancer. BMC Cancer 2020, 20, 265. [Google Scholar] [CrossRef] [PubMed]
- Sciacovelli, M.; Gonçalves, E.; Johnson, T.I.; Zecchini, V.R.; da Costa, A.S.; Gaude, E.; Drubbel, A.V.; Theobald, S.J.; Abbo, S.R.; Tran, M.G.; et al. Fumarate is an epigenetic modifier that elicits epithelial-to-mesenchymal transition. Nature 2016, 537, 544–547, Erratum in: Nature 2016, 540, 150. [Google Scholar] [CrossRef] [PubMed]
- George, B.; Verma, R.; Soofi, A.A.; Garg, P.; Zhang, J.; Park, T.J.; Giardino, L.; Ryzhova, L.; Johnstone, D.B.; Wong, H.; et al. Crk1/2-dependent signaling is necessary for podocyte foot process spreading in mouse models of glomerular disease. J. Clin. Investig. 2012, 122, 674–692. [Google Scholar] [CrossRef] [Green Version]
- George, B.; Fan, Q.; Dlugos, C.P.; Soofi, A.A.; Zhang, J.; Verma, R.; Park, T.J.; Wong, H.; Curran, T.; Nihalani, D.; et al. Crk1/2 and CrkL form a hetero-oligomer and functionally complement each other during podocyte morphogenesis. Kidney Int. 2014, 85, 1382–1394. [Google Scholar] [CrossRef] [Green Version]
- Möller-Kerutt, A.; Rodriguez-Gatica, J.E.; Wacker, K.; Bhatia, R.; Siebrasse, J.P.; Boon, N.; Van Marck, V.; Boor, P.; Kubitscheck, U.; Wijnholds, J.; et al. Crumbs2 is an essential slit diaphragm protein of the renal filtration barrier. J. Am. Soc. Nephrol. 2021, 32, 1053–1070. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [Green Version]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [Green Version]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene set knowledge discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [Green Version]
- Schulze, U.; Vollenbröker, B.; Braun, D.A.; Van Le, T.; Granado, D.; Kremerskothen, J.; Fränzel, B.; Klosowski, R.; Barth, J.; Fufezan, C.; et al. The Vac14-interaction network is linked to regulators of the endolysosomal and autophagic pathway. Mol. Cell. Proteom. 2014, 13, 1397–1411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desnick, R.J.; Allen, K.Y.; Desnick, S.J. Fabry’s disease: Enzymatic diagnosis of hemizygotes and heterozygotes: Alpha-galactosidase activities in plasma, serum, urine, and leukocytes. J. Lab. Clin. Med. 1973, 81, 157–171. [Google Scholar] [PubMed]
- Mayes, J.S.; Scheerer, J.B.; Sifers, R.N.; Donaldson, M.L. Differential assay for lysosomal alpha-galactosidases in human tissues and its application to Fabry’s disease. Clin. Chim. Acta 1981, 112, 247–251. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jehn, U.; Bayraktar, S.; Pollmann, S.; Van Marck, V.; Weide, T.; Pavenstädt, H.; Brand, E.; Lenders, M. α-Galactosidase a Deficiency in Fabry Disease Leads to Extensive Dysregulated Cellular Signaling Pathways in Human Podocytes. Int. J. Mol. Sci. 2021, 22, 11339. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222111339
Jehn U, Bayraktar S, Pollmann S, Van Marck V, Weide T, Pavenstädt H, Brand E, Lenders M. α-Galactosidase a Deficiency in Fabry Disease Leads to Extensive Dysregulated Cellular Signaling Pathways in Human Podocytes. International Journal of Molecular Sciences. 2021; 22(21):11339. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222111339
Chicago/Turabian StyleJehn, Ulrich, Samet Bayraktar, Solvey Pollmann, Veerle Van Marck, Thomas Weide, Hermann Pavenstädt, Eva Brand, and Malte Lenders. 2021. "α-Galactosidase a Deficiency in Fabry Disease Leads to Extensive Dysregulated Cellular Signaling Pathways in Human Podocytes" International Journal of Molecular Sciences 22, no. 21: 11339. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222111339