
Isoform-Specific Effects of Apolipoprotein E on Hydrogen Peroxide-Induced Apoptosis in Human Induced Pluripotent Stem Cell (iPSC)-Derived Cortical Neurons


Abstract
:1. Introduction
2. Results
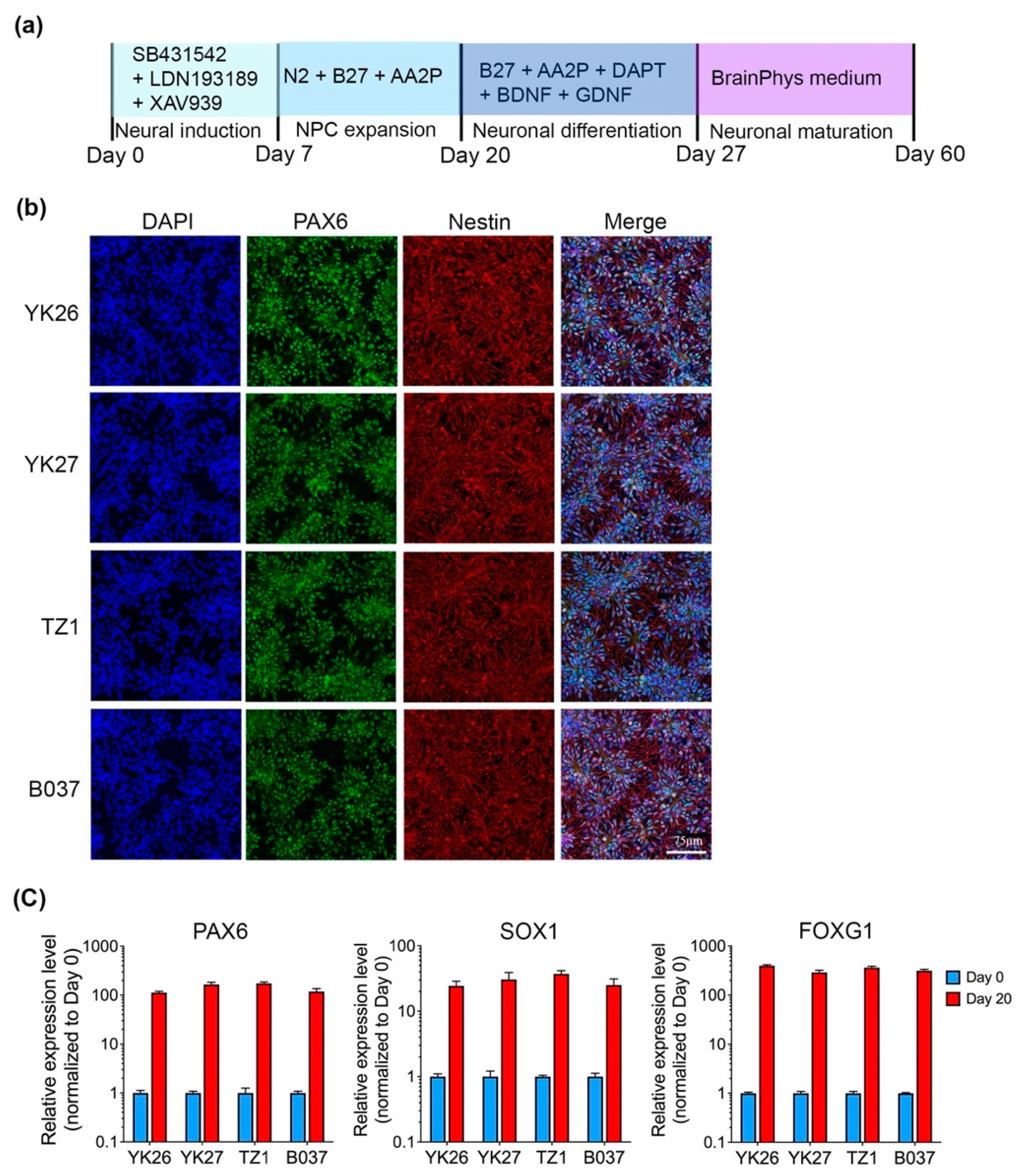
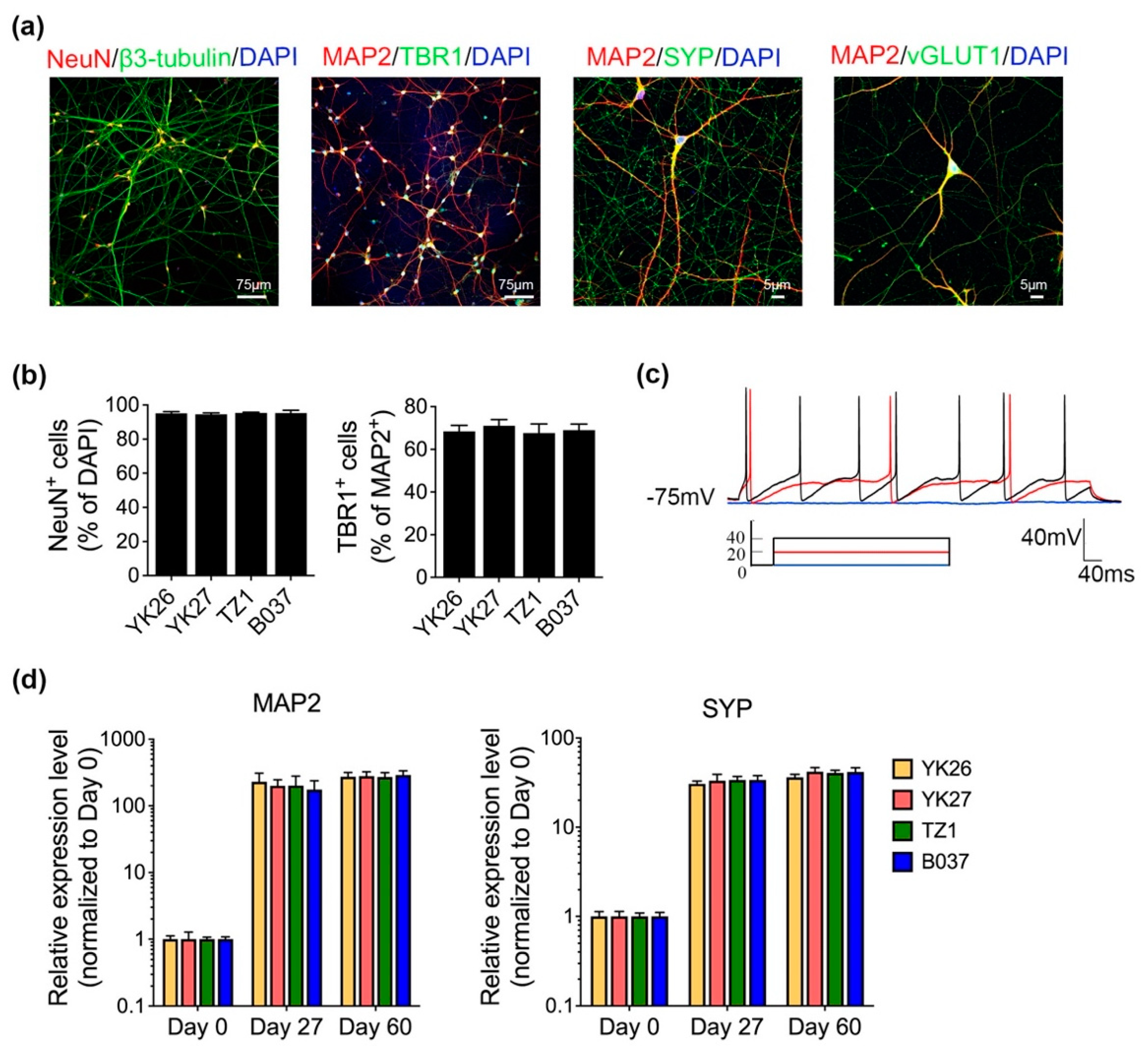
2.1. Generation and Characterization of Human iPSC-Derived Cortical Neurons
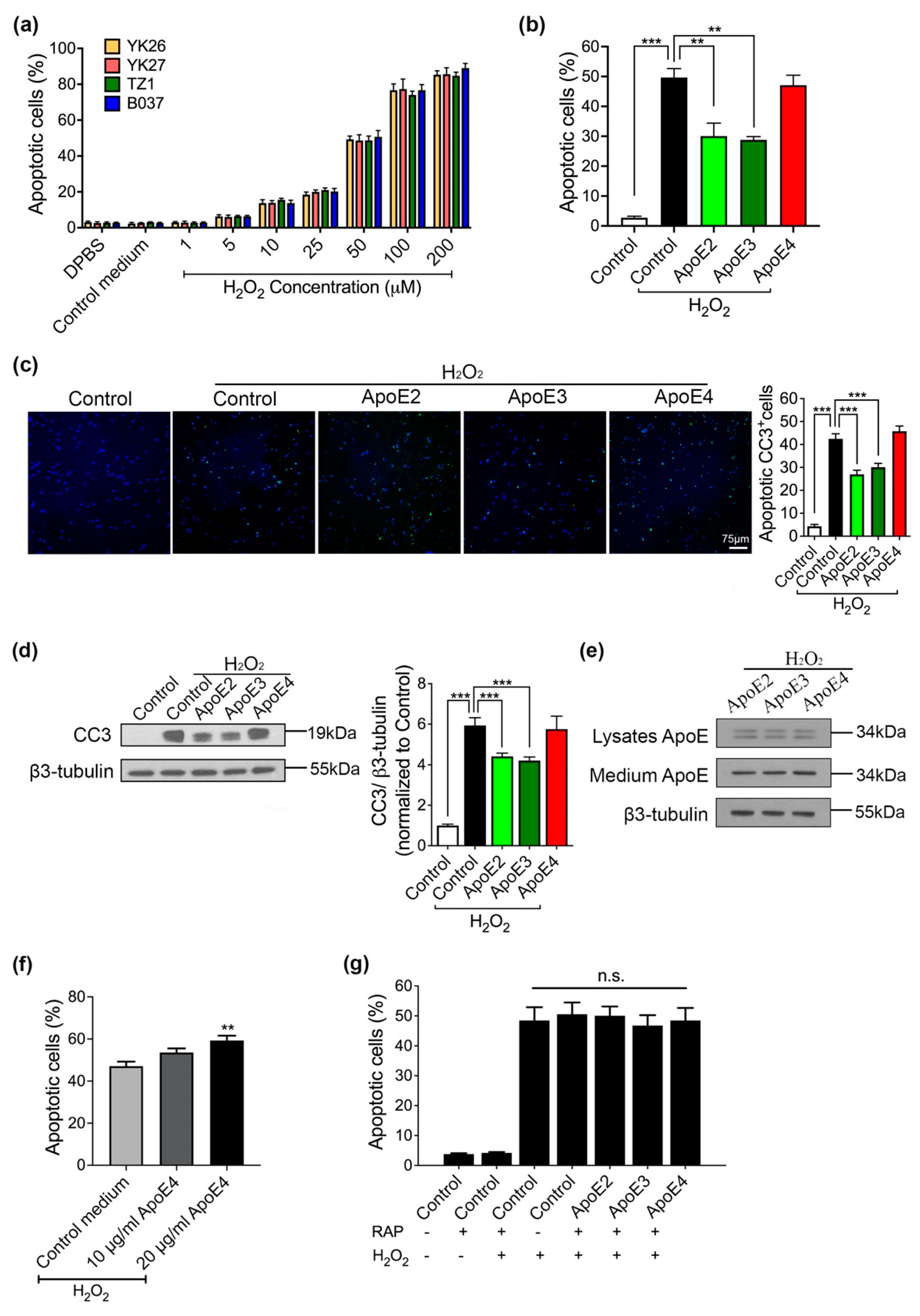
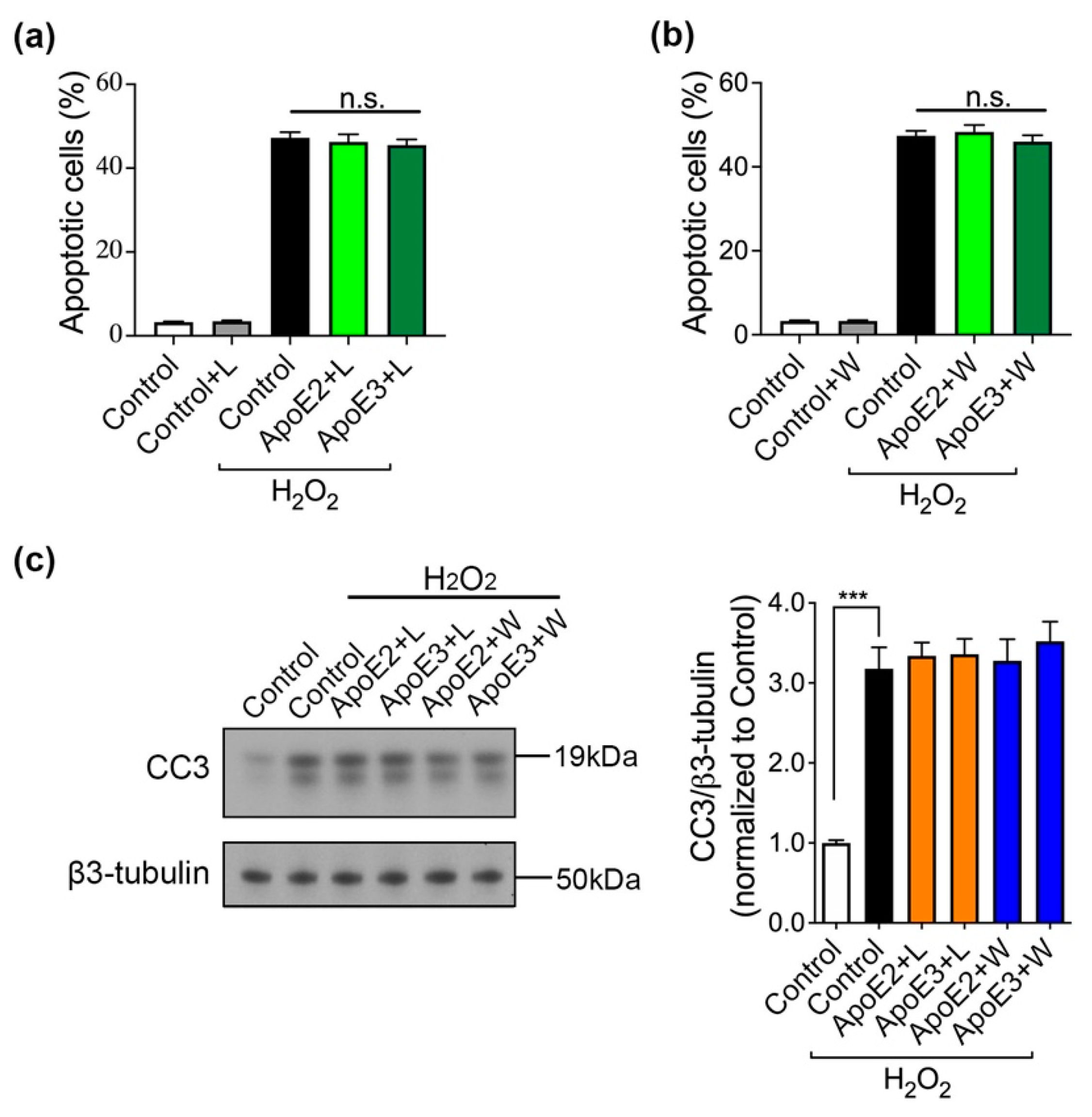
2.2. ApoE2 and ApoE3 Prevent Neurons from H2O2-Induced Apoptosis via Binding to ApoE Receptor(s)
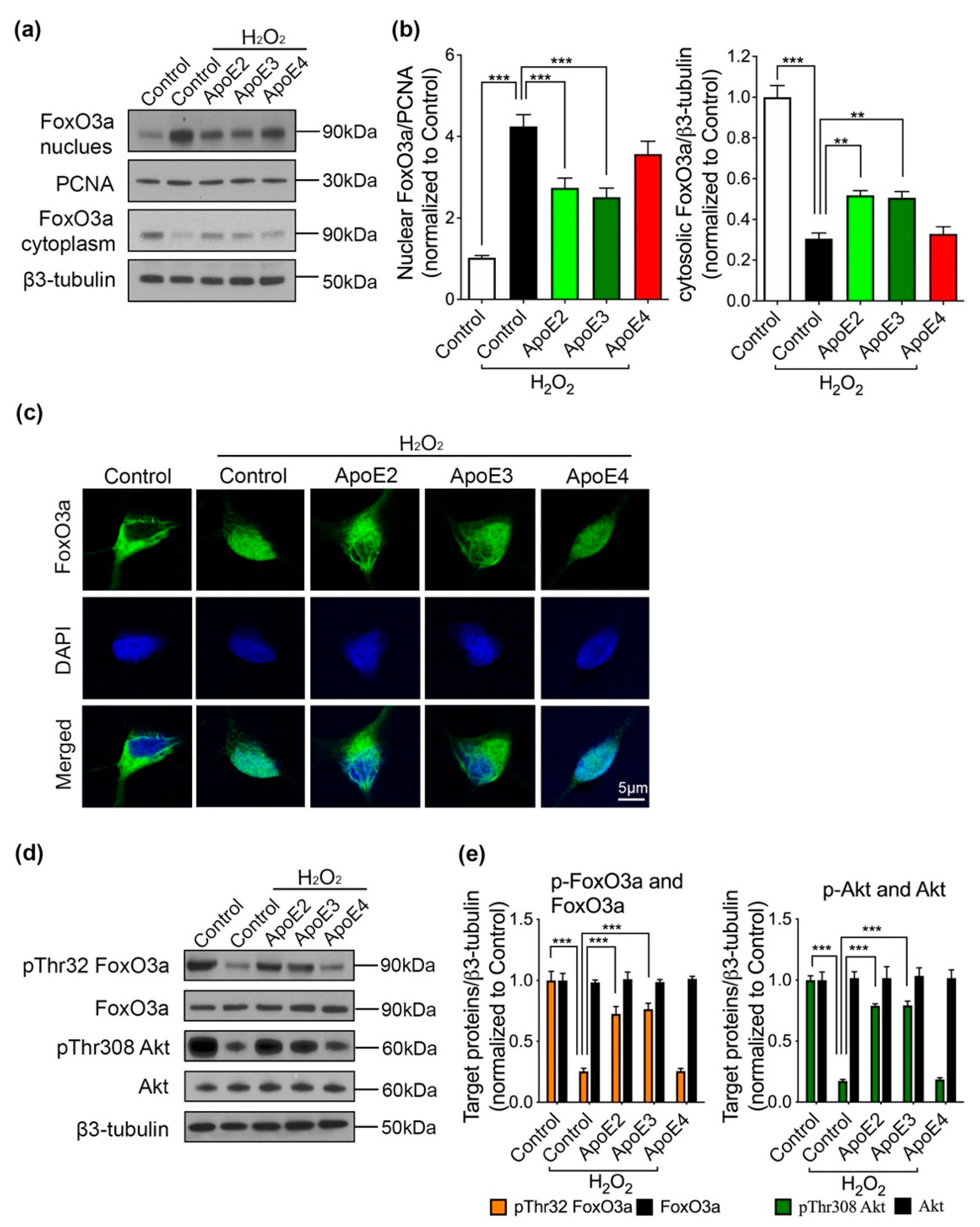
2.3. ApoE2 and ApoE3 Regulate Akt/FoxO3a Signaling Pathway in the Presence of H2O2
2.4. PI3K Mediates ApoE2 and ApoE3 Neuroprotection against Apoptosis
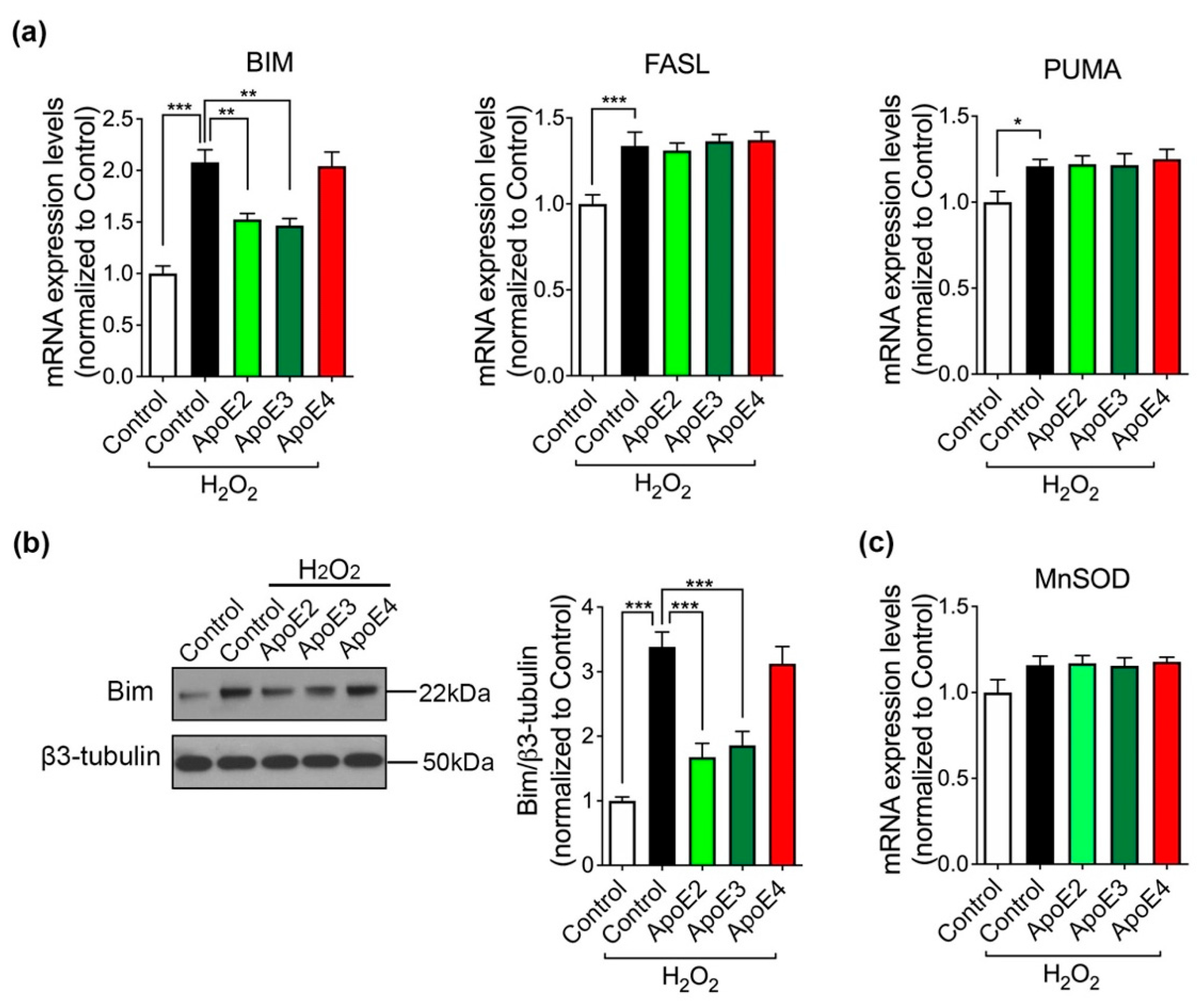
2.5. Bim, A Downstream Pro-Apoptotic Effector of FoxO3a, Is Regulated by ApoE2 and ApoE3
3. Discussion
4. Materials and Methods
4.1. Apolipoprotein E Preparation
4.2. Antibodies
4.3. iPSC Culture and In Vitro Differentiation of Embryoid Bodies (EBs)
4.4. Generation of Cortical Neurons from Human Induced Pluripotent Stem Cells (iPSCs)
4.5. Electrophysiology
4.6. Cell Apoptosis Measurement
4.7. Immunofluorescence (IF)
4.8. Quantitative Real Time PCR
4.9. Western Blot Analysis
4.10. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Corder, E.H.; Saunders, A.M.; Risch, N.J.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C., Jr.; Rimmler, J.B.; Locke, P.A.; Conneally, P.M.; Schmader, K.E.; et al. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat. Genet. 1994, 7, 180–184. [Google Scholar] [CrossRef]
- Holtzman, D.M.; Herz, J.; Bu, G. Apolipoprotein E and Apolipoprotein E Receptors: Normal Biology and Roles in Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrano-Pozo, A.; Das, S.; Hyman, B.T. APOE and Alzheimer’s disease: Advances in genetics, pathophysiology, and therapeutic approaches. Lancet Neurol. 2021, 20, 68–80. [Google Scholar] [CrossRef]
- Radi, E.; Formichi, P.; Battisti, C.; Federico, A. Apoptosis and Oxidative Stress in Neurodegenerative Diseases. J. Alzheimer’s Dis. 2014, 42, S125–S152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedlander, R.M. Apoptosis and Caspases in Neurodegenerative Diseases. N. Engl. J. Med. 2003, 348, 1365–1375. [Google Scholar] [CrossRef]
- Lee, J.-C.; Son, Y.-O.; Choi, K.-C.; Jang, Y.-S. Hydrogen peroxide induces apoptosis of BJAB cells due to formation of hydroxyl radicals via intracellular iron-mediated Fenton chemistry in glucose oxidase-mediated oxidative stress. Mol. Cells 2006, 22, 21–29. [Google Scholar]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef]
- Guo, X.-D.; Sun, G.-L.; Zhou, T.-T.; Wang, Y.-Y.; Xu, X.; Shi, X.-F.; Zhu, Z.-Y.; Rukachaisirikul, V.; Hu, L.-H.; Shen, X. LX2343 alleviates cognitive impairments in AD model rats by inhibiting oxidative stress-induced neuronal apoptosis and tauopathy. Acta Pharmacol. Sin. 2017, 38, 1104–1119. [Google Scholar] [CrossRef] [Green Version]
- Park, G.; Nhan, H.S.; Tyan, S.-H.; Kawakatsu, Y.; Zhang, C.; Navarro, M.; Koo, E.H. Caspase Activation and Caspase-Mediated Cleavage of APP Is Associated with Amyloid β-Protein-Induced Synapse Loss in Alzheimer’s Disease. Cell Rep. 2020, 31, 107839. [Google Scholar] [CrossRef] [PubMed]
- Van Der Horst, A.; Burgering, B.M. Stressing the role of FoxO proteins in lifespan and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 440–450. [Google Scholar] [CrossRef]
- Shi, Y.; Initiative, A.D.N.; Yamada, K.; Liddelow, S.A.; Smith, S.T.; Zhao, L.; Luo, W.; Tsai, R.M.; Spina, S.; Grinberg, L.T.; et al. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 2017, 549, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [Green Version]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt Promotes Cell Survival by Phosphorylating and Inhibiting a Forkhead Transcription Factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef] [Green Version]
- Biswas, S.C.; Shi, Y.; Vonsattel, J.-P.G.; Leung, C.L.; Troy, C.M.; Greene, L.A. Bim Is Elevated in Alzheimer’s Disease Neurons and Is Required for beta-Amyloid-Induced Neuronal Apoptosis. J. Neurosci. 2007, 27, 893–900. [Google Scholar] [CrossRef] [PubMed]
- Malishev, R.; Nandi, S.; Śmiłowicz, D.; Bakavayev, S.; Engel, S.; Bujanover, N.; Gazit, R.; Metzler-Nolte, N.; Jelinek, R. Interactions between BIM Protein and Beta-Amyloid May Reveal a Crucial Missing Link between Alzheimer’s Disease and Neuronal Cell Death. ACS Chem. Neurosci. 2019, 10, 3555–3564. [Google Scholar] [CrossRef] [PubMed]
- Sunayama, J.; Tsuruta, F.; Masuyama, N.; Gotoh, Y. JNK antagonizes Akt-mediated survival signals by phosphorylating 14-3-3. J. Cell Biol. 2005, 170, 295–304. [Google Scholar] [CrossRef] [Green Version]
- Plascencia-Villa, G.; Perry, G. Preventive and Therapeutic Strategies in Alzheimer’s Disease: Focus on Oxidative Stress, Redox Metals, and Ferroptosis. Antioxidants Redox Signal. 2021, 34, 591–610. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Mattson, M.P. Apolipoprotein E and oxidative stress in brain with relevance to Alzheimer’s disease. Neurobiol. Dis. 2020, 138, 104795. [Google Scholar] [CrossRef]
- Barnham, K.J.; Masters, C.L.; Bush, A.I. Neurodegenerative diseases and oxidatives stress. Nat. Rev. Drug Discov. 2004, 3, 205–214. [Google Scholar] [CrossRef]
- Huang, Y.-W.A.; Zhou, B.; Wernig, M.; Südhof, T.C. ApoE2, ApoE3, and ApoE4 Differentially Stimulate APP Transcription and Aβ Secretion. Cell 2017, 168, 427–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riddell, D.R.; Zhou, H.; Atchison, K.; Warwick, H.K.; Atkinson, P.J.; Jefferson, J.; Xu, L.; Aschmies, S.; Kirksey, Y.; Hu, Y.; et al. Impact of Apolipoprotein E (ApoE) Polymorphism on Brain ApoE Levels. J. Neurosci. 2008, 28, 11445–11453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, H.; Campenot, R.B.; Vance, D.E.; Vance, J.E. Apolipoprotein E-Containing Lipoproteins Protect Neurons from Apoptosis via a Signaling Pathway Involving Low-Density Lipoprotein Receptor-Related Protein-1. J. Neurosci. 2007, 27, 1933–1941. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhao, J.; Tikhanovich, I.; Kuravi, S.; Helzberg, J.; Dorko, K.; Roberts, B.; Kumer, S.; A Weinman, S. Serine 574 phosphorylation alters transcriptional programming of FOXO3 by selectively enhancing apoptotic gene expression. Cell Death Differ. 2015, 23, 583–595. [Google Scholar] [CrossRef] [Green Version]
- Cantley, L.C. The role of phosphoinositide 3-kinase in human disease. Harvey Lect. 2006, 100, 103–122. [Google Scholar]
- Dávila, D.; Torres-Aleman, I. Neuronal Death by Oxidative Stress Involves Activation of FOXO3 through a Two-Arm Pathway That Activates Stress Kinases and Attenuates Insulin-like Growth Factor I Signaling. Mol. Biol. Cell 2008, 19, 2014–2025. [Google Scholar] [CrossRef] [Green Version]
- Wong, H.-K.; Veremeyko, T.; Patel, N.; Lemere, C.A.; Walsh, D.M.; Esau, C.; Vanderburg, C.; Krichevsky, A.M. De-repression of FOXO3a death axis by microRNA-132 and -212 causes neuronal apoptosis in Alzheimer’s disease. Hum. Mol. Genet. 2013, 22, 3077–3092. [Google Scholar] [CrossRef] [Green Version]
- Butterfield, D.A.; Halliwell, B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 2019, 20, 148–160. [Google Scholar] [CrossRef]
- Lane-Donovan, C.; Herz, J. ApoE, ApoE Receptors, and the Synapse in Alzheimer’s Disease. Trends Endocrinol. Metab. 2017, 28, 273–284. [Google Scholar] [CrossRef] [Green Version]
- Conejerogoldberg, C.; Gomar, J.; Bascarán, M.T.B.; Hyde, T.M.; Kleinman, J.E.; Herman, M.M.; Chen, S.; Davies, P.J.A.; E Goldberg, T. APOE2 enhances neuroprotection against Alzheimer’s disease through multiple molecular mechanisms. Mol. Psychiatry 2014, 19, 1243–1250. [Google Scholar] [CrossRef]
- Huang, Y.; Mahley, R.W. Apolipoprotein E: Structure and function in lipid metabolism, neurobiology, and Alzheimer’s diseases. Neurobiol. Dis. 2014, 72 Pt A, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Getz, G.S.; Reardon, C.A. Apoprotein E as a lipid transport and signaling protein in the blood, liver, and artery wall. J. Lipid Res. 2009, 50, S156–S161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minta, K.; Brinkmalm, G.; Janelidze, S.; Sjödin, S.; Portelius, E.; Stomrud, E.; Zetterberg, H.; Blennow, K.; Hansson, O.; Andreasson, U. Quantification of total apolipoprotein E and its isoforms in cerebrospinal fluid from patients with neurodegenerative diseases. Alzheimer’s Res. Ther. 2020, 12, 19. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Morillo, E.; Hansson, O.; Atagi, Y.; Bu, G.; Minthon, L.; Diamandis, E.; Nielsen, H.M. Total apolipoprotein E levels and specific isoform composition in cerebrospinal fluid and plasma from Alzheimer’s disease patients and controls. Acta Neuropathol. 2014, 127, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Baker-Nigh, A.T.; Mawuenyega, K.G.; Bollinger, J.G.; Ovod, V.; Kasten, T.; Franklin, E.E.; Liao, F.; Jiang, H.; Holtzman, D.; Cairns, N.J.; et al. Human Central Nervous System (CNS) ApoE Isoforms Are Increased by Age, Differentially Altered by Amyloidosis, and Relative Amounts Reversed in the CNS Compared with Plasma. J. Biol. Chem. 2016, 291, 27204–27218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Fu, Y.; Yamazaki, Y.; Ren, Y.; Davis, M.D.; Liu, C.-C.; Lu, W.; Wang, X.; Chen, K.; Cherukuri, Y.; et al. APOE4 exacerbates synapse loss and neurodegeneration in Alzheimer’s disease patient iPSC-derived cerebral organoids. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef]
- Wadhwani, A.R.; Affaneh, A.; Van Gulden, S.; Kessler, J.A. Neuronal apolipoprotein E4 increases cell death and phosphorylated tau release in alzheimer disease. Ann. Neurol. 2019, 85, 726–739. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, Y.; Jiang, H.; Niikura, T.; Ito, Y.; Hagiwara, A.; Umezawa, K.; Abe, Y.; Murayama, Y.; Nishimoto, I. Neuronal Apoptosis by Apolipoprotein E4 through Low-Density Lipoprotein Receptor-Related Protein and Heterotrimeric GTPases. J. Neurosci. 2000, 20, 8401–8409. [Google Scholar] [CrossRef]
- Persson, T.; Lattanzio, F.; Calvo-Garrido, J.; Rimondini, R.; Rubio-Rodrigo, M.; Sundström, E.; Maioli, S.; Sandebring-Matton, A.; Cedazo-Mínguez, Á. Apolipoprotein E4 Elicits Lysosomal Cathepsin D Release, Decreased Thioredoxin-1 Levels, and Apoptosis. J. Alzheimer’s Dis. 2017, 56, 601–617. [Google Scholar] [CrossRef] [Green Version]
- Hoe, H.-S.; Harris, D.C.; Rebeck, G.W. Multiple pathways of apolipoprotein E signaling in primary neurons. J. Neurochem. 2005, 93, 145–155. [Google Scholar] [CrossRef]
- Qiu, Z.; Crutcher, K.; Hyman, B.; Rebeck, G. ApoE isoforms affect neuronal N-methyl-d-aspartate calcium responses and toxicity via receptor-mediated processes. Neuroscience 2003, 122, 291–303. [Google Scholar] [CrossRef]
- Manolopoulos, K.; Klotz, L.-O.; Korsten, P.; Bornstein, S.R.; Barthel, A. Linking Alzheimer’s disease to insulin resistance: The FoxO response to oxidative stress. Mol. Psychiatry 2010, 15, 1046–1052. [Google Scholar] [CrossRef] [Green Version]
- Ruffels, J.; Griffin, M.; Dickenson, J.M. Activation of ERK1/2, JNK and PKB by hydrogen peroxide in human SH-SY5Y neuroblastoma cells: Role of ERK1/2 in H2O2-induced cell death. Eur. J. Pharmacol. 2004, 483, 163–173. [Google Scholar] [CrossRef]
- Song, J.; Zhang, W.; Wang, J.; Yang, H.; Zhou, Q.; Wang, H.; Li, L.; Du, G. Inhibition of FOXO3a/BIM signaling pathway contributes to the protective effect of salvianolic acid A against cerebral ischemia/reperfusion injury. Acta Pharm. Sin. B 2019, 9, 505–515. [Google Scholar] [CrossRef]
- Zhang, X.; Tang, N.; Hadden, T.J.; Rishi, A.K. Akt, FoxO and regulation of apoptosis. Biochim. Biophys. Acta 2011, 1813, 1978–1986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saha, P.; Biswas, S.C. Amyloid-β induced astrocytosis and astrocyte death: Implication of FoxO3a–Bim–caspase3 death signaling. Mol. Cell. Neurosci. 2015, 68, 203–211. [Google Scholar] [CrossRef]
- Sanphui, P.; Biswas, S.C. FoxO3a is activated and executes neuron death via Bim in response to β-amyloid. Cell Death Dis. 2013, 4, e625. [Google Scholar] [CrossRef] [PubMed]
- Hatters, D.; Peters-Libeu, C.A.; Weisgraber, K.H. Apolipoprotein E structure: Insights into function. Trends Biochem. Sci. 2006, 31, 445–454. [Google Scholar] [CrossRef]
- Nguyen, D.; Dhanasekaran, P.; Nickel, M.; Nakatani, R.; Saito, H.; Phillips, M.C.; Lund-Katz, S. Molecular Basis for the Differences in Lipid and Lipoprotein Binding Properties of Human Apolipoproteins E3 and E4. Biochemistry 2010, 49, 10881–10889. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, J.; Kouiavskaia, D.; Migliorini, M.; Robinson, S.; Saenko, E.L.; Gorlatova, N.; Li, D.; Lawrence, D.; Hyman, B.T.; Weisgraber, K.H.; et al. The apoE isoform binding properties of the VLDL receptor reveal marked differences from LRP and the LDL receptor. J. Lipid Res. 2005, 46, 1721–1731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, F.; Yoon, H.; Kim, J. Apolipoprotein E metabolism and functions in brain and its role in Alzheimer’s disease. Curr. Opin. Lipidol. 2016, 28, 60–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Kirwan, P.; Livesey, F.J. Directed differentiation of human pluripotent stem cells to cerebral cortex neurons and neural networks. Nat. Protoc. 2012, 7, 1836–1846. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| iPSC Line | Gender | Age at Biopsy | APOE Genotype | Vendor | Diagnosis |
|---|---|---|---|---|---|
| YK26 | Female | Adult | APOE3/E3 | UCONN HEALTH | Non-dementia |
| YK27 | Female | Adult | APOE3/E3 | UCONN HEALTH | Non-dementia |
| TZ1 | Female | Fetal | APOE3/E3 | UCONN HEALTH | Non-dementia |
| BONi037-A (B037) | Female | 75–79 | APOE3/E3 | EBiSC | Non-dementia |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, H.; Zheng, W.; Li, C.; Xu, H. Isoform-Specific Effects of Apolipoprotein E on Hydrogen Peroxide-Induced Apoptosis in Human Induced Pluripotent Stem Cell (iPSC)-Derived Cortical Neurons. Int. J. Mol. Sci. 2021, 22, 11582. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222111582
Gao H, Zheng W, Li C, Xu H. Isoform-Specific Effects of Apolipoprotein E on Hydrogen Peroxide-Induced Apoptosis in Human Induced Pluripotent Stem Cell (iPSC)-Derived Cortical Neurons. International Journal of Molecular Sciences. 2021; 22(21):11582. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222111582
Chicago/Turabian StyleGao, Huiling, Wei Zheng, Cheng Li, and He Xu. 2021. "Isoform-Specific Effects of Apolipoprotein E on Hydrogen Peroxide-Induced Apoptosis in Human Induced Pluripotent Stem Cell (iPSC)-Derived Cortical Neurons" International Journal of Molecular Sciences 22, no. 21: 11582. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222111582