
Activation of ERK and p38 Reduces AZD8055-Mediated Inhibition of Protein Synthesis in Hepatocellular Carcinoma HepG2 Cell Line

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. The mTOR Inhibitor AZD8055 Was Selected during the Screening Process as an Anticancer Drug with Inhibitory Effects on Protein Translation
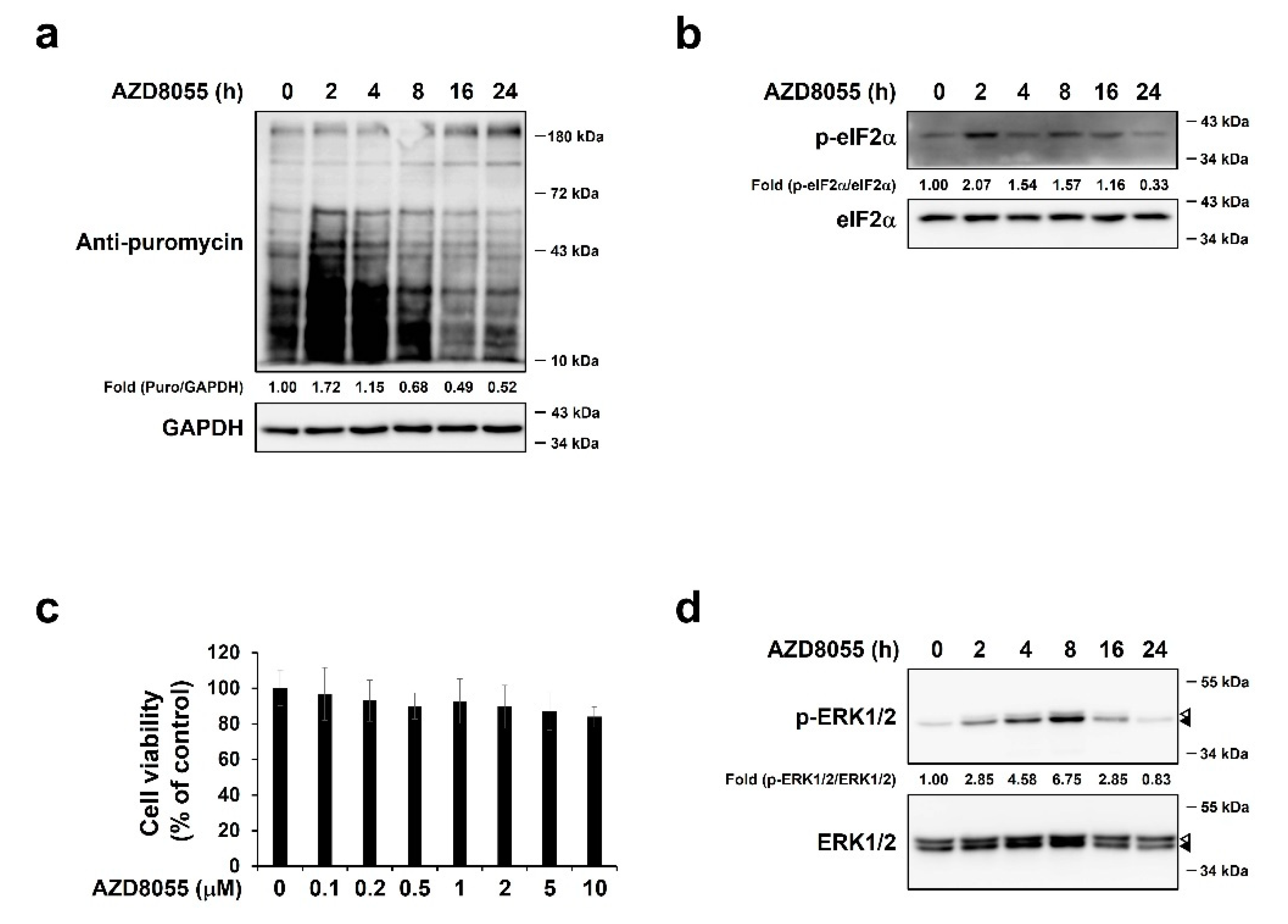
2.2. AZD8055 Inhibits Protein Synthesis and Up-Regulates Phosphorylation of ERK1/2 in Hepatocellular Carcinoma HepG2 Cells
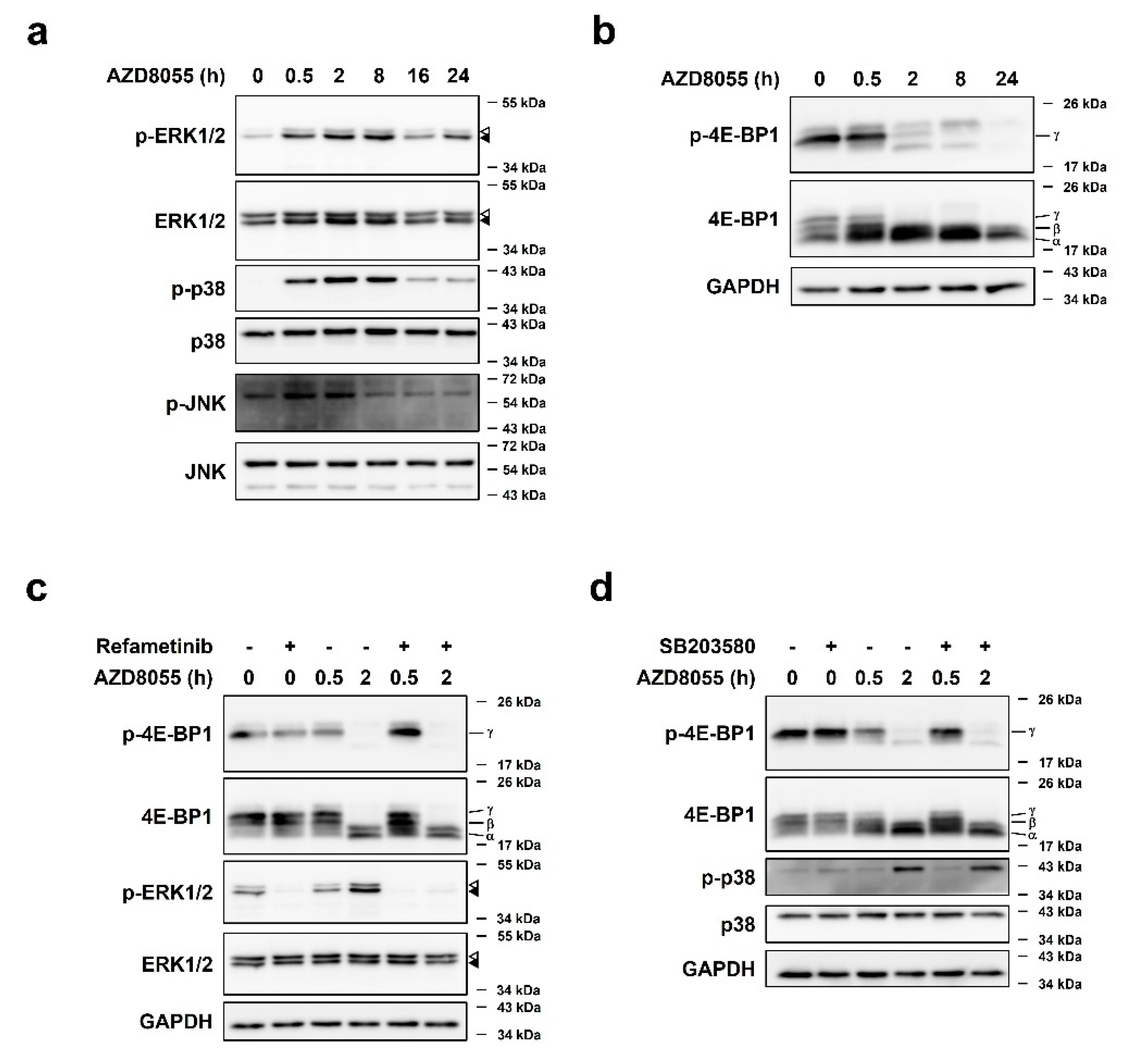
2.3. AZD8055 Up-Regulates Phosphorylation of ERK1/2 and p38 and Independently Down-Regulates Phosphorylation of 4E-BP1
2.4. AZD8055-Mediated ERK1/2 Up-Regulation Was Associated with AZD8055-Mediated Inhibition of Protein Synthesis
2.5. Activation of p38 by AZD8055 Treatment Was Associated with AZD8055-Mediated Inhibition of Protein Synthesis
2.6. AZD8055 Regulates the 4E-BP1 mRNA Pool by Up-Regulating ERK1/2 and p38 Pathways
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Lines and Cell Culture
4.3. Puromycin Incorporation Assay and Analysis
4.4. Measurement of Cell Viability
4.5. Immunoblot Analysis
4.6. Reverse-Transcription Polymerase Chain Reaction (RT-PCR) and Real-Time Quantitative PCR (qPCR) Primers
4.7. Statistical Analysis
5. Conclusions
- Anticancer drugs that inhibit protein synthesis were selected in MEFs through screening using puromycin incorporation assay.
- AZD8055 inhibits translation in hepatocellular carcinoma HepG2 cell lines.
- AZD8055 inhibits 4E-BP1 phosphorylation and induces ERK1/2 and p38 phosphorylation, which are independent of each other.
- Combined treatment of AZD8055 with refametinib or SB203580 has a synergistic effect on translational inhibition than AZD8055 treatment alone.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Martin, D.; Nguyen, Q.; Molinolo, A.; Gutkind, J.S. Accumulation of dephosphorylated 4EBP after mTOR inhibition with rapamycin is sufficient to disrupt paracrine transformation by the KSHV vGPCR oncogene. Oncogene 2014, 33, 2405–2412. [Google Scholar] [CrossRef] [Green Version]
- Bhat, M.; Robichaud, N.; Hulea, L.; Sonenberg, N.; Pelletier, J.; Topisirovic, I. Targeting the translation machinery in cancer. Nat. Rev. Drug Discov. 2015, 14, 261–278. [Google Scholar] [CrossRef]
- Gingras, A.C.; Raught, B.; Gygi, S.P.; Niedzwiecka, A.; Miron, M.; Burley, S.K.; Polakiewicz, R.D.; Wyslouch-Cieszynska, A.; Aebersold, R.; Sonenberg, N. Hierarchical phosphorylation of the translation inhibitor 4E-BP1. Genes Dev. 2001, 15, 2852–2864. [Google Scholar] [CrossRef]
- Liu, G.; Zhang, Y.; Bode, A.M.; Ma, W.Y.; Dong, Z. Phosphorylation of 4E-BP1 is mediated by the p38/MSK1 pathway in response to UVB irradiation. J. Biol. Chem. 2002, 277, 8810–8816. [Google Scholar] [CrossRef] [Green Version]
- Batool, A.; Aashaq, S.; Andrabi, K.I. Reappraisal to the study of 4E-BP1 as an mTOR substrate—A normative critique. Eur. J. Cell Biol. 2017, 96, 325–336. [Google Scholar] [CrossRef]
- Gingras, A.C.; Gygi, S.P.; Raught, B.; Polakiewicz, R.D.; Abraham, R.T.; Hoekstra, M.F.; Aebersold, R.; Sonenberg, N. Regulation of 4E-BP1 phosphorylation: A novel two-step mechanism. Genes Dev. 1999, 13, 1422–1437. [Google Scholar] [CrossRef]
- Herbert, T.P.; Tee, A.R.; Proud, C.G. The extracellular signal-regulated kinase pathway regulates the phosphorylation of 4E-BP1 at multiple sites. J. Biol. Chem. 2002, 277, 11591–11596. [Google Scholar] [CrossRef] [Green Version]
- Johnson, G.L.; Lapadat, R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 2002, 298, 1911–1912. [Google Scholar] [CrossRef] [Green Version]
- Bardwell, L. Mechanisms of MAPK signalling specificity. Biochem. Soc. Trans. 2006, 34, 837–841. [Google Scholar] [CrossRef] [Green Version]
- Darling, N.J.; Balmanno, K.; Cook, S.J. ERK1/2 signalling protects against apoptosis following endoplasmic reticulum stress but cannot provide long-term protection against BAX/BAK-independent cell death. PLoS ONE 2017, 12, e0184907. [Google Scholar] [CrossRef]
- Roberts, P.J.; Der, C.J. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 2007, 26, 3291–3310. [Google Scholar] [CrossRef] [Green Version]
- Cai, B.; Chang, S.H.; Becker, E.B.; Bonni, A.; Xia, Z. p38 MAP kinase mediates apoptosis through phosphorylation of BimEL at Ser-65. J. Biol. Chem. 2006, 281, 25215–25222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coulthard, L.R.; White, D.E.; Jones, D.L.; McDermott, M.F.; Burchill, S.A. p38(MAPK): Stress responses from molecular mechanisms to therapeutics. Trends Mol. Med. 2009, 15, 369–379. [Google Scholar] [CrossRef] [Green Version]
- Kato, H.; Nakajima, S.; Saito, Y.; Takahashi, S.; Katoh, R.; Kitamura, M. mTORC1 serves ER stress-triggered apoptosis via selective activation of the IRE1-JNK pathway. Cell Death Differ. 2012, 19, 310–320. [Google Scholar] [CrossRef] [Green Version]
- Verheij, M.; Ruiter, G.A.; Zerp, S.F.; van Blitterswijk, W.J.; Fuks, Z.; Haimovitz-Friedman, A.; Bartelink, H. The role of the stress-activated protein kinase (SAPK/JNK) signaling pathway in radiation-induced apoptosis. Radiother. Oncol. J. Eur. Soc. Ther. Radiol. Oncol. 1998, 47, 225–232. [Google Scholar] [CrossRef]
- Estrada, Y.; Dong, J.; Ossowski, L. Positive crosstalk between ERK and p38 in melanoma stimulates migration and in vivo proliferation. Pigment. Cell Melanoma Res. 2009, 22, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [Green Version]
- Populo, H.; Lopes, J.M.; Soares, P. The mTOR signalling pathway in human cancer. Int. J. Mol. Sci. 2012, 13, 1886–1918. [Google Scholar] [CrossRef] [PubMed]
- Albert, S.; Serova, M.; Dreyer, C.; Sablin, M.P.; Faivre, S.; Raymond, E. New inhibitors of the mammalian target of rapamycin signaling pathway for cancer. Expert Opin. Investig. Drugs 2010, 19, 919–930. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [Green Version]
- O’Reilly, K.E.; Rojo, F.; She, Q.B.; Solit, D.; Mills, G.B.; Smith, D.; Lane, H.; Hofmann, F.; Hicklin, D.J.; Ludwig, D.L.; et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006, 66, 1500–1508. [Google Scholar] [CrossRef] [Green Version]
- Soares, H.P.; Ni, Y.; Kisfalvi, K.; Sinnett-Smith, J.; Rozengurt, E. Different patterns of Akt and ERK feedback activation in response to rapamycin, active-site mTOR inhibitors and metformin in pancreatic cancer cells. PLoS ONE 2013, 8, e57289. [Google Scholar] [CrossRef] [Green Version]
- Chresta, C.M.; Davies, B.R.; Hickson, I.; Harding, T.; Cosulich, S.; Critchlow, S.E.; Vincent, J.P.; Ellston, R.; Jones, D.; Sini, P.; et al. AZD8055 is a potent, selective, and orally bioavailable ATP-competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer Res. 2010, 70, 288–298. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Echeverria, C. Allosteric and ATP-competitive kinase inhibitors of mTOR for cancer treatment. Bioorganic Med. Chem. Lett. 2010, 20, 4308–4312. [Google Scholar] [CrossRef]
- Chapuis, N.; Tamburini, J.; Green, A.S.; Vignon, C.; Bardet, V.; Neyret, A.; Pannetier, M.; Willems, L.; Park, S.; Macone, A.; et al. Dual inhibition of PI3K and mTORC1/2 signaling by NVP-BEZ235 as a new therapeutic strategy for acute myeloid leukemia. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2010, 16, 5424–5435. [Google Scholar] [CrossRef] [Green Version]
- Feldman, M.E.; Apsel, B.; Uotila, A.; Loewith, R.; Knight, Z.A.; Ruggero, D.; Shokat, K.M. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 2009, 7, e38. [Google Scholar] [CrossRef]
- Thoreen, C.C.; Kang, S.A.; Chang, J.W.; Liu, Q.; Zhang, J.; Gao, Y.; Reichling, L.J.; Sim, T.; Sabatini, D.M.; Gray, N.S. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J. Biol. Chem. 2009, 284, 8023–8032. [Google Scholar] [CrossRef] [Green Version]
- Blaser, B.; Waselle, L.; Dormond-Meuwly, A.; Dufour, M.; Roulin, D.; Demartines, N.; Dormond, O. Antitumor activities of ATP-competitive inhibitors of mTOR in colon cancer cells. BMC Cancer 2012, 12, 86. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Lee, C.H.; Tseng, B.Y.; Tsai, Y.H.; Tsai, H.W.; Yao, C.L.; Tseng, S.H. AZD8055 Exerts Antitumor Effects on Colon Cancer Cells by Inhibiting mTOR and Cell-cycle Progression. Anticancer Res. 2018, 38, 1445–1454. [Google Scholar] [CrossRef]
- Willems, L.; Chapuis, N.; Puissant, A.; Maciel, T.T.; Green, A.S.; Jacque, N.; Vignon, C.; Park, S.; Guichard, S.; Herault, O.; et al. The dual mTORC1 and mTORC2 inhibitor AZD8055 has anti-tumor activity in acute myeloid leukemia. Leukemia 2012, 26, 1195–1202. [Google Scholar] [CrossRef] [Green Version]
- Ewald, F.; Norz, D.; Grottke, A.; Bach, J.; Herzberger, C.; Hofmann, B.T.; Nashan, B.; Jucker, M. Vertical Targeting of AKT and mTOR as Well as Dual Targeting of AKT and MEK Signaling Is Synergistic in Hepatocellular Carcinoma. J. Cancer 2015, 6, 1195–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.M.; Guo, C.L.; Shi, J.J.; Xu, Y.C.; Chen, Y.; Shen, Y.Y.; Su, Y.; Ding, J.; Meng, L.H. HSP90 inhibitor AUY922 abrogates up-regulation of RTKs by mTOR inhibitor AZD8055 and potentiates its antiproliferative activity in human breast cancer. Int. J. Cancer 2014, 135, 2462–2474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, F.; Zhang, Y.D.; Geng, L.; Zhang, P.; Wang, G.Y.; Liu, Y. mTOR Inhibition Induces EGFR Feedback Activation in Association with Its Resistance to Human Pancreatic Cancer. Int. J. Mol. Sci. 2015, 16, 3267–3282. [Google Scholar] [CrossRef] [Green Version]
- Bailey, S.T.; Zhou, B.; Damrauer, J.S.; Krishnan, B.; Wilson, H.L.; Smith, A.M.; Li, M.; Yeh, J.J.; Kim, W.Y. mTOR inhibition induces compensatory, therapeutically targetable MEK activation in renal cell carcinoma. PLoS ONE 2014, 9, e104413. [Google Scholar] [CrossRef] [Green Version]
- Hoang, B.; Benavides, A.; Shi, Y.; Yang, Y.; Frost, P.; Gera, J.; Lichtenstein, A. The PP242 mammalian target of rapamycin (mTOR) inhibitor activates extracellular signal-regulated kinase (ERK) in multiple myeloma cells via a target of rapamycin complex 1 (TORC1)/eukaryotic translation initiation factor 4E (eIF-4E)/RAF pathway and activation is a mechanism of resistance. J. Biol. Chem. 2012, 287, 21796–21805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The integrated stress response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef] [Green Version]
- Elvira, R.; Cha, S.J.; Noh, G.M.; Kim, K.; Han, J. PERK-Mediated eIF2alpha Phosphorylation Contributes to The Protection of Dopaminergic Neurons from Chronic Heat Stress in Drosophila. Int. J. Mol. Sci. 2020, 21, 845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cano-Gonzalez, A.; Mauro-Lizcano, M.; Iglesias-Serret, D.; Gil, J.; Lopez-Rivas, A. Involvement of both caspase-8 and Noxa-activated pathways in endoplasmic reticulum stress-induced apoptosis in triple-negative breast tumor cells. Cell Death Dis. 2018, 9, 134. [Google Scholar] [CrossRef] [Green Version]
- Petigny-Lechartier, C.; Duboc, C.; Jebahi, A.; Louis, M.H.; Abeilard, E.; Denoyelle, C.; Gauduchon, P.; Poulain, L.; Villedieu, M. The mTORC1/2 Inhibitor AZD8055 Strengthens the Efficiency of the MEK Inhibitor Trametinib to Reduce the Mcl-1/[Bim and Puma] ratio and to Sensitize Ovarian Carcinoma Cells to ABT-737. Mol. Cancer Ther. 2017, 16, 102–115. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.Q.; Toyoda, H.; Qi, L.; Morimoto, M.; Hanaki, R.; Iwamoto, S.; Komada, Y.; Hirayama, M. Induction of MEK/ERK activity by AZD8055 confers acquired resistance in neuroblastoma. Biochem. Biophys. Res. Commun. 2018, 499, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.K.; Park, J.I. MEK1/2 Inhibitors: Molecular Activity and Resistance Mechanisms. Semin. Oncol. 2015, 42, 849–862. [Google Scholar] [CrossRef] [Green Version]
- Monick, M.M.; Powers, L.S.; Gross, T.J.; Flaherty, D.M.; Barrett, C.W.; Hunninghake, G.W. Active ERK contributes to protein translation by preventing JNK-dependent inhibition of protein phosphatase 1. J. Immunol. 2006, 177, 1636–1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rolli-Derkinderen, M.; Machavoine, F.; Baraban, J.M.; Grolleau, A.; Beretta, L.; Dy, M. ERK and p38 inhibit the expression of 4E-BP1 repressor of translation through induction of Egr-1. J. Biol. Chem. 2003, 278, 18859–18867. [Google Scholar] [CrossRef] [Green Version]
- Yanagiya, A.; Suyama, E.; Adachi, H.; Svitkin, Y.V.; Aza-Blanc, P.; Imataka, H.; Mikami, S.; Martineau, Y.; Ronai, Z.A.; Sonenberg, N. Translational homeostasis via the mRNA cap-binding protein, eIF4E. Mol. Cell 2012, 46, 847–858. [Google Scholar] [CrossRef] [Green Version]
- Cope, C.L.; Gilley, R.; Balmanno, K.; Sale, M.J.; Howarth, K.D.; Hampson, M.; Smith, P.D.; Guichard, S.M.; Cook, S.J. Adaptation to mTOR kinase inhibitors by amplification of eIF4E to maintain cap-dependent translation. J. Cell Sci. 2014, 127, 788–800. [Google Scholar] [CrossRef] [Green Version]
- Banko, J.L.; Poulin, F.; Hou, L.; DeMaria, C.T.; Sonenberg, N.; Klann, E. The translation repressor 4E-BP2 is critical for eIF4F complex formation, synaptic plasticity, and memory in the hippocampus. J. Neurosci. Off. J. Soc. Neurosci. 2005, 25, 9581–9590. [Google Scholar] [CrossRef]
- Tsukiyama-Kohara, K.; Vidal, S.M.; Gingras, A.C.; Glover, T.W.; Hanash, S.M.; Heng, H.; Sonenberg, N. Tissue distribution, genomic structure, and chromosome mapping of mouse and human eukaryotic initiation factor 4E-binding proteins 1 and 2. Genomics 1996, 38, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Tamburini, J.; Green, A.S.; Bardet, V.; Chapuis, N.; Park, S.; Willems, L.; Uzunov, M.; Ifrah, N.; Dreyfus, F.; Lacombe, C.; et al. Protein synthesis is resistant to rapamycin and constitutes a promising therapeutic target in acute myeloid leukemia. Blood 2009, 114, 1618–1627. [Google Scholar] [CrossRef] [Green Version]
- Graff, J.R.; Konicek, B.W.; Vincent, T.M.; Lynch, R.L.; Monteith, D.; Weir, S.N.; Schwier, P.; Capen, A.; Goode, R.L.; Dowless, M.S.; et al. Therapeutic suppression of translation initiation factor eIF4E expression reduces tumor growth without toxicity. J. Clin. Investig. 2007, 117, 2638–2648. [Google Scholar] [CrossRef]
- Schmieder, R.; Puehler, F.; Neuhaus, R.; Kissel, M.; Adjei, A.A.; Miner, J.N.; Mumberg, D.; Ziegelbauer, K.; Scholz, A. Allosteric MEK1/2 inhibitor refametinib (BAY 86-9766) in combination with sorafenib exhibits antitumor activity in preclinical murine and rat models of hepatocellular carcinoma. Neoplasia 2013, 15, 1161–1171. [Google Scholar] [CrossRef]
- Lim, H.Y.; Heo, J.; Choi, H.J.; Lin, C.Y.; Yoon, J.H.; Hsu, C.; Rau, K.M.; Poon, R.T.; Yeo, W.; Park, J.W.; et al. A phase II study of the efficacy and safety of the combination therapy of the MEK inhibitor refametinib (BAY 86-9766) plus sorafenib for Asian patients with unresectable hepatocellular carcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 5976–5985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huynh, H.; Ong, R.; Goh, K.Y.; Lee, L.Y.; Puehler, F.; Scholz, A.; Politz, O.; Mumberg, D.; Ziegelbauer, K. Sorafenib/MEK inhibitor combination inhibits tumor growth and the Wnt/betacatenin pathway in xenograft models of hepatocellular carcinoma. Int. J. Oncol. 2019, 54, 1123–1133. [Google Scholar] [CrossRef] [PubMed]
- Holt, S.V.; Logie, A.; Davies, B.R.; Alferez, D.; Runswick, S.; Fenton, S.; Chresta, C.M.; Gu, Y.; Zhang, J.; Wu, Y.L.; et al. Enhanced apoptosis and tumor growth suppression elicited by combination of MEK (selumetinib) and mTOR kinase inhibitors (AZD8055). Cancer Res. 2012, 72, 1804–1813. [Google Scholar] [CrossRef] [Green Version]
- Fey, D.; Croucher, D.R.; Kolch, W.; Kholodenko, B.N. Crosstalk and signaling switches in mitogen-activated protein kinase cascades. Front. Physiol. 2012, 3, 355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muranen, T.; Selfors, L.M.; Hwang, J.; Gallegos, L.L.; Coloff, J.L.; Thoreen, C.C.; Kang, S.A.; Sabatini, D.M.; Mills, G.B.; Brugge, J.S. ERK and p38 MAPK Activities Determine Sensitivity to PI3K/mTOR Inhibition via Regulation of MYC and YAP. Cancer Res. 2016, 76, 7168–7180. [Google Scholar] [CrossRef] [Green Version]
- Aguirre-Ghiso, J.A.; Estrada, Y.; Liu, D.; Ossowski, L. ERKMAPK activity as a determinant of tumor growth and dormancy; Regulation by p38(SAPK). Cancer Res. 2003, 63, 1684–1695. [Google Scholar]
- Gingras, A.C.; Kennedy, S.G.; O’Leary, M.A.; Sonenberg, N.; Hay, N. 4E-BP1, a repressor of mRNA translation, is phosphorylated and inactivated by the Akt(PKB) signaling pathway. Genes Dev. 1998, 12, 502–513. [Google Scholar] [CrossRef]
- Graff, J.R.; Konicek, B.W.; Carter, J.H.; Marcusson, E.G. Targeting the eukaryotic translation initiation factor 4E for cancer therapy. Cancer Res. 2008, 68, 631–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- She, Q.B.; Halilovic, E.; Ye, Q.; Zhen, W.; Shirasawa, S.; Sasazuki, T.; Solit, D.B.; Rosen, N. 4E-BP1 is a key effector of the oncogenic activation of the AKT and ERK signaling pathways that integrates their function in tumors. Cancer Cell 2010, 18, 39–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ducker, G.S.; Atreya, C.E.; Simko, J.P.; Hom, Y.K.; Matli, M.R.; Benes, C.H.; Hann, B.; Nakakura, E.K.; Bergsland, E.K.; Donner, D.B.; et al. Incomplete inhibition of phosphorylation of 4E-BP1 as a mechanism of primary resistance to ATP-competitive mTOR inhibitors. Oncogene 2014, 33, 1590–1600. [Google Scholar] [CrossRef] [Green Version]
- Alain, T.; Morita, M.; Fonseca, B.D.; Yanagiya, A.; Siddiqui, N.; Bhat, M.; Zammit, D.; Marcus, V.; Metrakos, P.; Voyer, L.A.; et al. eIF4E/4E-BP Ratio Predicts the Efficacy of mTOR Targeted Therapies. Cancer Res. 2012, 72, 6468–6476. [Google Scholar] [CrossRef] [Green Version]
- Fadden, P.; Haystead, T.A.; Lawrence, J.C., Jr. Identification of phosphorylation sites in the translational regulator, PHAS-I, that are controlled by insulin and rapamycin in rat adipocytes. J. Biol. Chem. 1997, 272, 10240–10247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, T.A.; Kong, X.; Saltiel, A.R.; Blackshear, P.J.; Lawrence, J.C., Jr. Control of PHAS-I by insulin in 3T3-L1 adipocytes. Synthesis, degradation, and phosphorylation by a rapamycin-sensitive and mitogen-activated protein kinase-independent pathway. J. Biol. Chem. 1995, 270, 18531–18538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elia, A.; Constantinou, C.; Clemens, M.J. Effects of protein phosphorylation on ubiquitination and stability of the translational inhibitor protein 4E-BP1. Oncogene 2008, 27, 811–822. [Google Scholar] [CrossRef] [Green Version]
- De Benedetti, A.; Graff, J.R. eIF-4E expression and its role in malignancies and metastases. Oncogene 2004, 23, 3189–3199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, M.; Cho, J.; Cho, W.S.; Shin, G.C.; Lee, K. The Glucosamine-Mediated Induction of CHOP Reduces the Expression of Inflammatory Cytokines by Modulating JNK and NF-kappa B in LPS-Stimulated RAW264.7 Cells. Genes Genom. 2009, 31, 251–260. [Google Scholar] [CrossRef]
- Han, J.; Back, S.H.; Hur, J.; Lin, Y.H.; Gildersleeve, R.; Shan, J.; Yuan, C.L.; Krokowski, D.; Wang, S.; Hatzoglou, M.; et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 2013, 15, 481–490. [Google Scholar] [CrossRef]
- Shin, J.I.; Jeon, Y.J.; Lee, S.; Lee, Y.G.; Kim, J.B.; Lee, K. G-Protein-Coupled Receptor 120 Mediates DHA-Induced Apoptosis by Regulating IP3R, ROS and, ER Stress Levels in Cisplatin-Resistant Cancer Cells. Mol. Cells 2019, 42, 252–261. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.I.; Jeon, Y.J.; Lee, S.; Lee, Y.G.; Kim, J.B.; Kwon, H.C.; Kim, S.H.; Kim, I.; Lee, K.; Han, Y.S. Apoptotic and Anti-Inflammatory Effects of Eupatorium japonicum Thunb. in Rheumatoid Arthritis Fibroblast-Like Synoviocytes. BioMed Res. Int. 2018, 2018, 1383697. [Google Scholar] [CrossRef] [Green Version]
- Liang, S.; Guo, R.; Zhang, Z.; Liu, D.; Xu, H.; Xu, Z.; Wang, X.; Yang, L. Upregulation of the eIF4E signaling pathway contributes to the progression of gastric cancer, and targeting eIF4E by perifosine inhibits cell growth. Oncol. Rep. 2013, 29, 2422–2430. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jee, H.-y.; Lee, Y.-G.; Lee, S.; Elvira, R.; Seo, H.-e.; Lee, J.-Y.; Han, J.; Lee, K. Activation of ERK and p38 Reduces AZD8055-Mediated Inhibition of Protein Synthesis in Hepatocellular Carcinoma HepG2 Cell Line. Int. J. Mol. Sci. 2021, 22, 11824. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222111824
Jee H-y, Lee Y-G, Lee S, Elvira R, Seo H-e, Lee J-Y, Han J, Lee K. Activation of ERK and p38 Reduces AZD8055-Mediated Inhibition of Protein Synthesis in Hepatocellular Carcinoma HepG2 Cell Line. International Journal of Molecular Sciences. 2021; 22(21):11824. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222111824
Chicago/Turabian StyleJee, Ha-yeon, Yoon-Gyeong Lee, Sol Lee, Rosalie Elvira, Hye-eun Seo, Ji-Yeon Lee, Jaeseok Han, and Kyungho Lee. 2021. "Activation of ERK and p38 Reduces AZD8055-Mediated Inhibition of Protein Synthesis in Hepatocellular Carcinoma HepG2 Cell Line" International Journal of Molecular Sciences 22, no. 21: 11824. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222111824