
Species Phylogeny versus Gene Trees: A Case Study of an Incongruent Data Matrix Based on Paphiopedilum Pfitz. (Orchidaceae)

 , , and
, , and
Abstract
:1. Introduction
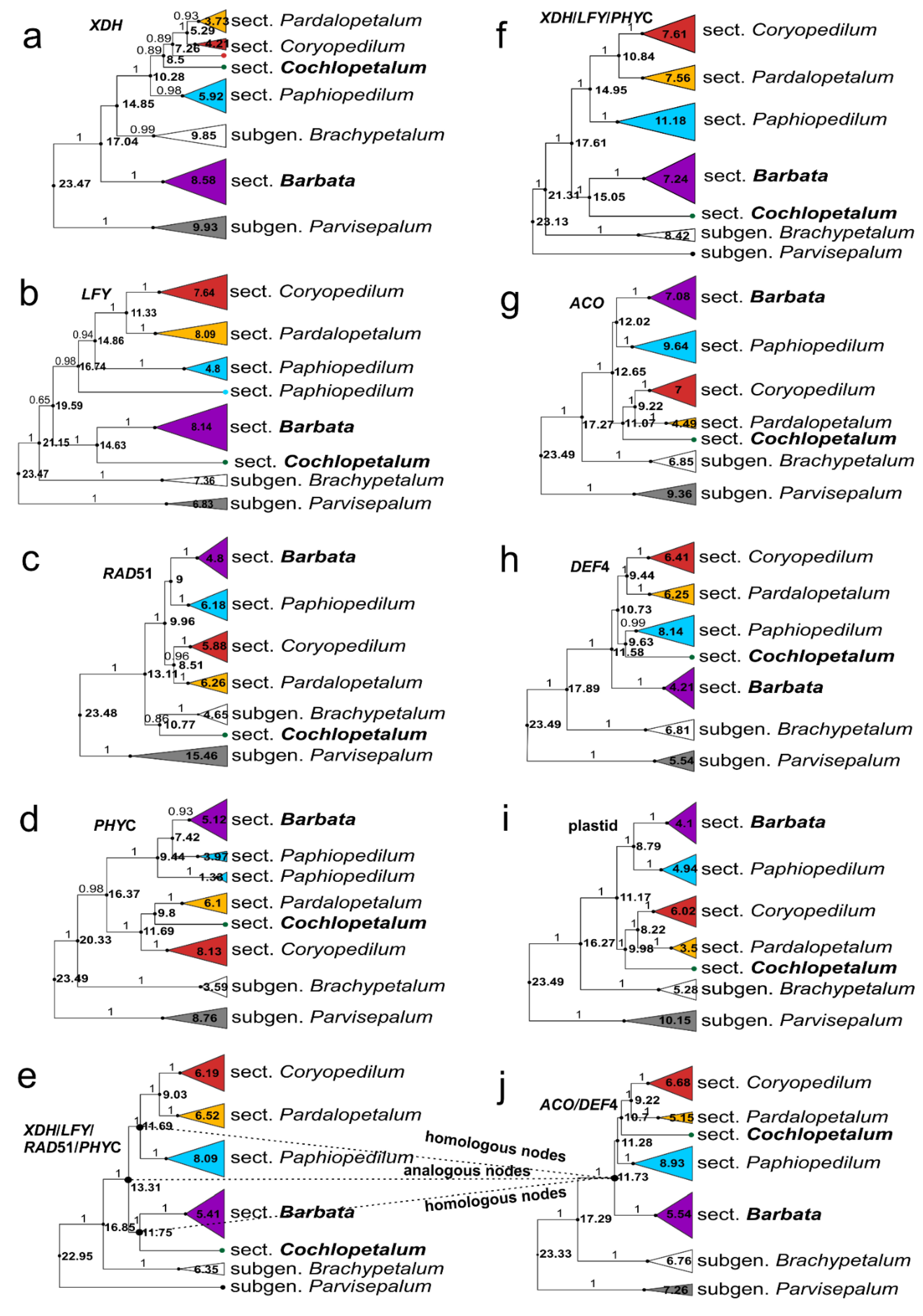
2. Results
2.1. Molecular Clock Phylogeny
2.2. Molecular Phylogenetics and Network Analysis
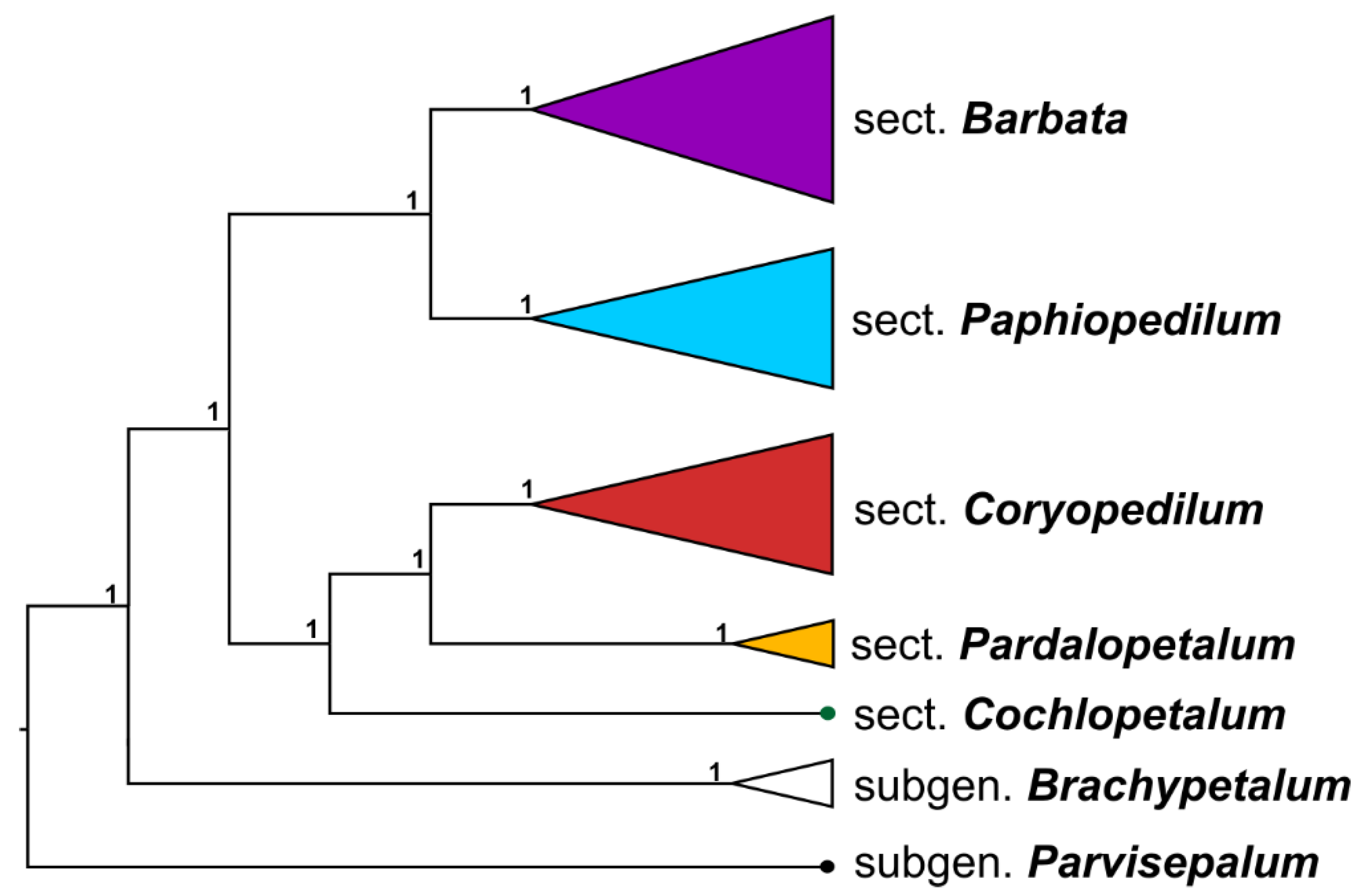
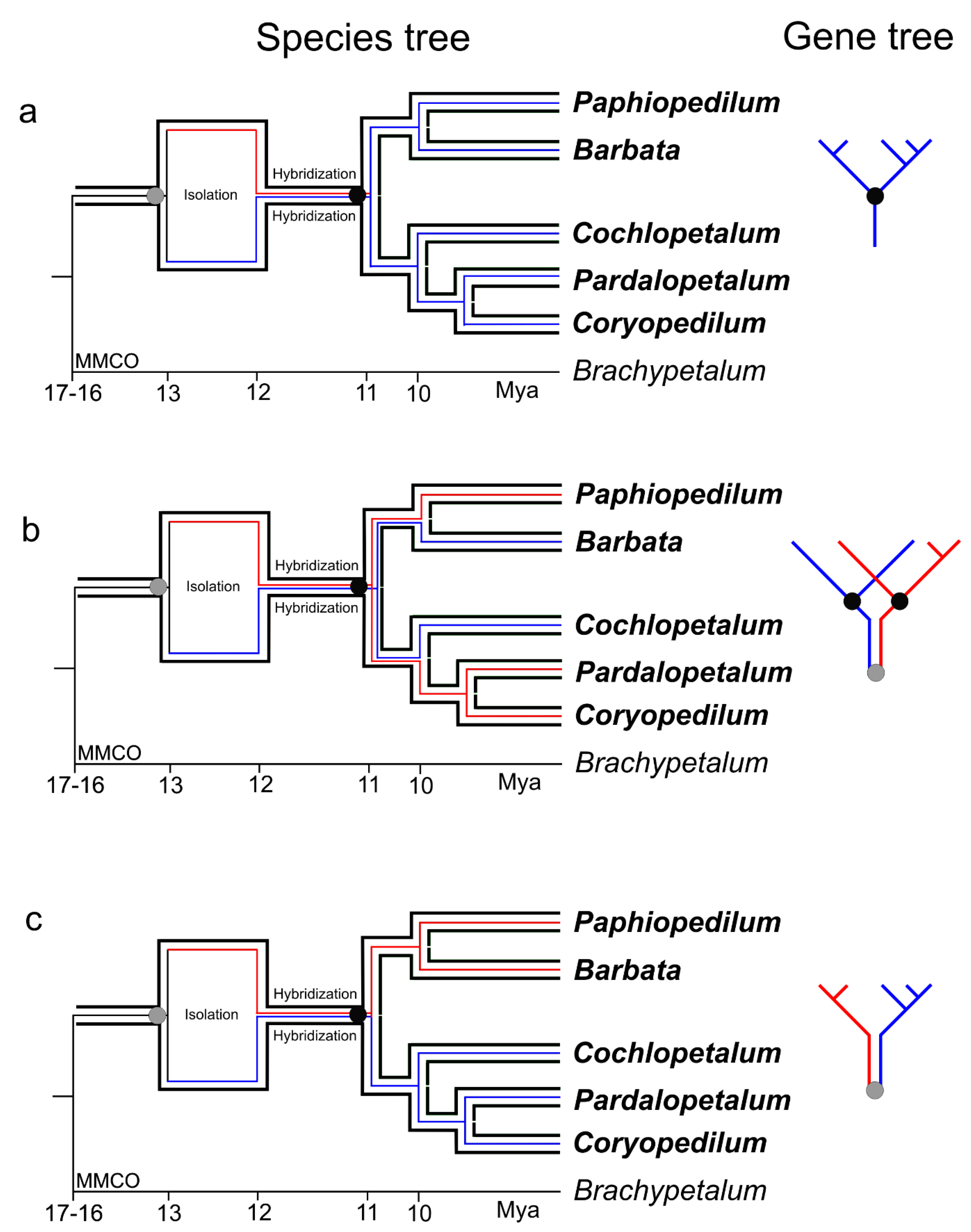
3. Discussion
3.1. Molecular Dating
3.2. Geological: Climate Changes through Miocene
3.3. Post-Hybridization Scenario
3.4. Reconstruction of the Phylogeny
4. Materials and Methods
4.1. DNA Isolation, Amplification, and Sequencing
4.2. Molecular Phylogenetic and Time Divergence Analysis
4.3. Phylogenetic Network Analysis
4.4. Using Nuclear Genes for Inferring Phylogeny
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rieseberg, L.H.; Ellstrand, N.C.; Arnold, M. What Can Molecular and Morphological Markers Tell Us About Plant Hybridization? Crit. Rev. Plant Sci. 1993, 12, 213–241. [Google Scholar] [CrossRef]
- Maddison, W.P. Gene Trees in Species Trees. Syst. Biol. 1997, 46, 523–536. [Google Scholar] [CrossRef]
- Frajman, B.; Eggens, F.; Oxelman, B. Hybrid Origins and Homoploid Reticulate Evolution within Heliosperma (Sileneae, Caryophyllaceae)—A Multigene Phylogenetic Approach with Relative Dating. Syst. Biol. 2009, 58, 328–345. [Google Scholar] [CrossRef] [Green Version]
- Maureira-Butler, I.J.; Pfeil, B.E.; Muangprom, A.; Osborn, T.C.; Doyle, J.J. The Reticulate History of Medicago (Fabaceae). Syst. Biol. 2008, 57, 466–482. [Google Scholar] [CrossRef] [Green Version]
- Joly, S.; McLenachan, P.A.; Lockhart, P.J. A Statistical Approach for Distinguishing Hybridization and Incomplete Lineage Sorting. Am. Nat. 2009, 174, E54–E70. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Than, C.; Degnan, J.H.; Nakhleh, L. Coalescent Histories on Phylogenetic Networks and Detection of Hybridization Despite Incomplete Lineage Sorting. Syst. Biol. 2011, 60, 138–149. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Degnan, J.H.; Nakhleh, L. The Probability of a Gene Tree Topology within a Phylogenetic Network with Applications to Hybridization Detection. PLoS Genet. 2012, 8, e1002660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cribb, P.J. The Genus Paphiopedilum, 2nd ed.; Natural History Publications (Borneo) in association with Royal Botanic Gardens; Kew: Kota Kinabalu, Malaysia, 1998. [Google Scholar]
- Brieger, F.G.; Maatsch, R.; Senghas, R. Unterfamilie: Cypripedioideae in Die Orchideen; Schlechter, R., Ed.; Paul Parey: Berlin, Germany, 1971; pp. 161–185. [Google Scholar]
- Karasawa, K.; Saito, K. A revision of the genus Paphiopedilum (Orchidaceae). Bull. Hiroshima Bot. Gard. 1982, 5, 1–69. [Google Scholar]
- Hennessy, E.F.; Cribb, P.; Mathew, B. The Genus Paphiopedilum. Kew Bull. 2000, 55, 249. [Google Scholar] [CrossRef]
- Braem, G.J.; Baker, C.O.; Baker, M.L. The Genus Paphiopedilum—Natural History and Cultivation; Botanical Publishers: Kissimmee, FL, USA, 1998; Volume 2. [Google Scholar]
- Braem, G.J.; Chiron, G. Paphiopedilum; Tropicalia: Lyon, France, 2003. [Google Scholar]
- Chochai, A.; Leitch, I.J.; Ingrouille, M.J.; Fay, M.F. Molecular phylogenetics of Paphiopedilum (Cypripedioideae; Orchidaceae) based on nuclear ribosomal ITS and plastid sequences. Bot. J. Linn. Soc. 2012, 170, 176–196. [Google Scholar] [CrossRef] [Green Version]
- Górniak, M.; Szlachetko, D.L.; Kowalkowska, A.K.; Bohdanowicz, J.; Canh, C.X. Taxonomic placement of Paphiopedilum canhii (Cypripedioideae; Orchidaceae) based on cytological, molecular and micromorphological evidence. Mol. Phylogenet. Evol. 2014, 70, 429–441. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.-Y.; Luo, Y.-B.; Liu, Z.-J.; Wang, X.-Q. Reticulate evolution and sea-level fluctuations together drove species diversification of slipper orchids (Paphiopedilum) in South-East Asia. Mol. Ecol. 2015, 24, 2838–2855. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.-Y.; Yang, J.-X.; Bai, M.-Z.; Zhang, G.-Q.; Liu, Z.-J. The chloroplast genome evolution of Venus slipper (Paphiopedilum): IR expansion, SSC contraction, and highly rearranged SSC regions. BMC Plant Biol. 2021, 21, 1–14. [Google Scholar] [CrossRef]
- Hall, R. The palaeogeography of Sundaland and Wallacea since the Late Jurassic. J. Limnol. 2013, 72, e1. [Google Scholar] [CrossRef]
- Morley, R.J. Palynological evidence for Tertiary plant dispersals in the SE Asian region in relation to plate tectonics and climate. In Biogeography and Geological Evolution of SE Asia; Hall, R., Holloway, D.J., Eds.; Backhuys: Leiden, The Netherlands, 1990; pp. 211–234. [Google Scholar]
- Woodruff, D.S. Neogene marine transgressions, palaeogeography and biogeographic transitions on the Thai-Malay Peninsula. J. Biogeogr. 2003, 30, 551–567. [Google Scholar] [CrossRef] [Green Version]
- Cannon, C.H.; Manos, P.S. Phylogeography of the Southeast Asian stone oaks (Lithocarpus). J. Biogeogr. 2003, 30, 211–226. [Google Scholar] [CrossRef]
- Su, Y.C.; Saunders, R.M. Evolutionary divergence times in the Annonaceae: Evidence of a late Miocene origin of Pseuduvaria in Sundaland with subsequent diversification in New Guinea. BMC Evol. Biol. 2009, 9, 153. [Google Scholar] [CrossRef] [Green Version]
- Thomas, D.C.; Hughes, M.; Phutthai, T.; Ardi, W.H.; Rajbhandary, S.; Rubite, R.; Twyford, A.D.; Richardson, J.E. West to east dispersal and subsequent rapid diversification of the mega-diverse genus Begonia (Begoniaceae) in the Malesian archipelago. J. Biogeogr. 2011, 39, 98–113. [Google Scholar] [CrossRef]
- Nauheimer, L.; Boyce, P.C.; Renner, S.S. Giant taro and its relatives: A phylogeny of the large genus Alocasia (Araceae) sheds light on Miocene floristic exchange in the Malesian region. Mol. Phylogenetics Evol. 2012, 63, 43–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Bruyn, M.; Stelbrink, B.; Morley, R.J.; Hall, R.; Carvalho, G.R.; Cannon, C.; Bergh, G.D.V.D.; Meijaard, E.; Metcalfe, I.; Boitani, L.; et al. Borneo and Indochina are Major Evolutionary Hotspots for Southeast Asian Biodiversity. Syst. Biol. 2014, 63, 879–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zachos, J. Trends, Rhythms, and Aberrations in Global Climate 65 Ma to Present. Science 2001, 292, 686–693. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Wang, Q.; An, Z.; Qiang, X.; Dong, J.; Chang, H.; Zhang, M.; Guo, X. Mid-Miocene climatic optimum: Clay mineral evidence from the red clay succession, Longzhong Basin, Northern China. Palaeogeogr. Palaeoclim. Palaeoecol. 2017, 512, 46–55. [Google Scholar] [CrossRef]
- Tsai, C.-C.; Liao, P.-C.; Ko, Y.-Z.; Chen, C.-H.; Chiang, Y.-C. Phylogeny and Historical Biogeography of Paphiopedilum Pfitzer (Orchidaceae) Based on Nuclear and Plastid DNA. Front. Plant Sci. 2020, 11, 126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, T.; Albert, V.A. Dynamic distribution patterns of ribosomal DNA and chromosomal evolution in Paphiopedilum, a lady’s slipper orchid. BMC Plant Biol. 2011, 11, 126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müntzing, A. Outlines to a genetic monograph of the genus galeopsis. Hereditas 1930, 13, 185–341. [Google Scholar] [CrossRef]
- Stebbins, G.L. The hybrid origin of microspecies in the Elymus glaucus complex. Cytologia 1957, 36, 336–340. [Google Scholar]
- Grant, V. The Regulation of Recombination in Plants. Cold Spring Harb. Symp. Quant. Biol. 1958, 23, 337–363. [Google Scholar] [CrossRef]
- Buerkle, A.; Morris, R.J.; Asmussen, M.A.; Rieseberg, L. The likelihood of homoploid hybrid speciation. Heredity 2000, 84, 441–451. [Google Scholar] [CrossRef]
- Folk, R.A.; Soltis, P.S.; Soltis, D.E.; Guralnick, R. New prospects in the detection and comparative analysis of hybridization in the tree of life. Am. J. Bot. 2018, 105, 364–375. [Google Scholar] [CrossRef] [Green Version]
- Szlachetko, D.L.; Kolanowska, M.; Muller, F.; Vannini, J.; Rojek, J.; Górniak, M. First Guatemalan record of natural hybridisation between Neotropical species of the Lady’s Slipper orchid (Orchidaceae, Cypripedioideae). PeerJ 2017, 5, e4162. [Google Scholar] [CrossRef]
- Pelser, P.B.; Kennedy, A.H.; Tepe, E.; Shidler, J.B.; Nordenstam, B.; Kadereit, J.W.; Watson, L.E. Patterns and causes of incongruence between plastid and nuclear Senecioneae (Asteraceae) phylogenies. Am. J. Bot. 2010, 97, 856–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chase, M.W.; Hills, H.H. Silica gel: An ideal material for field preservation of leaf samples for DNA studies. TAXON 1991, 40, 215–220. [Google Scholar] [CrossRef]
- Casadaban, M.J.; Cohen, S.N. Analysis of gene control signals by DNA fusion and cloning in Escherichia coli. J. Mol. Biol. 1980, 138, 179–207. [Google Scholar] [CrossRef]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.-Y.; Luo, Y.-B.; Liu, Z.-J.; Wang, X.-Q. Evolution and Biogeography of the Slipper Orchids: Eocene Vicariance of the Conduplicate Genera in the Old and New World Tropics. PLoS ONE 2012, 7, e38788. [Google Scholar] [CrossRef] [Green Version]
- Ramírez, S.R.; Gravendeel, B.; Singer, R.B.; Marshall, C.R.; Pierce, N.E. Dating the origin of the Orchidaceae from a fossil orchid with its pollinator. Nature 2007, 448, 1042–1045. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Gouy, M.; Guindon, S.; Gascuel, O. SeaView Version 4: A Multiplatform Graphical User Interface for Sequence Alignment and Phylogenetic Tree Building. Mol. Biol. Evol. 2009, 27, 221–224. [Google Scholar] [CrossRef] [Green Version]
- Gernhard, T. The conditioned reconstructed process. J. Theor. Biol. 2008, 253, 769–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nylander, J.A.A. MrModeltest v2. Program Distributed by the Author; Evolutionary Biology Centre, Uppsala University: Uppsala, Sweden, 2004. [Google Scholar]
- Rambaut, A.; Drummond, A.J. Tracer v1.6. 2014. Available online: http://beast.bio.ed.ac.uk/Tracer (accessed on 8 September 2021).
- Kass, R.E.; Raftery, A.E. Bayes Factors. J. Am. Stat. Assoc. 1995, 90, 773–795. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Hoehna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian Phylogenetic Inference and Model Choice Across a Large Model Space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Than, C.; Ruths, D.A.; Nakhleh, L. PhyloNet: A software package for analyzing and reconstructing reticulate evolutionary relationships. BMC Bioinform. 2008, 9, 322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, D.; Yu, Y.; Zhu, J.; Nakhleh, L. Inferring Phylogenetic Networks Using PhyloNet. Syst. Biol. 2018, 67, 735–740. [Google Scholar] [CrossRef]
- Wen, D.; Yu, Y.; Nakhleh, L. Correction: Bayesian Inference of Reticulate Phylogenies under the Multispecies Network Coalescent. PLoS Genet. 2017, 13, e1006598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huson, D.H.; Scornavacca, C. Dendroscope 3: An Interactive Tool for Rooted Phylogenetic Trees and Networks. Syst. Biol. 2012, 61, 1061–1067. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Matrix | Model Comparison | Log Marginal Likelihood | Bayes Factor (2 BF) | Evidence against H0 |
|---|---|---|---|---|
| ACO | Relaxed/B and D # vs. Strict/B and D * | −4499.32 vs. −4496.78 | 5.08 | Weak |
| DEF4 | Relaxed/B and D # vs. Strict/B and D * | −3701.28 vs. −3700.92 | 0.72 | Inconclusive |
| PHYC | Relaxed/B and D # vs. Strict/B and D * | −1665.58 vs. −1662.93 | 5.3 | Weak |
| XDH | Relaxed/B and D # vs. Strict/B and D * | −2143.99 vs. −2143.49 | 1.0 | Inconclusive |
| LFY | Strict/B and D # vs. Relaxed/B and D * | −12,001.96 vs. −11,972.86 | 58.2 | Very strong |
| RAD51 | Relaxed/B and D # vs. Strict/B and D * | −2908.64 vs. −2906.61 | 4.06 | Weak |
| plastid | Strict/B and D # vs. Relaxed/B and D * | −15,868.13 vs. −15,848.6 | 39.06 | Very strong |
| Matrix | ACO | DEF4 | ACO/DEF4 | PHYC | XDH | LFY | RAD51 | XDH/LFY/ RAD51/ PHYC | Plastid | Nuclear Combined Referring to Table 1 |
|---|---|---|---|---|---|---|---|---|---|---|
| No. of taxa | 22 | 23 | 21 | 23 | 23 | 21 | 23 | 20 | 23 | 19 |
| Included positions in matrix | 1393 | 1241 | 2634 | 799 | 909 | 3368 | 892 | 5948 | 8731 | 13,117 |
| Variable site | 296 | 246 | 518 | 55 | 101 | 862 | 193 | 1154 | 442 | 2093 |
| Parsimony-uninformative sites | 163 | 128 | 281 | 21 | 63 | 377 | 113 | 581 | 182 | 1270 |
| Parsimony-informative sites | 133 | 118 | 237 | 34 | 38 | 485 | 80 | 573 | 260 | 823 |
| Consistency index (CI) | 0.9 | 0.95 | 0.91 | 0.9 | 0.95 | 0.8 | 0.88 | 0.8 | 0.88 | 0.9 |
| Retention index (RI) | 0.88 | 0.95 | 0.89 | 0.94 | 0.96 | 0.79 | 0.86 | 0.79 | 0.91 | 0.82 |
| Taxa/Markers | ACO | DEF4 | PHYC | XDH_1 | XDH_2 | LFY_1 | LFY_2 | RAD51_1 | RAD51_2 |
|---|---|---|---|---|---|---|---|---|---|
| Coryopedilum | |||||||||
| Pardalopetalum | |||||||||
| Cochlopetalum | |||||||||
| Paphiopedilum | |||||||||
| Barbata | |||||||||
| Brachypetalum | |||||||||
| Parvisepalum |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Górniak, M.; Szlachetko, D.L.; Olędrzyńska, N.; Naczk, A.M.; Mieszkowska, A.; Boss, L.; Ziętara, M.S. Species Phylogeny versus Gene Trees: A Case Study of an Incongruent Data Matrix Based on Paphiopedilum Pfitz. (Orchidaceae). Int. J. Mol. Sci. 2021, 22, 11393. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222111393
Górniak M, Szlachetko DL, Olędrzyńska N, Naczk AM, Mieszkowska A, Boss L, Ziętara MS. Species Phylogeny versus Gene Trees: A Case Study of an Incongruent Data Matrix Based on Paphiopedilum Pfitz. (Orchidaceae). International Journal of Molecular Sciences. 2021; 22(21):11393. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222111393
Chicago/Turabian StyleGórniak, Marcin, Dariusz L. Szlachetko, Natalia Olędrzyńska, Aleksandra M. Naczk, Agata Mieszkowska, Lidia Boss, and Marek S. Ziętara. 2021. "Species Phylogeny versus Gene Trees: A Case Study of an Incongruent Data Matrix Based on Paphiopedilum Pfitz. (Orchidaceae)" International Journal of Molecular Sciences 22, no. 21: 11393. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222111393