
Targeting Folate Metabolism Is Selectively Cytotoxic to Glioma Stem Cells and Effectively Cooperates with Differentiation Therapy to Eliminate Tumor-Initiating Cells in Glioma Xenografts

 ,
,  and
and 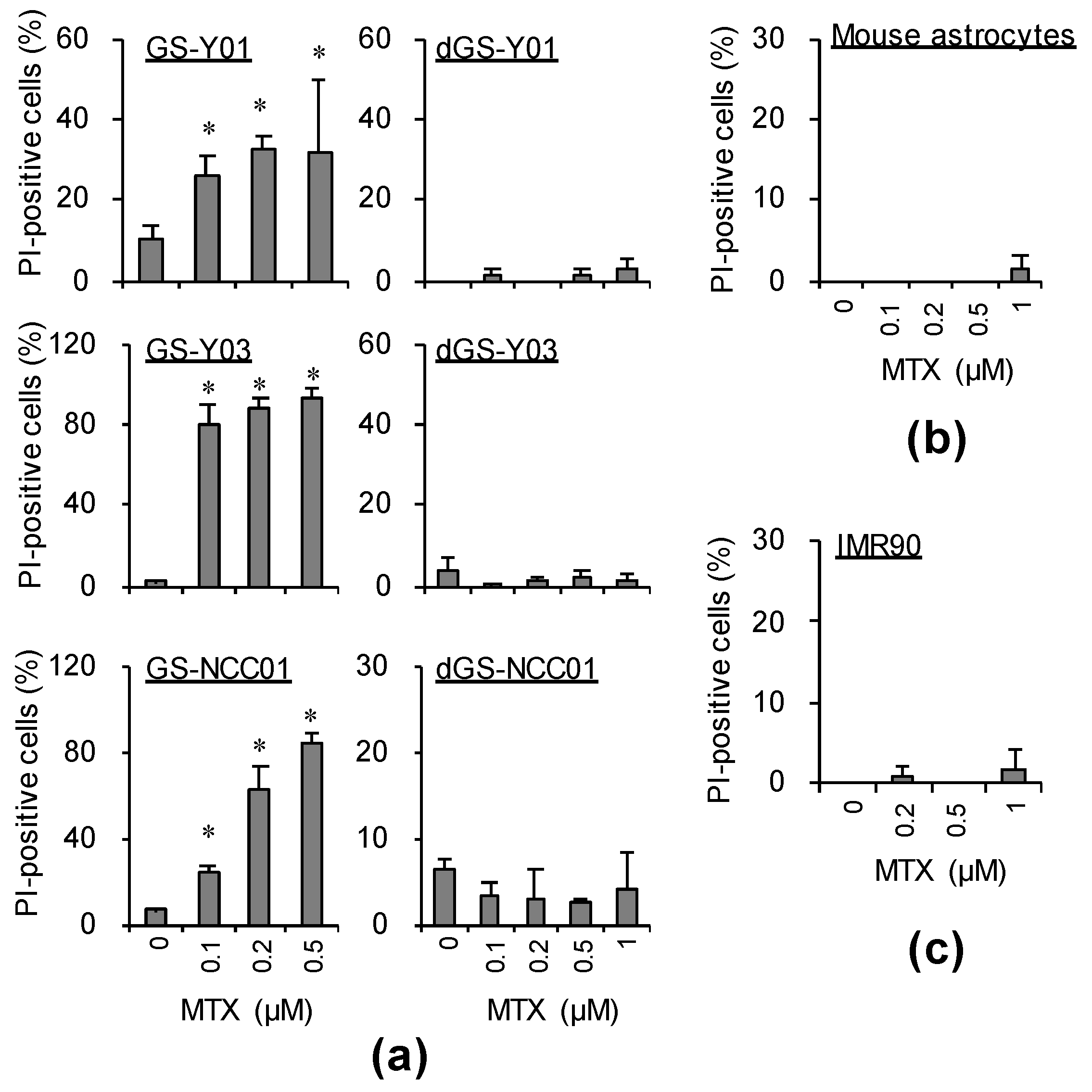{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
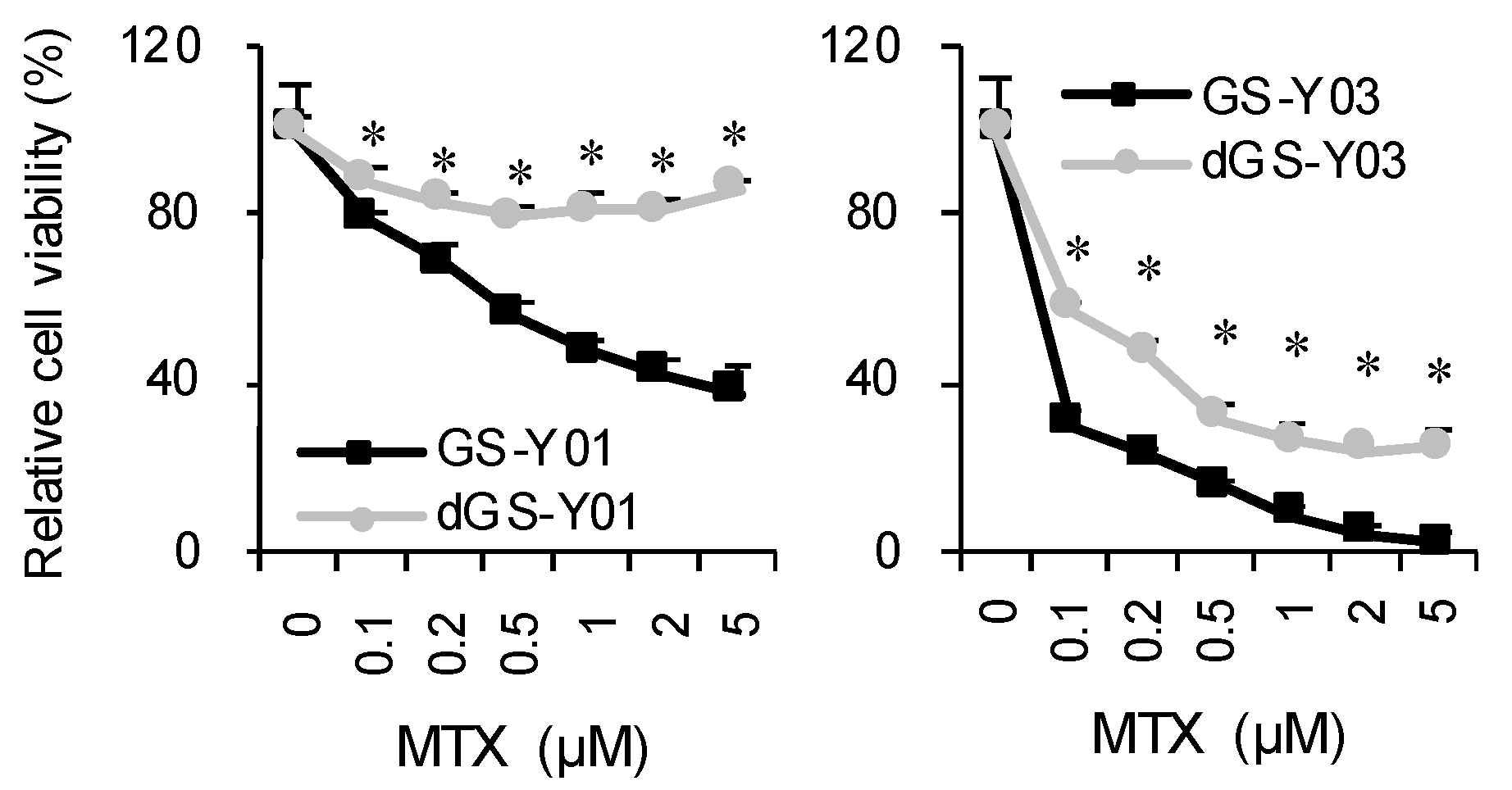
2.1. MTX Exhibits GSC-Selective Cytotoxic Effects
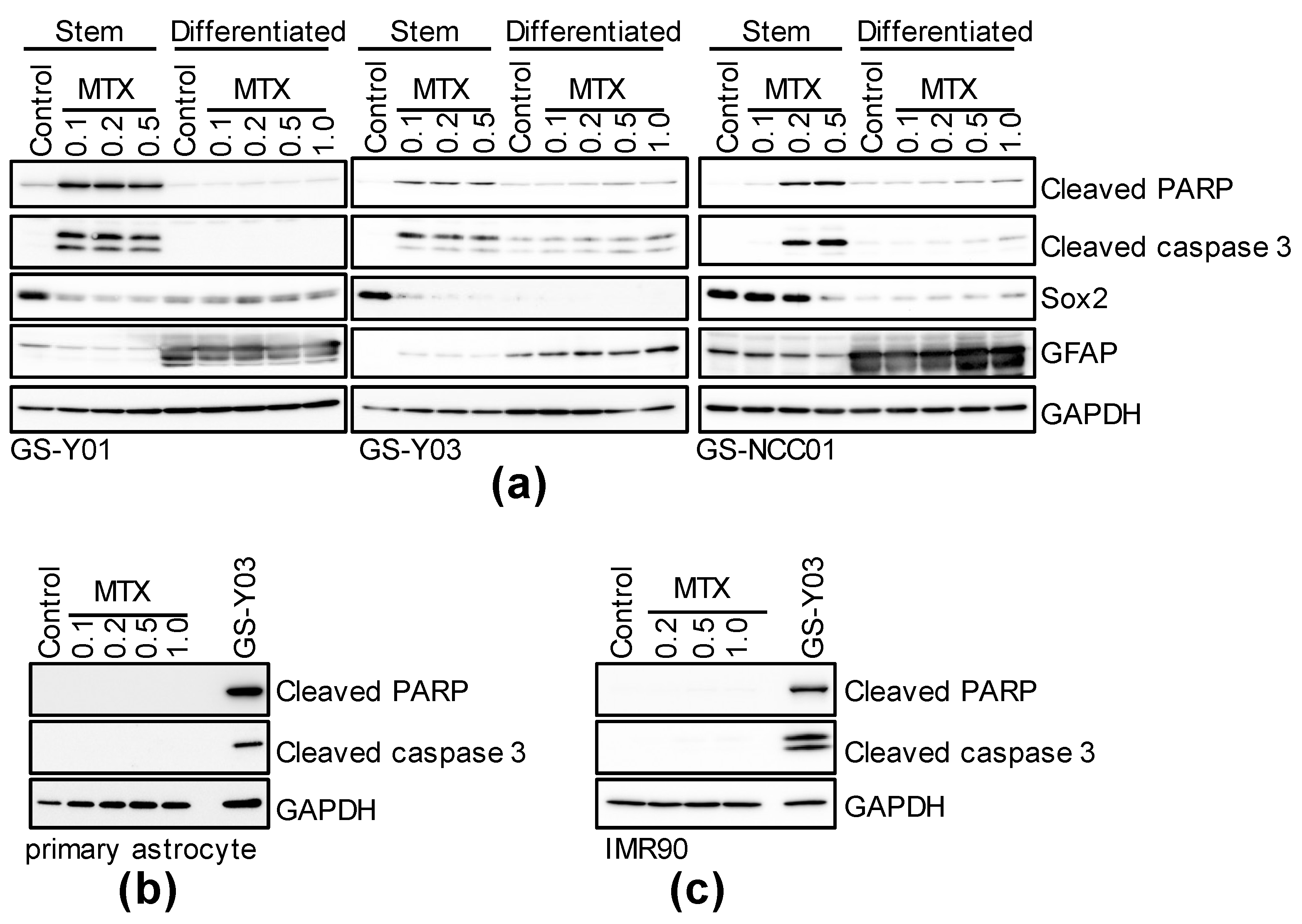
2.2. Folate Antagonists Induces Apoptotic Cell Death of GSCs
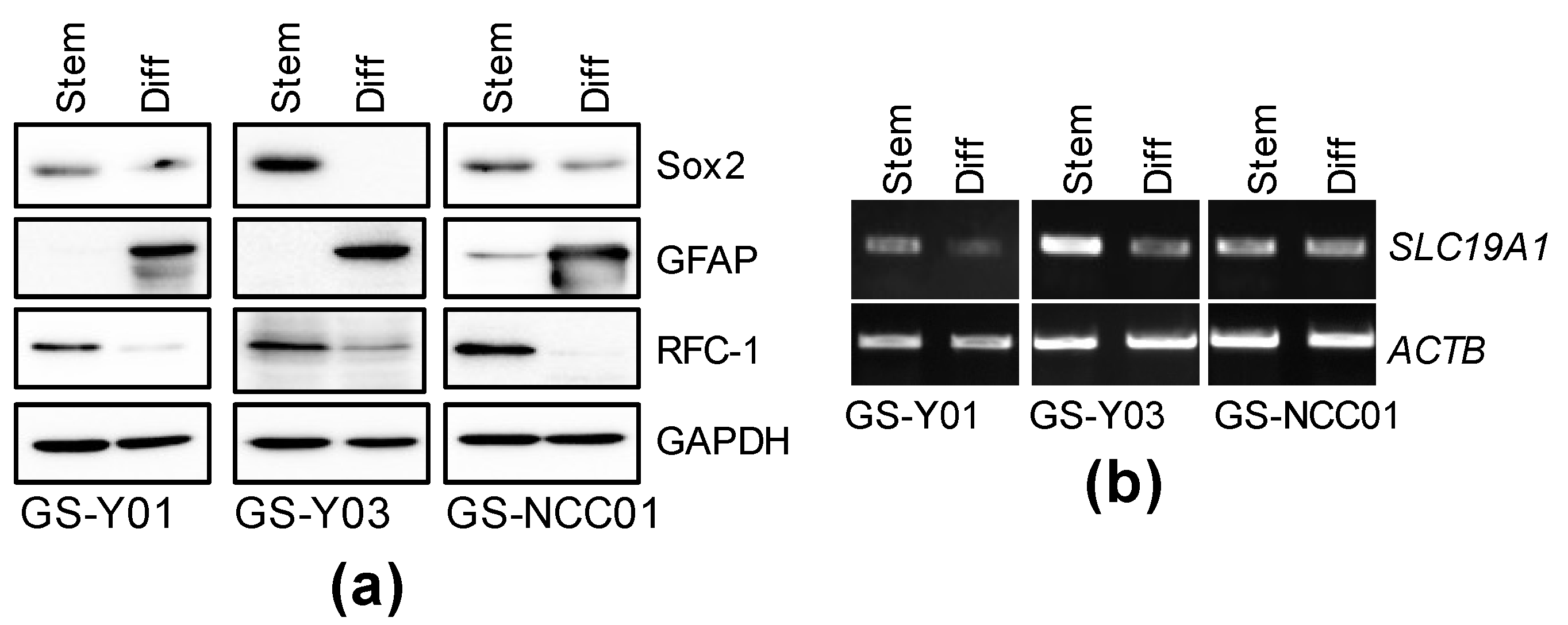
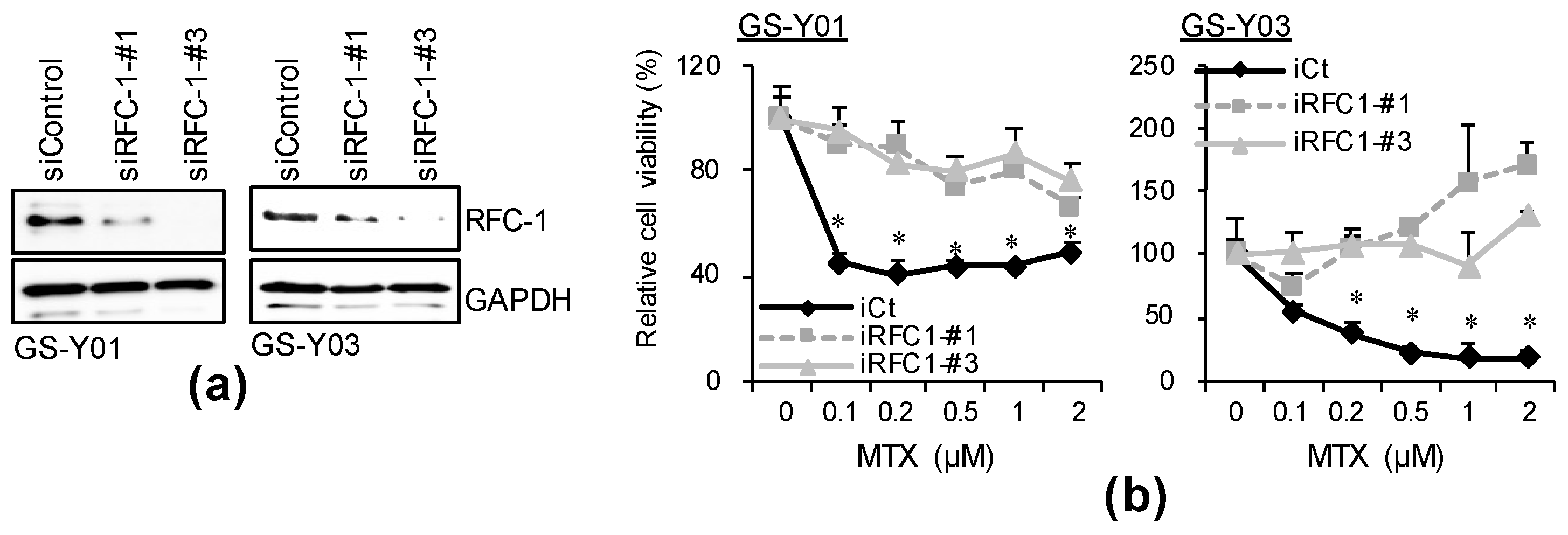
2.3. GSC-Selective Cytotoxic Effects of MTX Are Dependent on RFC-1/SLC19A1
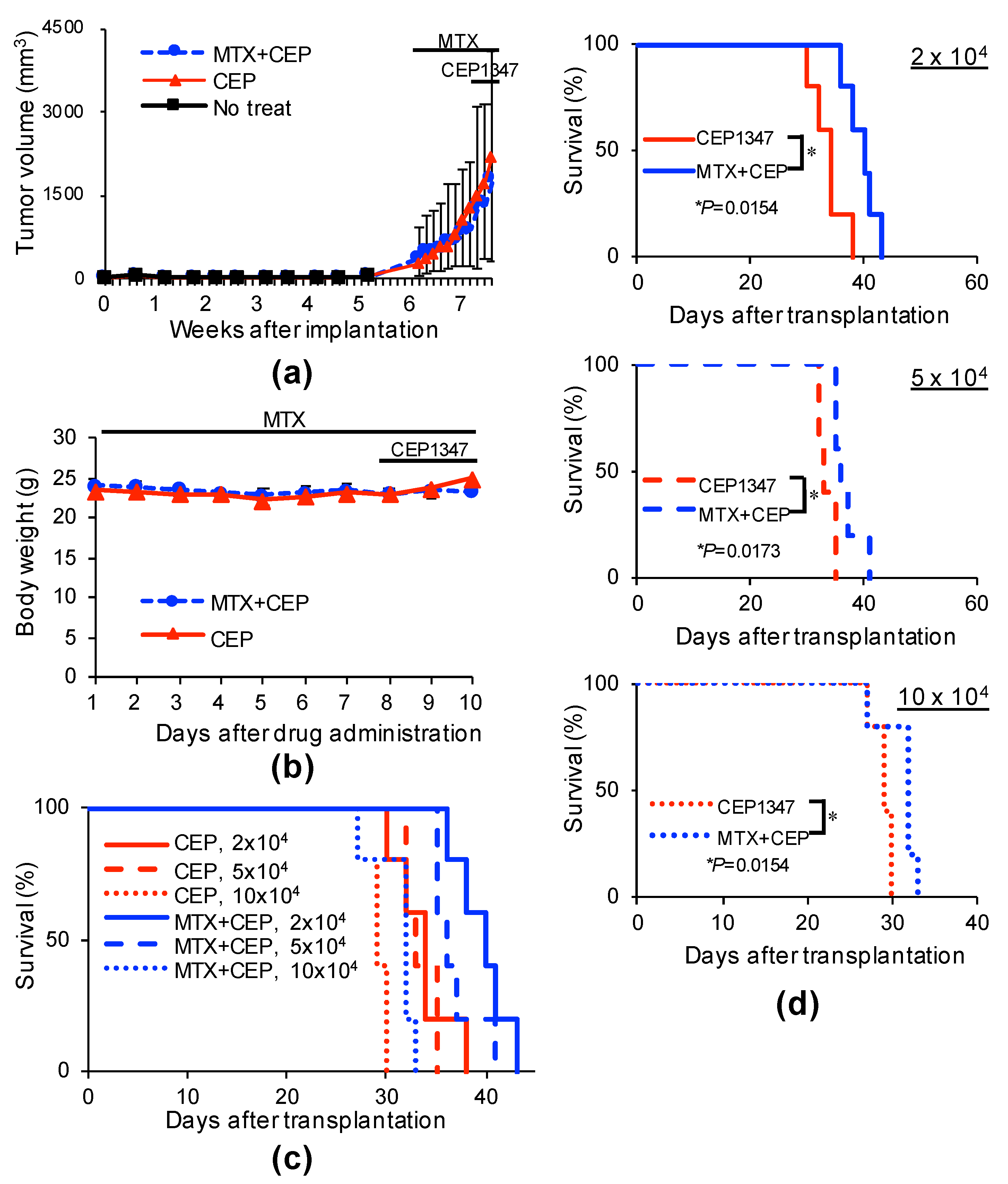
2.4. MTX Alters the GSC-Inhibitory Effects of Differentiation Induction In Vivo
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Cell Culture
4.3. Propidium Iodide Incorporation Assay
4.4. Cell Viability Assay
4.5. Immunoblotting
4.6. Reverse Transcription-PCR Analysis
4.7. Gene Silencing by siRNA
4.8. Mouse Studies
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Matsuda, K.; Sato, A.; Okada, M.; Shibuya, K.; Seino, S.; Suzuki, K.; Watanabe, E.; Narita, Y.; Shibui, S.; Kayama, T.; et al. Targeting JNK for therapeutic depletion of stem-like glioblastoma cells. Sci. Rep. 2012, 2, 516. [Google Scholar] [CrossRef] [Green Version]
- Okada, M.; Kuramoto, K.; Takeda, H.; Watarai, H.; Sakaki, H.; Seino, S.; Seino, M.; Suzuki, S.; Kitanaka, C. The novel JNK inhibitor AS602801 inhibits cancer stem cells in vitro and in vivo. Oncotarget 2016, 7, 27021–27032. [Google Scholar] [CrossRef] [Green Version]
- Okada, M.; Takeda, H.; Sakaki, H.; Kuramoto, K.; Suzuki, S.; Sanomachi, T.; Togashi, K.; Seino, S.; Kitanaka, C. Repositioning CEP-1347, a chemical agent originally developed for the treatment of Parkinson’s disease, as an anti-cancer stem cell drug. Oncotarget 2017, 8, 94872–94882. [Google Scholar] [CrossRef] [Green Version]
- Sato, A.; Sunayama, J.; Okada, M.; Watanabe, E.; Seino, S.; Shibuya, K.; Suzuki, K.; Narita, Y.; Shibui, S.; Kayama, T.; et al. Glioma-initiating cell elimination by metformin activation of FOXO3 via AMPK. Stem Cells Transl. Med. 2012, 1, 811–824. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, K.; Okada, M.; Suzuki, S.; Seino, M.; Seino, S.; Takeda, H.; Kitanaka, C. Targeting the facilitative glucose transporter GLUT1 inhibits the self-renewal and tumor-initiating capacity of cancer stem cells. Oncotarget 2015, 6, 651–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaushik, I.; Ramachandran, S.; Prasad, S.; Srivastava, S.K. Drug rechanneling: A novel paradigm for cancer treatment. Semin Cancer Biol. 2021, 68, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Kuramoto, K.; Suzuki, S.; Sakaki, H.; Takeda, H.; Sanomachi, T.; Seino, S.; Narita, Y.; Kayama, T.; Kitanaka, C.; Okada, M. Licochalcone A specifically induces cell death in glioma stem cells via mitochondrial dysfunction. FEBS Open Bio. 2017, 7, 835–844. [Google Scholar] [CrossRef]
- Kuramoto, K.; Yamamoto, M.; Suzuki, S.; Sanomachi, T.; Togashi, K.; Seino, S.; Kitanaka, C.; Okada, M. Verteporfin inhibits oxidative phosphorylation and induces cell death specifically in glioma stem cells. FEBS J. 2020, 287, 2023–2036. [Google Scholar] [CrossRef]
- Yu, J.; Zhou, P. The advances of methotrexate resistance in rheumatoid arthritis. Inflammopharmacology 2020, 28, 1183–1193. [Google Scholar] [CrossRef] [PubMed]
- Shams, S.; Martinez, J.M.; Dawson, J.R.D.; Flores, J.; Gabriel, M.; Garcia, G.; Guevara, A.; Murray, K.; Pacifici, N.; Vargas, M.V.; et al. The Therapeutic Landscape of Rheumatoid Arthritis: Current State and Future Directions. Front. Pharmacol. 2021, 12, 680043. [Google Scholar] [CrossRef]
- Pui, C.H.; Evans, W.E. Treatment of acute lymphoblastic leukemia. N. Engl. J. Med. 2006, 354, 166–178. [Google Scholar] [CrossRef] [PubMed]
- Rusthoven, J.J.; Eisenhauer, E.; Butts, C.; Gregg, R.; Dancey, J.; Fisher, B.; Iglesias, J. Multitargeted antifolate LY231514 as first-line chemotherapy for patients with advanced non-small-cell lung cancer: A phase II study. National Cancer Institute of Canada Clinical Trials Group. J. Clin. Oncol. 1999, 17, 1194. [Google Scholar] [CrossRef]
- Vogelzang, N.J.; Rusthoven, J.J.; Symanowski, J.; Denham, C.; Kaukel, E.; Ruffie, P.; Gatzemeier, U.; Boyer, M.; Emri, S.; Manegold, C.; et al. Phase III study of pemetrexed in combination with cisplatin versus cisplatin alone in patients with malignant pleural mesothelioma. J. Clin. Oncol. 2003, 21, 2636–2644. [Google Scholar] [CrossRef] [PubMed]
- Alam, C.; Kondo, M.; O’Connor, D.L.; Bendayan, R. Clinical Implications of Folate Transport in the Central Nervous System. Trends Pharmacol. Sci. 2020, 41, 349–361. [Google Scholar] [CrossRef]
- Sirotnak, F.M.; Moccio, D.M.; Dorick, D.M. Optimization of high-dose methotrexate with leucovorin rescue therapy in the L1210 leukemia and sarcoma 180 murine tumor models. Cancer Res. 1978, 38, 345–353. [Google Scholar] [PubMed]
- Sirotnak, F.M.; Otter, G.M.; Schmid, F.A. Markedly improved efficacy of edatrexate compared to methotrexate in a high-dose regimen with leucovorin rescue against metastatic murine solid tumors. Cancer Res. 1993, 53, 587–591. [Google Scholar]
- Tedeschi, P.M.; Kathari, Y.K.; Farooqi, I.N.; Bertino, J.R. Leucovorin rescue allows effective high-dose pralatrexate treatment and an increase in therapeutic index in mesothelioma xenografts. Cancer Chemother Pharmacol. 2014, 74, 1029–1032. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.V.; Vanner, R.; Dirks, P.; Eaves, C.J. Cancer stem cells: An evolving concept. Nat. Rev. Cancer 2012, 12, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Maroney, A.C.; Finn, J.P.; Connors, T.J.; Durkin, J.T.; Angeles, T.; Gessner, G.; Xu, Z.; Meyer, S.L.; Savage, M.J.; Greene, L.A.; et al. Cep-1347 (KT7515), a semisynthetic inhibitor of the mixed lineage kinase family. J. Biol. Chem. 2001, 276, 25302–25308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kremer, J.M.; Galivan, J.; Streckfuss, A.; Kamen, B. Methotrexate metabolism analysis in blood and liver of rheumatoid arthritis patients. Association with hepatic folate deficiency and formation of polyglutamates. Arthritis Rheum 1986, 29, 832–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagawa, K.; Kudoh, S.; Matsui, K.; Negoro, S.; Yamamoto, N.; Latz, J.E.; Adachi, S.; Fukuoka, M. A phase I study of pemetrexed (LY231514) supplemented with folate and vitamin B12 in Japanese patients with solid tumours. Br. J. Cancer 2006, 95, 677–682. [Google Scholar] [CrossRef] [Green Version]
- Ferreri, A.J.; Reni, M.; Villa, E. Therapeutic management of primary central nervous system lymphoma: Lessons from prospective trials. Ann. Oncol. 2000, 11, 927–937. [Google Scholar] [CrossRef] [PubMed]
- Figueiró, F.; de Oliveira, C.P.; Rockenbach, L.; Mendes, F.B.; Bergamin, L.S.; Jandrey, E.H.; Edelweiss, M.I.; Guterres, S.S.; Pohlmann, A.R.; Battastini, A.M. Pharmacological Improvement and Preclinical Evaluation of Methotrexate-Loaded Lipid-Core Nanocapsules in a Glioblastoma Model. J. Biomed. Nanotechnol. 2015, 11, 1808–1818. [Google Scholar] [CrossRef]
- Bertino, J.R.; Göker, E.; Gorlick, R.; Li, W.W.; Banerjee, D. Resistance mechanisms to methotrexate in tumors. Stem Cells 1996, 14, 5–9. [Google Scholar] [CrossRef]
- Dirkse, A.; Golebiewska, A.; Buder, T.; Nazarov, P.V.; Muller, A.; Poovathingal, S.; Brons, N.H.C.; Leite, S.; Sauvageot, N.; Sarkisjan, D.; et al. Stem cell-associated heterogeneity in Glioblastoma results from intrinsic tumor plasticity shaped by the microenvironment. Nat. Commun. 2019, 10, 1787. [Google Scholar] [CrossRef]
- Hussein, M.; Chai, D.C.; Kyama, C.M.; Mwenda, J.M.; Palmer, S.S.; Gotteland, J.P.; D’Hooghe, T.M. c-Jun NH2-terminal kinase inhibitor bentamapimod reduces induced endometriosis in baboons: An assessor-blind placebo-controlled randomized study. Fertil Steril 2016, 105, 815–824.e815. [Google Scholar] [CrossRef] [Green Version]
- Palmer, S.S.; Altan, M.; Denis, D.; Tos, E.G.; Gotteland, J.P.; Osteen, K.G.; Bruner-Tran, K.L.; Nataraja, S.G. Bentamapimod (JNK Inhibitor AS602801) Induces Regression of Endometriotic Lesions in Animal Models. Reprod. Sci. 2016, 23, 11–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ducker, G.S.; Rabinowitz, J.D. One-Carbon Metabolism in Health and Disease. Cell Metab. 2017, 25, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Zgheib, R.; Battaglia-Hsu, S.F.; Hergalant, S.; Quéré, M.; Alberto, J.M.; Chéry, C.; Rouyer, P.; Gauchotte, G.; Guéant, J.L.; Namour, F. Folate can promote the methionine-dependent reprogramming of glioblastoma cells towards pluripotency. Cell Death Dis. 2019, 10, 596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konno, M.; Asai, A.; Kawamoto, K.; Nishida, N.; Satoh, T.; Doki, Y.; Mori, M.; Ishii, H. The one-carbon metabolism pathway highlights therapeutic targets for gastrointestinal cancer (Review). Int. J. Oncol. 2017, 50, 1057–1063. [Google Scholar] [CrossRef] [Green Version]
- Fawal, M.A.; Jungas, T.; Davy, A. Inhibition of DHFR targets the self-renewing potential of brain tumor initiating cells. Cancer Lett. 2021, 503, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Bowman, R.L.; Wang, Q.; Carro, A.; Verhaak, R.G.; Squatrito, M. GlioVis data portal for visualization and analysis of brain tumor expression datasets. Neuro. Oncol. 2017, 19, 139–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alata Jimenez, N.; Torres Pérez, S.A.; Sánchez-Vásquez, E.; Fernandino, J.I.; Strobl-Mazzulla, P.H. Folate deficiency prevents neural crest fate by disturbing the epigenetic Sox2 repression on the dorsal neural tube. Dev. Biol. 2018, 444 (Suppl. 1), S193–S201. [Google Scholar] [CrossRef]
- Mohanty, V.; Shah, A.; Allender, E.; Siddiqui, M.R.; Monick, S.; Ichi, S.; Mania-Farnell, B.; McLone, D.G.; Tomita, T.; Mayanil, C.S. Folate Receptor Alpha Upregulates Oct4, Sox2 and Klf4 and Downregulates miR-138 and miR-let-7 in Cranial Neural Crest Cells. Stem Cells 2016, 34, 2721–2732. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Okada, M.; Suzuki, S.; Togashi, K.; Sugai, A.; Yamamoto, M.; Kitanaka, C. Targeting Folate Metabolism Is Selectively Cytotoxic to Glioma Stem Cells and Effectively Cooperates with Differentiation Therapy to Eliminate Tumor-Initiating Cells in Glioma Xenografts. Int. J. Mol. Sci. 2021, 22, 11633. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222111633
Okada M, Suzuki S, Togashi K, Sugai A, Yamamoto M, Kitanaka C. Targeting Folate Metabolism Is Selectively Cytotoxic to Glioma Stem Cells and Effectively Cooperates with Differentiation Therapy to Eliminate Tumor-Initiating Cells in Glioma Xenografts. International Journal of Molecular Sciences. 2021; 22(21):11633. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222111633
Chicago/Turabian StyleOkada, Masashi, Shuhei Suzuki, Keita Togashi, Asuka Sugai, Masahiro Yamamoto, and Chifumi Kitanaka. 2021. "Targeting Folate Metabolism Is Selectively Cytotoxic to Glioma Stem Cells and Effectively Cooperates with Differentiation Therapy to Eliminate Tumor-Initiating Cells in Glioma Xenografts" International Journal of Molecular Sciences 22, no. 21: 11633. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222111633