
Pathogenic Basis of Thromboinflammation and Endothelial Injury in COVID-19: Current Findings and Therapeutic Implications


Abstract
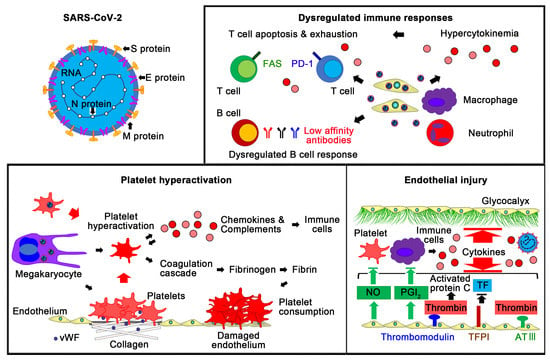
:
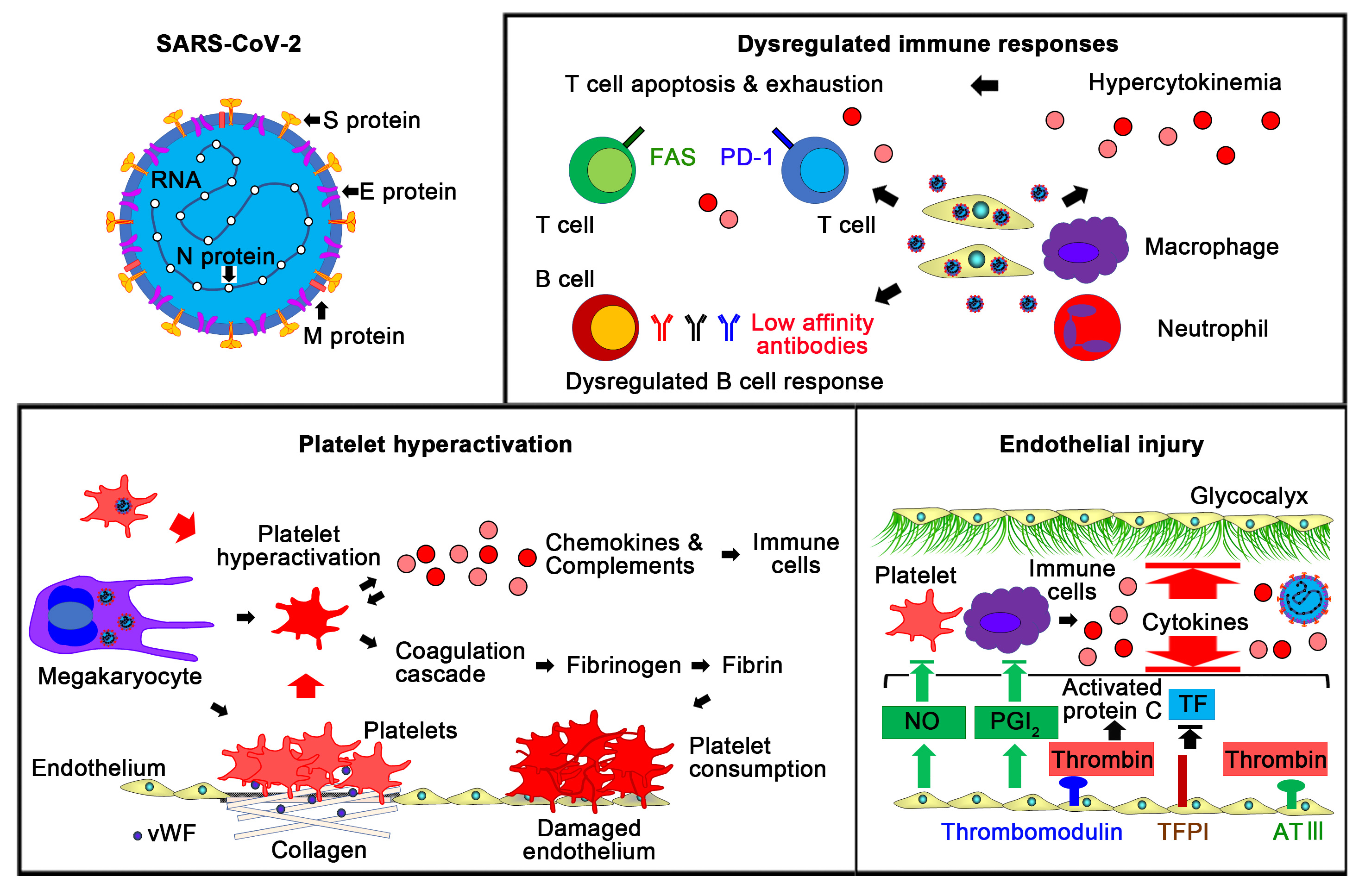{kind=link}
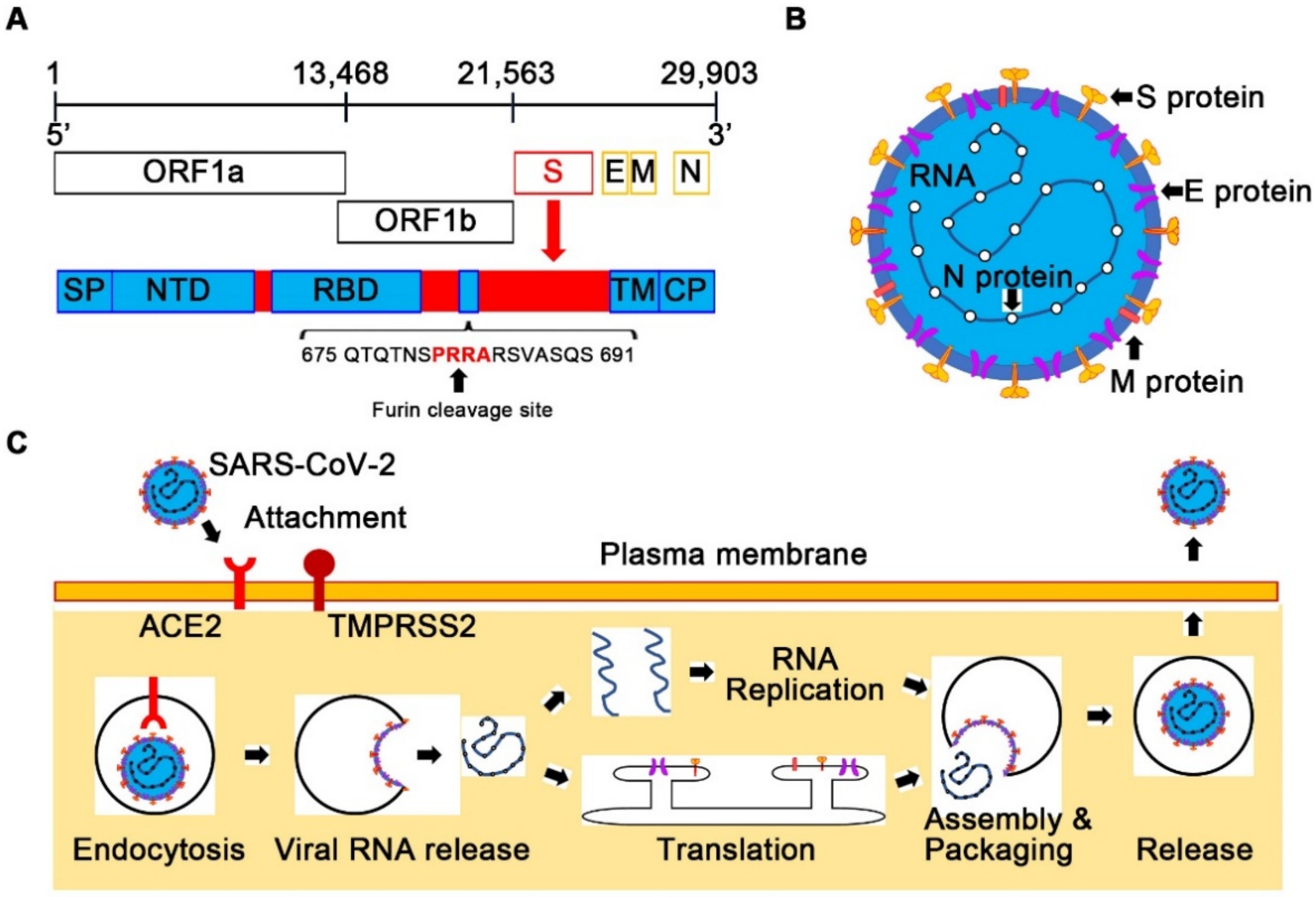{kind=link}
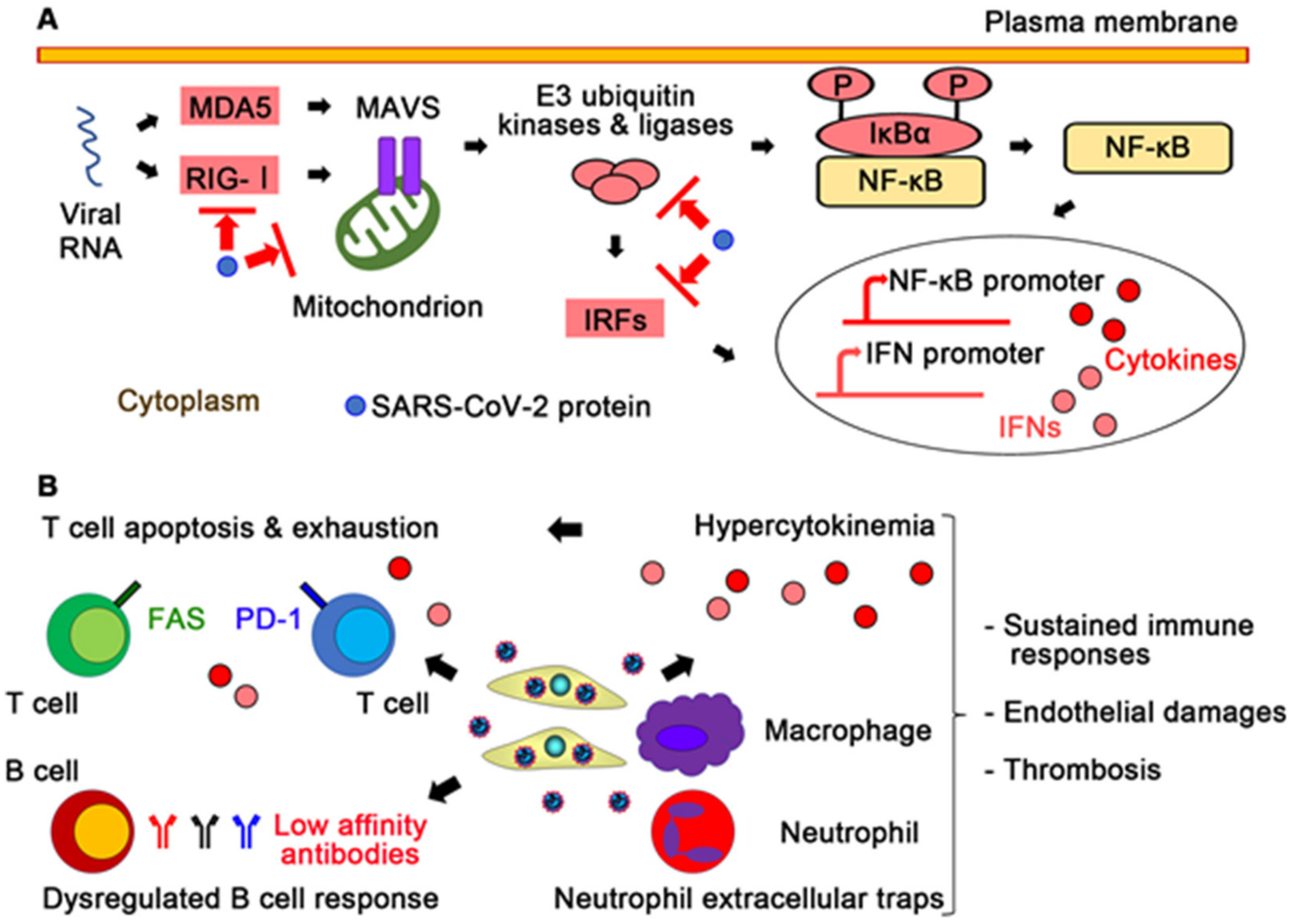{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Genome Composition, Structure, and Lifecycle of SARS-CoV-2
3. Dysregulated Immune Responses in COVID-19
3.1. The Innate Immune System and COVID-19
3.2. The Adaptive Immune System and COVID-19
4. Platelet Activation and the Coagulation Cascade in COVID-19
4.1. Platelet Dysfunction and COVID-19
4.2. Coagulation Cascade and COVID-19
5. Endothelial Dysfunction in COVID-19
5.1. ACE2 and Endothelial Dysfunction in COVID-19
5.2. The Immune System and Endothelial Dysfunction in COVID-19
6. Cardiovascular Risk Factors and COVID-19
7. Therapeutic Approaches Targeting Thromboinflammation and Endothelial Injury in COVID-19
7.1. Targeting Dysregulated Immune Responses
7.2. Targeting Platelet Hyperactivation and Coagulation Cascade
7.3. Targeting Endothelial Dysfunction
8. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Hou, Y.J.; Okuda, K.; Edwards, C.E.; Martinez, D.R.; Asakura, T.; Dinnon, K.H., 3rd; Kato, T.; Lee, R.E.; Yount, B.L.; Mascenik, T.M.; et al. SARS-CoV-2 Reverse Genetics Reveals a Variable Infection Gradient in the Respiratory Tract. Cell 2020, 182, 429–446.e14. [Google Scholar] [CrossRef] [PubMed]
- Perico, L.; Benigni, A.; Casiraghi, F.; Ng, L.F.P.; Renia, L.; Remuzzi, G. Immunity, endothelial injury and complement-induced coagulopathy in COVID-19. Nat. Rev. Nephrol. 2021, 17, 46–64. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021, 19, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.X.; Tyagi, T.; Jain, K.; Gu, V.W.; Lee, S.H.; Hwa, J.M.; Kwan, J.M.; Krause, D.S.; Lee, A.I.; Halene, S.; et al. Thrombocytopathy and endotheliopathy: Crucial contributors to COVID-19 thromboinflammation. Nat. Rev. Cardiol. 2021, 18, 194–209. [Google Scholar] [CrossRef]
- Cui, S.; Chen, S.; Li, X.; Liu, S.; Wang, F. Prevalence of venous thromboembolism in patients with severe novel coronavirus pneumonia. J. Thromb. Haemost. 2020, 18, 1421–1424. [Google Scholar] [CrossRef]
- Ren, B.; Yan, F.; Deng, Z.; Zhang, S.; Xiao, L.; Wu, M.; Cai, L. Extremely High Incidence of Lower Extremity Deep Venous Thrombosis in 48 Patients With Severe COVID-19 in Wuhan. Circulation 2020, 142, 181–183. [Google Scholar] [CrossRef]
- Zhang, L.; Feng, X.; Zhang, D.; Jiang, C.; Mei, H.; Wang, J.; Zhang, C.; Li, H.; Xia, X.; Kong, S.; et al. Deep Vein Thrombosis in Hospitalized Patients With COVID-19 in Wuhan, China: Prevalence, Risk Factors, and Outcome. Circulation 2020, 142, 114–128. [Google Scholar] [CrossRef]
- Klok, F.A.; Kruip, M.; Van der Meer, N.J.M.; Arbous, M.S.; Gommers, D.; Kant, K.M.; Kaptein, F.H.J.; van Paassen, J.; Stals, M.A.M.; Huisman, M.V.; et al. Confirmation of the high cumulative incidence of thrombotic complications in critically ill ICU patients with COVID-19: An updated analysis. Thromb. Res. 2020, 191, 148–150. [Google Scholar] [CrossRef] [PubMed]
- Klok, F.A.; Kruip, M.; Van der Meer, N.J.M.; Arbous, M.S.; Gommers, D.; Kant, K.M.; Kaptein, F.H.J.; van Paassen, J.; Stals, M.A.M.; Huisman, M.V.; et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb. Res. 2020, 191, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Middeldorp, S.; Coppens, M.; Van Haaps, T.F.; Foppen, M.; Vlaar, A.P.; Muller, M.C.A.; Bouman, C.C.S.; Beenen, L.F.M.; Kootte, R.S.; Heijmans, J.; et al. Incidence of venous thromboembolism in hospitalized patients with COVID-19. J. Thromb. Haemost. 2020, 18, 1995–2002. [Google Scholar] [CrossRef]
- Poissy, J.; Goutay, J.; Caplan, M.; Parmentier, E.; Duburcq, T.; Lassalle, F.; Jeanpierre, E.; Rauch, A.; Labreuche, J.; Susen, S.; et al. Pulmonary Embolism in Patients With COVID-19: Awareness of an Increased Prevalence. Circulation 2020, 142, 184–186. [Google Scholar] [CrossRef]
- Helms, J.; Tacquard, C.; Severac, F.; Leonard-Lorant, I.; Ohana, M.; Delabranche, X.; Merdji, H.; Clere-Jehl, R.; Schenck, M.; Fagot Gandet, F.; et al. High risk of thrombosis in patients with severe SARS-CoV-2 infection: A multicenter prospective cohort study. Intensive Care Med. 2020, 46, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Nahum, J.; Morichau-Beauchant, T.; Daviaud, F.; Echegut, P.; Fichet, J.; Maillet, J.M.; Thierry, S. Venous Thrombosis Among Critically Ill Patients With Coronavirus Disease 2019 (COVID-19). JAMA Netw. Open 2020, 3, e2010478. [Google Scholar] [CrossRef]
- Lodigiani, C.; Iapichino, G.; Carenzo, L.; Cecconi, M.; Ferrazzi, P.; Sebastian, T.; Kucher, N.; Studt, J.D.; Sacco, C.; Bertuzzi, A.; et al. Venous and arterial thromboembolic complications in COVID-19 patients admitted to an academic hospital in Milan, Italy. Thromb. Res. 2020, 191, 9–14. [Google Scholar] [CrossRef]
- Mai, V.; Tan, B.K.; Mainbourg, S.; Potus, F.; Cucherat, M.; Lega, J.C.; Provencher, S. Venous thromboembolism in COVID-19 compared to non-COVID-19 cohorts: A systematic review with meta-analysis. Vascul. Pharmacol. 2021, 139, 106882. [Google Scholar] [CrossRef]
- Stefanini, G.G.; Montorfano, M.; Trabattoni, D.; Andreini, D.; Ferrante, G.; Ancona, M.; Metra, M.; Curello, S.; Maffeo, D.; Pero, G.; et al. ST-Elevation Myocardial Infarction in Patients With COVID-19: Clinical and Angiographic Outcomes. Circulation 2020, 141, 2113–2116. [Google Scholar] [CrossRef]
- Bangalore, S.; Sharma, A.; Slotwiner, A.; Yatskar, L.; Harari, R.; Shah, B.; Ibrahim, H.; Friedman, G.H.; Thompson, C.; Alviar, C.L.; et al. ST-Segment Elevation in Patients with Covid-19—A Case Series. N. Engl. J. Med. 2020, 382, 2478–2480. [Google Scholar] [CrossRef]
- Roffi, M.; Guagliumi, G.; Ibanez, B. The Obstacle Course of Reperfusion for ST-Segment-Elevation Myocardial Infarction in the COVID-19 Pandemic. Circulation 2020, 141, 1951–1953. [Google Scholar] [CrossRef] [PubMed]
- DeBaun, M.R. Initiating adjunct low-dose hydroxyurea therapy for stroke prevention in children with SCA during the COVID-19 pandemic. Blood 2020, 135, 1997–1999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oxley, T.J.; Mocco, J.; Majidi, S.; Kellner, C.P.; Shoirah, H.; Singh, I.P.; De Leacy, R.A.; Shigematsu, T.; Ladner, T.R.; Yaeger, K.A.; et al. Large-Vessel Stroke as a Presenting Feature of Covid-19 in the Young. N. Engl. J. Med. 2020, 382, e60. [Google Scholar] [CrossRef] [PubMed]
- Bellosta, R.; Luzzani, L.; Natalini, G.; Pegorer, M.A.; Attisani, L.; Cossu, L.G.; Ferrandina, C.; Fossati, A.; Conti, E.; Bush, R.L.; et al. Acute limb ischemia in patients with COVID-19 pneumonia. J. Vasc. Surg. 2020, 72, 1864–1872. [Google Scholar] [CrossRef] [PubMed]
- Wichmann, D.; Sperhake, J.P.; Lutgehetmann, M.; Steurer, S.; Edler, C.; Heinemann, A.; Heinrich, F.; Mushumba, H.; Kniep, I.; Schroder, A.S.; et al. Autopsy Findings and Venous Thromboembolism in Patients With COVID-19: A Prospective Cohort Study. Ann. Intern. Med. 2020, 173, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Dolhnikoff, M.; Duarte-Neto, A.N.; De Almeida Monteiro, R.A.; Da Silva, L.F.F.; De Oliveira, E.P.; Saldiva, P.H.N.; Mauad, T.; Negri, E.M. Pathological evidence of pulmonary thrombotic phenomena in severe COVID-19. J. Thromb. Haemost. 2020, 18, 1517–1519. [Google Scholar] [CrossRef] [Green Version]
- Carsana, L.; Sonzogni, A.; Nasr, A.; Rossi, R.S.; Pellegrinelli, A.; Zerbi, P.; Rech, R.; Colombo, R.; Antinori, S.; Corbellino, M.; et al. Pulmonary post-mortem findings in a series of COVID-19 cases from northern Italy: A two-centre descriptive study. Lancet Infect. Dis. 2020, 20, 1135–1140. [Google Scholar] [CrossRef]
- Menter, T.; Haslbauer, J.D.; Nienhold, R.; Savic, S.; Hopfer, H.; Deigendesch, N.; Frank, S.; Turek, D.; Willi, N.; Pargger, H.; et al. Postmortem examination of COVID-19 patients reveals diffuse alveolar damage with severe capillary congestion and variegated findings in lungs and other organs suggesting vascular dysfunction. Histopathology 2020, 77, 198–209. [Google Scholar] [CrossRef]
- Fox, S.E.; Akmatbekov, A.; Harbert, J.L.; Li, G.; Quincy Brown, J.; Vander Heide, R.S. Pulmonary and cardiac pathology in African American patients with COVID-19: An autopsy series from New Orleans. Lancet Respir. Med. 2020, 8, 681–686. [Google Scholar] [CrossRef]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef]
- Rapkiewicz, A.V.; Mai, X.; Carsons, S.E.; Pittaluga, S.; Kleiner, D.E.; Berger, J.S.; Thomas, S.; Adler, N.M.; Charytan, D.M.; Gasmi, B.; et al. Megakaryocytes and platelet-fibrin thrombi characterize multi-organ thrombosis at autopsy in COVID-19: A case series. EClinicalMedicine 2020, 24, 100434. [Google Scholar] [CrossRef] [PubMed]
- Peiris, J.S.; Yuen, K.Y.; Osterhaus, A.D.; Stohr, K. The severe acute respiratory syndrome. N. Engl. J. Med. 2003, 349, 2431–2441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zumla, A.; Hui, D.S.; Perlman, S. Middle East respiratory syndrome. Lancet 2015, 386, 995–1007. [Google Scholar] [CrossRef] [Green Version]
- De Wit, E.; Van Doremalen, N.; Falzarano, D.; Munster, V.J. SARS and MERS: Recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 2016, 14, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Goyal, P.; Choi, J.J.; Pinheiro, L.C.; Schenck, E.J.; Chen, R.; Jabri, A.; Satlin, M.J.; Campion, T.R., Jr.; Nahid, M.; Ringel, J.B.; et al. Clinical Characteristics of Covid-19 in New York City. N. Engl. J. Med. 2020, 382, 2372–2374. [Google Scholar] [CrossRef] [PubMed]
- Grasselli, G.; Greco, M.; Zanella, A.; Albano, G.; Antonelli, M.; Bellani, G.; Bonanomi, E.; Cabrini, L.; Carlesso, E.; Castelli, G.; et al. COVID-19 Lombardy ICU Network. Risk Factors Associated With Mortality Among Patients With COVID-19 in Intensive Care Units in Lombardy, Italy. JAMA Intern. Med. 2020, 180, 1345–1355. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Ou, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.L.; Hui, D.S.C.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef]
- Blair, P.; Rex, S.; Vitseva, O.; Beaulieu, L.; Tanriverdi, K.; Chakrabarti, S.; Hayashi, C.; Genco, C.A.; Iafrati, M.; Freedman, J.E. Stimulation of Toll-like receptor 2 in human platelets induces a thromboinflammatory response through activation of phosphoinositide 3-kinase. Circ. Res. 2009, 104, 346–354. [Google Scholar] [CrossRef] [Green Version]
- Engelmann, B.; Massberg, S. Thrombosis as an intravascular effector of innate immunity. Nat. Rev. Immunol. 2013, 13, 34–45. [Google Scholar] [CrossRef]
- Guo, L.; Rondina, M.T. The Era of Thromboinflammation: Platelets Are Dynamic Sensors and Effector Cells During Infectious Diseases. Front. Immunol. 2019, 10, 2204. [Google Scholar] [CrossRef] [Green Version]
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef] [PubMed]
- Wrobel, A.G.; Benton, D.J.; Xu, P.; Roustan, C.; Martin, S.R.; Rosenthal, P.B.; Skehel, J.J.; Gamblin, S.J. SARS-CoV-2 and bat RaTG13 spike glycoprotein structures inform on virus evolution and furin-cleavage effects. Nat. Struct. Mol. Biol. 2020, 27, 763–767. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.F.; Kok, K.H.; Zhu, Z.; Chu, H.; To, K.K.; Yuan, S.; Yuen, K.Y. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect. 2020, 9, 221–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sungnak, W.; Huang, N.; Becavin, C.; Berg, M.; Queen, R.; Litvinukova, M.; Talavera-Lopez, C.; Maatz, H.; Reichart, D.; Sampaziotis, F.; et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat. Med. 2020, 26, 681–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukassen, S.; Chua, R.L.; Trefzer, T.; Kahn, N.C.; Schneider, M.A.; Muley, T.; Winter, H.; Meister, M.; Veith, C.; Boots, A.W.; et al. SARS-CoV-2 receptor ACE2 and TMPRSS2 are primarily expressed in bronchial transient secretory cells. EMBO J. 2020, 39, e105114. [Google Scholar] [CrossRef] [PubMed]
- Lucas, C.; Wong, P.; Klein, J.; Castro, T.B.R.; Silva, J.; Sundaram, M.; Ellingson, M.K.; Mao, T.; Oh, J.E.; Israelow, B.; et al. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature 2020, 584, 463–469. [Google Scholar] [CrossRef]
- Giamarellos-Bourboulis, E.J.; Netea, M.G.; Rovina, N.; Akinosoglou, K.; Antoniadou, A.; Antonakos, N.; Damoraki, G.; Gkavogianni, T.; Adami, M.E.; Katsaounou, P.; et al. Complex Immune Dysregulation in COVID-19 Patients with Severe Respiratory Failure. Cell Host Microbe 2020, 27, 992–1000.e3. [Google Scholar] [CrossRef]
- Mathew, D.; Giles, J.R.; Baxter, A.E.; Oldridge, D.A.; Greenplate, A.R.; Wu, J.E.; Alanio, C.; Kuri-Cervantes, L.; Pampena, M.B.; D’Andrea, K.; et al. Deep immune profiling of COVID-19 patients reveals distinct immunotypes with therapeutic implications. Science 2020, 369, eabc8511. [Google Scholar] [CrossRef]
- McKechnie, J.L.; Blish, C.A. The Innate Immune System: Fighting on the Front Lines or Fanning the Flames of COVID-19? Cell Host Microbe 2020, 27, 863–869. [Google Scholar] [CrossRef]
- Higashikuni, Y.; Tanaka, K.; Kato, M.; Nureki, O.; Hirata, Y.; Nagai, R.; Komuro, I.; Sata, M. Toll-like receptor-2 mediates adaptive cardiac hypertrophy in response to pressure overload through interleukin-1beta upregulation via nuclear factor kappaB activation. J. Am. Heart Assoc. 2013, 2, e000267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vabret, N.; Britton, G.J.; Gruber, C.; Hegde, S.; Kim, J.; Kuksin, M.; Levantovsky, R.; Malle, L.; Moreira, A.; Park, M.D.; et al. Immunology of COVID-19: Current State of the Science. Immunity 2020, 52, 910–941. [Google Scholar] [CrossRef] [PubMed]
- Thoms, M.; Buschauer, R.; Ameismeier, M.; Koepke, L.; Denk, T.; Hirschenberger, M.; Kratzat, H.; Hayn, M.; Mackens-Kiani, T.; Cheng, J.; et al. Structural basis for translational shutdown and immune evasion by the Nsp1 protein of SARS-CoV-2. Science 2020, 369, 1249–1255. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.W.; Zhang, H.N.; Meng, Q.F.; Xie, J.; Li, Y.; Chen, H.; Zheng, Y.X.; Wang, X.N.; Qi, H.; Zhang, J.; et al. SARS-CoV-2 Orf9b suppresses type I interferon responses by targeting TOM70. Cell Mol. Immunol. 2020, 17, 998–1000. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, C.; Horner, S.M. MAVS Coordination of Antiviral Innate Immunity. J. Virol. 2015, 89, 6974–6977. [Google Scholar] [CrossRef] [Green Version]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef]
- Hadjadj, J.; Yatim, N.; Barnabei, L.; Corneau, A.; Boussier, J.; Smith, N.; Pere, H.; Charbit, B.; Bondet, V.; Chenevier-Gobeaux, C.; et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 2020, 369, 718–724. [Google Scholar] [CrossRef]
- Li, J.; Guo, M.; Tian, X.; Wang, X.; Yang, X.; Wu, P.; Liu, C.; Xiao, Z.; Qu, Y.; Yin, Y.; et al. Virus-Host Interactome and Proteomic Survey Reveal Potential Virulence Factors Influencing SARS-CoV-2 Pathogenesis. Med 2021, 2, 99–112.e7. [Google Scholar] [CrossRef]
- Trouillet-Assant, S.; Viel, S.; Gaymard, A.; Pons, S.; Richard, J.C.; Perret, M.; Villard, M.; Brengel-Pesce, K.; Lina, B.; Mezidi, M.; et al. Type I IFN immunoprofiling in COVID-19 patients. J. Allergy Clin. Immunol. 2020, 146, 206–208.e2. [Google Scholar] [CrossRef]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J.; HLH Across Speciality Collaboration, UK. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033–1034. [Google Scholar] [CrossRef]
- Tan, L.; Kang, X.; Zhang, B.; Zheng, S.; Liu, B.; Yu, T.; Yang, F.; Wang, Q.; Miao, H. Plasma therapy cured a COVID-19 patient with long duration of viral shedding for 49 days: The clinical features, laboratory tests, plasma therapy, and implications for public health management. MedComm 2020, 1, 77–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bost, P.; Giladi, A.; Liu, Y.; Bendjelal, Y.; Xu, G.; David, E.; Blecher-Gonen, R.; Cohen, M.; Medaglia, C.; Li, H.; et al. Host-Viral Infection Maps Reveal Signatures of Severe COVID-19 Patients. Cell 2020, 181, 1475–1488.e12. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Yi, X.; Sun, Y.; Bi, X.; Du, J.; Zhang, C.; Quan, S.; Zhang, F.; Sun, R.; Qian, L.; et al. Proteomic and Metabolomic Characterization of COVID-19 Patient Sera. Cell 2020, 182, 59–72.e15. [Google Scholar] [CrossRef]
- Barnes, B.J.; Adrover, J.M.; Baxter-Stoltzfus, A.; Borczuk, A.; Cools-Lartigue, J.; Crawford, J.M.; Dassler-Plenker, J.; Guerci, P.; Huynh, C.; Knight, J.S.; et al. Targeting potential drivers of COVID-19: Neutrophil extracellular traps. J. Exp. Med. 2020, 217, e20200652. [Google Scholar] [CrossRef] [PubMed]
- Radermecker, C.; Detrembleur, N.; Guiot, J.; Cavalier, E.; Henket, M.; d’Emal, C.; Vanwinge, C.; Cataldo, D.; Oury, C.; Delvenne, P.; et al. Neutrophil extracellular traps infiltrate the lung airway, interstitial, and vascular compartments in severe COVID-19. J. Exp. Med. 2020, 217, e20201012. [Google Scholar] [CrossRef]
- Iliadi, V.; Konstantinidou, I.; Aftzoglou, K.; Iliadis, S.; Konstantinidis, T.G.; Tsigalou, C. The Emerging Role of Neutrophils in the Pathogenesis of Thrombosis in COVID-19. Int. J. Mol. Sci. 2021, 22, 5368. [Google Scholar] [CrossRef]
- Chen, W.; Pan, J.Y. Anatomical and Pathological Observation and Analysis of SARS and COVID-19: Microthrombosis Is the Main Cause of Death. Biol. Proced. Online 2021, 23, 4. [Google Scholar] [CrossRef]
- Levi, M.; van der Poll, T. Coagulation and sepsis. Thromb. Res. 2017, 149, 38–44. [Google Scholar] [CrossRef]
- Teuwen, L.A.; Geldhof, V.; Pasut, A.; Carmeliet, P. COVID-19: The vasculature unleashed. Nat. Rev. Immunol. 2020, 20, 389–391. [Google Scholar] [CrossRef]
- Shi, Y.; Gauer, J.S.; Baker, S.R.; Philippou, H.; Connell, S.D.; Ariens, R.A.S. Neutrophils can promote clotting via FXI and impact clot structure via neutrophil extracellular traps in a distinctive manner in vitro. Sci. Rep. 2021, 11, 1718. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Xie, J.; Zhao, L.; Fei, X.; Zhang, H.; Tan, Y.; Nie, X.; Zhou, L.; Liu, Z.; Ren, Y.; et al. Alveolar macrophage dysfunction and cytokine storm in the pathogenesis of two severe COVID-19 patients. EBioMedicine 2020, 57, 102833. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yu, Y.; Xu, J.; Shu, H.; Xia, J.; Liu, H.; Wu, Y.; Zhang, L.; Yu, Z.; Fang, M.; et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: A single-centered, retrospective, observational study. Lancet Respir. Med. 2020, 8, 475–481. [Google Scholar] [CrossRef] [Green Version]
- Zheng, M.; Gao, Y.; Wang, G.; Song, G.; Liu, S.; Sun, D.; Xu, Y.; Tian, Z. Functional exhaustion of antiviral lymphocytes in COVID-19 patients. Cell. Mol. Immunol. 2020, 17, 533–535. [Google Scholar] [CrossRef] [Green Version]
- Diao, B.; Wang, C.; Tan, Y.; Chen, X.; Liu, Y.; Ning, L.; Chen, L.; Li, M.; Liu, Y.; Wang, G.; et al. Reduction and Functional Exhaustion of T Cells in Patients With Coronavirus Disease 2019 (COVID-19). Front. Immunol. 2020, 11, 827. [Google Scholar] [CrossRef] [PubMed]
- Bellesi, S.; Metafuni, E.; Hohaus, S.; Maiolo, E.; Marchionni, F.; D’Innocenzo, S.; La Sorda, M.; Ferraironi, M.; Ramundo, F.; Fantoni, M.; et al. Increased CD95 (Fas) and PD-1 expression in peripheral blood T lymphocytes in COVID-19 patients. Br. J. Haematol. 2020, 191, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Grifoni, A.; Weiskopf, D.; Ramirez, S.I.; Mateus, J.; Dan, J.M.; Moderbacher, C.R.; Rawlings, S.A.; Sutherland, A.; Premkumar, L.; Jadi, R.S.; et al. Targets of T Cell Responses to SARS-CoV-2 Coronavirus in Humans with COVID-19 Disease and Unexposed Individuals. Cell 2020, 181, 1489–1501.e15. [Google Scholar] [CrossRef]
- Mateus, J.; Grifoni, A.; Tarke, A.; Sidney, J.; Ramirez, S.I.; Dan, J.M.; Burger, Z.C.; Rawlings, S.A.; Smith, D.M.; Phillips, E.; et al. Selective and cross-reactive SARS-CoV-2 T cell epitopes in unexposed humans. Science 2020, 370, 89–94. [Google Scholar] [CrossRef]
- Kuri-Cervantes, L.; Pampena, M.B.; Meng, W.; Rosenfeld, A.M.; Ittner, C.A.G.; Weisman, A.R.; Agyekum, R.S.; Mathew, D.; Baxter, A.E.; Vella, L.A.; et al. Comprehensive mapping of immune perturbations associated with severe COVID-19. Sci. Immunol. 2020, 5, eabd7114. [Google Scholar] [CrossRef]
- Wilk, A.J.; Rustagi, A.; Zhao, N.Q.; Roque, J.; Martinez-Colon, G.J.; McKechnie, J.L.; Ivison, G.T.; Ranganath, T.; Vergara, R.; Hollis, T.; et al. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat. Med. 2020, 26, 1070–1076. [Google Scholar] [CrossRef]
- Long, Q.X.; Tang, X.J.; Shi, Q.L.; Li, Q.; Deng, H.J.; Yuan, J.; Hu, J.L.; Xu, W.; Zhang, Y.; Lv, F.J.; et al. Clinical and immunological assessment of asymptomatic SARS-CoV-2 infections. Nat. Med. 2020, 26, 1200–1204. [Google Scholar] [CrossRef] [PubMed]
- Amrun, S.N.; Lee, C.Y.; Lee, B.; Fong, S.W.; Young, B.E.; Chee, R.S.; Yeo, N.K.; Torres-Ruesta, A.; Carissimo, G.; Poh, C.M.; et al. Linear B-cell epitopes in the spike and nucleocapsid proteins as markers of SARS-CoV-2 exposure and disease severity. EBioMedicine 2020, 58, 102911. [Google Scholar] [CrossRef] [PubMed]
- Seydoux, E.; Homad, L.J.; MacCamy, A.J.; Parks, K.R.; Hurlburt, N.K.; Jennewein, M.F.; Akins, N.R.; Stuart, A.B.; Wan, Y.H.; Feng, J.; et al. Analysis of a SARS-CoV-2-Infected Individual Reveals Development of Potent Neutralizing Antibodies with Limited Somatic Mutation. Immunity 2020, 53, 98–105.e5. [Google Scholar] [CrossRef] [PubMed]
- Juno, J.A.; Tan, H.X.; Lee, W.S.; Reynaldi, A.; Kelly, H.G.; Wragg, K.; Esterbauer, R.; Kent, H.E.; Batten, C.J.; Mordant, F.L.; et al. Humoral and circulating follicular helper T cell responses in recovered patients with COVID-19. Nat. Med. 2020, 26, 1428–1434. [Google Scholar] [CrossRef] [PubMed]
- Robbiani, D.F.; Gaebler, C.; Muecksch, F.; Lorenzi, J.C.C.; Wang, Z.; Cho, A.; Agudelo, M.; Barnes, C.O.; Gazumyan, A.; Finkin, S.; et al. Convergent antibody responses to SARS-CoV-2 in convalescent individuals. Nature 2020, 584, 437–442. [Google Scholar] [CrossRef]
- Koupenova, M.; Freedman, J.E. Platelets and Immunity: Going Viral. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 1605–1607. [Google Scholar] [CrossRef]
- Taus, F.; Salvagno, G.; Cane, S.; Fava, C.; Mazzaferri, F.; Carrara, E.; Petrova, V.; Barouni, R.M.; Dima, F.; Dalbeni, A.; et al. Platelets Promote Thromboinflammation in SARS-CoV-2 Pneumonia. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2975–2989. [Google Scholar] [CrossRef]
- Zaid, Y.; Puhm, F.; Allaeys, I.; Naya, A.; Oudghiri, M.; Khalki, L.; Limami, Y.; Zaid, N.; Sadki, K.; Ben El Haj, R.; et al. Platelets Can Associate with SARS-Cov-2 RNA and Are Hyperactivated in COVID-19. Circ. Res. 2020, 127, 1404–1418. [Google Scholar] [CrossRef]
- Lindemann, S.; Tolley, N.D.; Dixon, D.A.; McIntyre, T.M.; Prescott, S.M.; Zimmerman, G.A.; Weyrich, A.S. Activated platelets mediate inflammatory signaling by regulated interleukin 1beta synthesis. J. Cell Biol. 2001, 154, 485–490. [Google Scholar] [CrossRef] [Green Version]
- Weyrich, A.S.; Dixon, D.A.; Pabla, R.; Elstad, M.R.; McIntyre, T.M.; Prescott, S.M.; Zimmerman, G.A. Signal-dependent translation of a regulatory protein, Bcl-3, in activated human platelets. Proc. Natl. Acad. Sci. USA 1998, 95, 5556–5561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chae, W.J.; Ehrlich, A.K.; Chan, P.Y.; Teixeira, A.M.; Henegariu, O.; Hao, L.; Shin, J.H.; Park, J.H.; Tang, W.H.; Kim, S.T.; et al. The Wnt Antagonist Dickkopf-1 Promotes Pathological Type 2 Cell-Mediated Inflammation. Immunity 2016, 44, 246–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maquet, J.; Lafaurie, M.; Sommet, A.; Moulis, G. Thrombocytopenia is independently associated with poor outcome in patients hospitalized for COVID-19. Br. J. Haematol. 2020, 190, e276–e279. [Google Scholar] [CrossRef] [PubMed]
- Al-Samkari, H.; Karp Leaf, R.S.; Dzik, W.H.; Carlson, J.C.T.; Fogerty, A.E.; Waheed, A.; Goodarzi, K.; Bendapudi, P.K.; Bornikova, L.; Gupta, S.; et al. COVID-19 and coagulation: Bleeding and thrombotic manifestations of SARS-CoV-2 infection. Blood 2020, 136, 489–500. [Google Scholar] [CrossRef]
- Favaloro, E.J.; Henry, B.M.; Lippi, G. Increased VWF and Decreased ADAMTS-13 in COVID-19: Creating a Milieu for (Micro)Thrombosis. Semin. Thromb. Hemost. 2021, 47, 400–418. [Google Scholar]
- Gavriilaki, E.; Asteris, P.G.; Touloumenidou, T.; Koravou, E.E.; Koutra, M.; Papayanni, P.G.; Karali, V.; Papalexandri, A.; Varelas, C.; Chatzopoulou, F.; et al. Genetic justification of severe COVID-19 using a rigorous algorithm. Clin. Immunol. 2021, 226, 108726. [Google Scholar] [CrossRef]
- Manne, B.K.; Denorme, F.; Middleton, E.A.; Portier, I.; Rowley, J.W.; Stubben, C.; Petrey, A.C.; Tolley, N.D.; Guo, L.; Cody, M.; et al. Platelet gene expression and function in patients with COVID-19. Blood 2020, 136, 1317–1329. [Google Scholar] [CrossRef] [PubMed]
- Rahman, N.T.; Schulz, V.P.; Wang, L.; Gallagher, P.G.; Denisenko, O.; Gualdrini, F.; Esnault, C.; Krause, D.S. MRTFA augments megakaryocyte maturation by enhancing the SRF regulatory axis. Blood Adv. 2018, 2, 2691–2703. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, Y.; Wang, X.; Yang, L.; Li, H.; Wang, Y.; Liu, M.; Zhao, X.; Xie, Y.; Yang, Y.; et al. SARS-CoV-2 binds platelet ACE2 to enhance thrombosis in COVID-19. J. Hematol. Oncol. 2020, 13, 120. [Google Scholar] [CrossRef]
- Koupenova, M.; Corkrey, H.A.; Vitseva, O.; Tanriverdi, K.; Somasundaran, M.; Liu, P.; Soofi, S.; Bhandari, R.; Godwin, M.; Parsi, K.M.; et al. SARS-CoV-2 Initiates Programmed Cell Death in Platelets. Circ. Res. 2021, 129, 631–646. [Google Scholar] [CrossRef]
- Koupenova, M.; Vitseva, O.; MacKay, C.R.; Beaulieu, L.M.; Benjamin, E.J.; Mick, E.; Kurt-Jones, E.A.; Ravid, K.; Freedman, J.E. Platelet-TLR7 mediates host survival and platelet count during viral infection in the absence of platelet-dependent thrombosis. Blood 2014, 124, 791–802. [Google Scholar] [CrossRef] [Green Version]
- Shen, S.; Zhang, J.; Fang, Y.; Lu, S.; Wu, J.; Zheng, X.; Deng, F. SARS-CoV-2 interacts with platelets and megakaryocytes via ACE2-independent mechanism. J. Hematol. Oncol. 2021, 14, 72. [Google Scholar] [CrossRef]
- Tang, W.H.; Stitham, J.; Jin, Y.; Liu, R.; Lee, S.H.; Du, J.; Atteya, G.; Gleim, S.; Spollett, G.; Martin, K.; et al. Aldose reductase-mediated phosphorylation of p53 leads to mitochondrial dysfunction and damage in diabetic platelets. Circulation 2014, 129, 1598–1609. [Google Scholar] [CrossRef] [Green Version]
- Tyagi, T.; Ahmad, S.; Gupta, N.; Sahu, A.; Ahmad, Y.; Nair, V.; Chatterjee, T.; Bajaj, N.; Sengupta, S.; Ganju, L.; et al. Altered expression of platelet proteins and calpain activity mediate hypoxia-induced prothrombotic phenotype. Blood 2014, 123, 1250–1260. [Google Scholar] [CrossRef] [Green Version]
- Melchinger, H.; Jain, K.; Tyagi, T.; Hwa, J. Role of Platelet Mitochondria: Life in a Nucleus-Free Zone. Front. Cardiovasc. Med. 2019, 6, 153. [Google Scholar] [CrossRef]
- Mackman, N.; Antoniak, S.; Wolberg, A.S.; Kasthuri, R.; Key, N.S. Coagulation Abnormalities and Thrombosis in Patients Infected With SARS-CoV-2 and Other Pandemic Viruses. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2033–2044. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Wu, D.; Guo, W.; Cao, Y.; Huang, D.; Wang, H.; Wang, T.; Zhang, X.; Chen, H.; Yu, H.; et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J. Clin. Investig. 2020, 130, 2620–2629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, T.; Wu, D.; Chen, H.; Yan, W.; Yang, D.; Chen, G.; Ma, K.; Xu, D.; Yu, H.; Wang, H.; et al. Clinical characteristics of 113 deceased patients with coronavirus disease 2019: Retrospective study. Br. Med. J. 2020, 368, m1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panigada, M.; Bottino, N.; Tagliabue, P.; Grasselli, G.; Novembrino, C.; Chantarangkul, V.; Pesenti, A.; Peyvandi, F.; Tripodi, A. Hypercoagulability of COVID-19 patients in intensive care unit: A report of thromboelastography findings and other parameters of hemostasis. J. Thromb. Haemost. 2020, 18, 1738–1742. [Google Scholar] [CrossRef] [PubMed]
- Turecek, P.L.; Johnsen, J.M.; Pipe, S.W.; O’Donnell, J.S.; The iPATH Study Group. Biological mechanisms underlying inter-individual variation in factor VIII clearance in haemophilia. Haemophilia 2020, 26, 575–583. [Google Scholar] [CrossRef]
- Foley, J.H.; Conway, E.M. Cross Talk Pathways Between Coagulation and Inflammation. Circ. Res. 2016, 118, 1392–1408. [Google Scholar] [CrossRef] [PubMed]
- Olson, J.D. D-dimer: An Overview of Hemostasis and Fibrinolysis, Assays, and Clinical Applications. Adv. Clin. Chem. 2015, 69, 1–46. [Google Scholar] [PubMed]
- Yau, J.W.; Teoh, H.; Verma, S. Endothelial cell control of thrombosis. BMC Cardiovasc. Disord. 2015, 15, 130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reitsma, S.; Slaaf, D.W.; Vink, H.; Van Zandvoort, M.A.; oude Egbrink, M.G. The endothelial glycocalyx: Composition, functions, and visualization. Pflugers Arch. 2007, 454, 345–359. [Google Scholar] [CrossRef] [Green Version]
- Pober, J.S.; Sessa, W.C. Evolving functions of endothelial cells in inflammation. Nat. Rev. Immunol. 2007, 7, 803–815. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, J.M.; Gonagle, D.M.; Ward, S.E.; Preston, R.J.S.; O’Donnell, J.S. Endothelial cells orchestrate COVID-19 coagulopathy. Lancet Haematol. 2020, 7, e553–e555. [Google Scholar] [CrossRef]
- Goshua, G.; Pine, A.B.; Meizlish, M.L.; Chang, C.H.; Zhang, H.; Bahel, P.; Baluha, A.; Bar, N.; Bona, R.D.; Burns, A.J.; et al. Endotheliopathy in COVID-19-associated coagulopathy: Evidence from a single-centre, cross-sectional study. Lancet Haematol. 2020, 7, e575–e582. [Google Scholar] [CrossRef]
- Pine, A.B.; Meizlish, M.L.; Goshua, G.; Chang, C.H.; Zhang, H.; Bishai, J.; Bahel, P.; Patel, A.; Gbyli, R.; Kwan, J.M.; et al. Circulating markers of angiogenesis and endotheliopathy in COVID-19. Pulm. Circ. 2020, 10, 2045894020966547. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Monteil, V.; Kwon, H.; Prado, P.; Hagelkruys, A.; Wimmer, R.A.; Stahl, M.; Leopoldi, A.; Garreta, E.; Hurtado Del Pozo, C.; Prosper, F.; et al. Inhibition of SARS-CoV-2 Infections in Engineered Human Tissues Using Clinical-Grade Soluble Human ACE2. Cell 2020, 181, 905–913.e7. [Google Scholar] [CrossRef]
- Liu, F.; Han, K.; Blair, R.; Kenst, K.; Qin, Z.; Upcin, B.; Worsdorfer, P.; Midkiff, C.C.; Mudd, J.; Belyaeva, E.; et al. SARS-CoV-2 Infects Endothelial Cells In Vivo and In Vitro. Front. Cell Infect. Microbiol. 2021, 11, 701278. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Stavrou, E.; Schmaier, A.A.; Grobe, N.; Morris, M.; Chen, A.; Nieman, M.T.; Adams, G.N.; LaRusch, G.; Zhou, Y.; et al. Angiotensin 1–7 and Mas decrease thrombosis in Bdkrb2-/- mice by increasing NO and prostacyclin to reduce platelet spreading and glycoprotein VI activation. Blood 2013, 121, 3023–3032. [Google Scholar] [CrossRef] [PubMed]
- Verdecchia, P.; Cavallini, C.; Spanevello, A.; Angeli, F. The pivotal link between ACE2 deficiency and SARS-CoV-2 infection. Eur. J. Intern. Med. 2020, 76, 14–20. [Google Scholar] [CrossRef]
- Lei, Y.; Zhang, J.; Schiavon, C.R.; He, M.; Chen, L.; Shen, H.; Zhang, Y.; Yin, Q.; Cho, Y.; Andrade, L.; et al. SARS-CoV-2 Spike Protein Impairs Endothelial Function via Downregulation of ACE 2. Circ. Res. 2021, 128, 1323–1326. [Google Scholar] [CrossRef] [PubMed]
- Garvin, M.R.; Alvarez, C.; Miller, J.I.; Prates, E.T.; Walker, A.M.; Amos, B.K.; Mast, A.E.; Justice, A.; Aronow, B.; Jacobson, D. A mechanistic model and therapeutic interventions for COVID-19 involving a RAS-mediated bradykinin storm. eLife 2020, 9, e59177. [Google Scholar] [CrossRef]
- Sodhi, C.P.; Wohlford-Lenane, C.; Yamaguchi, Y.; Prindle, T.; Fulton, W.B.; Wang, S.; McCray, P.B., Jr.; Chappell, M.; Hackam, D.J.; Jia, H. Attenuation of pulmonary ACE2 activity impairs inactivation of des-Arg(9) bradykinin/BKB1R axis and facilitates LPS-induced neutrophil infiltration. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 314, L17–L31. [Google Scholar] [CrossRef]
- Lipcsey, M.; Persson, B.; Eriksson, O.; Blom, A.M.; Fromell, K.; Hultstrom, M.; Huber-Lang, M.; Ekdahl, K.N.; Frithiof, R.; Nilsson, B. The Outcome of Critically Ill COVID-19 Patients Is Linked to Thromboinflammation Dominated by the Kallikrein/Kinin System. Front. Immunol. 2021, 12, 627579. [Google Scholar] [CrossRef] [PubMed]
- Noris, M.; Benigni, A.; Remuzzi, G. The case of complement activation in COVID-19 multiorgan impact. Kidney Int. 2020, 98, 314–322. [Google Scholar] [CrossRef]
- Messner, C.B.; Demichev, V.; Wendisch, D.; Michalick, L.; White, M.; Freiwald, A.; Textoris-Taube, K.; Vernardis, S.I.; Egger, A.S.; Kreidl, M.; et al. Ultra-High-Throughput Clinical Proteomics Reveals Classifiers of COVID-19 Infection. Cell Syst. 2020, 11, 11–24.e4. [Google Scholar] [CrossRef]
- Macor, P.; Durigutto, P.; Mangogna, A.; Bussani, R.; D’Errico, S.; Zanon, M.; Pozzi, N.; Meroni, P.; Tedesco, F. Multi-organ complement deposition in COVID-19 patients. medRxiv 2021, medRxiv:2021.01.07.21249116. [Google Scholar]
- Sinkovits, G.; Mezo, B.; Reti, M.; Muller, V.; Ivanyi, Z.; Gal, J.; Gopcsa, L.; Remenyi, P.; Szathmary, B.; Lakatos, B.; et al. Complement Overactivation and Consumption Predicts In-Hospital Mortality in SARS-CoV-2 Infection. Front. Immunol. 2021, 12, 663187. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Yuan, X.; Chen, H.; Chaturvedi, S.; Braunstein, E.M.; Brodsky, R.A. Direct activation of the alternative complement pathway by SARS-CoV-2 spike proteins is blocked by factor D inhibition. Blood 2020, 136, 2080–2089. [Google Scholar] [CrossRef] [PubMed]
- Ali, Y.M.; Ferrari, M.; Lynch, N.J.; Yaseen, S.; Dudler, T.; Gragerov, S.; Demopulos, G.; Heeney, J.L.; Schwaeble, W.J. Lectin Pathway Mediates Complement Activation by SARS-CoV-2 Proteins. Front. Immunol. 2021, 12, 714511. [Google Scholar] [CrossRef]
- Bumiller-Bini, V.; de Freitas Oliveira-Tore, C.; Carvalho, T.M.; Kretzschmar, G.C.; Goncalves, L.B.; Alencar, N.M.; Gasparetto Filho, M.A.; Beltrame, M.H.; Winter Boldt, A.B. MASPs at the crossroad between the complement and the coagulation cascades-the case for COVID-19. Genet. Mol. Biol. 2021, 44 (Suppl. 1), e20200199. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, O.; Hultstrom, M.; Persson, B.; Lipcsey, M.; Ekdahl, K.N.; Nilsson, B.; Frithiof, R. Mannose-Binding Lectin is Associated with Thrombosis and Coagulopathy in Critically Ill COVID-19 Patients. Thromb. Haemost. 2020, 120, 1720–1724. [Google Scholar] [PubMed]
- Skendros, P.; Mitsios, A.; Chrysanthopoulou, A.; Mastellos, D.C.; Metallidis, S.; Rafailidis, P.; Ntinopoulou, M.; Sertaridou, E.; Tsironidou, V.; Tsigalou, C.; et al. Complement and tissue factor-enriched neutrophil extracellular traps are key drivers in COVID-19 immunothrombosis. J. Clin. Investig. 2020, 130, 6151–6157. [Google Scholar] [CrossRef] [PubMed]
- Ramlall, V.; Thangaraj, P.M.; Meydan, C.; Foox, J.; Butler, D.; Kim, J.; May, B.; De Freitas, J.K.; Glicksberg, B.S.; Mason, C.E.; et al. Immune complement and coagulation dysfunction in adverse outcomes of SARS-CoV-2 infection. Nat. Med. 2020, 26, 1609–1615. [Google Scholar] [CrossRef] [PubMed]
- Ungvari, Z.; Tarantini, S.; Kiss, T.; Wren, J.D.; Giles, C.B.; Griffin, C.T.; Murfee, W.L.; Pacher, P.; Csiszar, A. Endothelial dysfunction and angiogenesis impairment in the ageing vasculature. Nat. Rev. Cardiol. 2018, 15, 555–565. [Google Scholar] [CrossRef]
- Nishimoto, S.; Fukuda, D.; Higashikuni, Y.; Tanaka, K.; Hirata, Y.; Murata, C.; Kim-Kaneyama, J.R.; Sato, F.; Bando, M.; Yagi, S.; et al. Obesity-induced DNA released from adipocytes stimulates chronic adipose tissue inflammation and insulin resistance. Sci. Adv. 2016, 2, e1501332. [Google Scholar] [CrossRef] [Green Version]
- Fukuda, D.; Nishimoto, S.; Aini, K.; Tanaka, A.; Nishiguchi, T.; Kim-Kaneyama, J.R.; Lei, X.F.; Masuda, K.; Naruto, T.; Tanaka, K.; et al. Toll-Like Receptor 9 Plays a Pivotal Role in Angiotensin II-Induced Atherosclerosis. J. Am. Heart Assoc. 2019, 8, e010860. [Google Scholar] [CrossRef] [Green Version]
- Oka, T.; Hikoso, S.; Yamaguchi, O.; Taneike, M.; Takeda, T.; Tamai, T.; Oyabu, J.; Murakawa, T.; Nakayama, H.; Nishida, K.; et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 2012, 485, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Csiszar, A.; Ungvari, Z.; Edwards, J.G.; Kaminski, P.; Wolin, M.S.; Koller, A.; Kaley, G. Aging-induced phenotypic changes and oxidative stress impair coronary arteriolar function. Circ. Res. 2002, 90, 1159–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donato, A.J.; Eskurza, I.; Silver, A.E.; Levy, A.S.; Pierce, G.L.; Gates, P.E.; Seals, D.R. Direct evidence of endothelial oxidative stress with aging in humans: Relation to impaired endothelium-dependent dilation and upregulation of nuclear factor-kappaB. Circ. Res. 2007, 100, 1659–1666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adler, A.; Messina, E.; Sherman, B.; Wang, Z.; Huang, H.; Linke, A.; Hintze, T.H. NAD(P)H oxidase-generated superoxide anion accounts for reduced control of myocardial O2 consumption by NO in old Fischer 344 rats. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H1015–H1022. [Google Scholar] [CrossRef] [PubMed]
- Ungvari, Z.; Tarantini, S.; Nyul-Toth, A.; Kiss, T.; Yabluchanskiy, A.; Csipo, T.; Balasubramanian, P.; Lipecz, A.; Benyo, Z.; Csiszar, A. Nrf2 dysfunction and impaired cellular resilience to oxidative stressors in the aged vasculature: From increased cellular senescence to the pathogenesis of age-related vascular diseases. Geroscience 2019, 41, 727–738. [Google Scholar] [CrossRef]
- Lakatta, E.G.; Levy, D. Arterial and cardiac aging: Major shareholders in cardiovascular disease enterprises: Part II: The aging heart in health: Links to heart disease. Circulation 2003, 107, 346–354. [Google Scholar] [CrossRef] [Green Version]
- Widlansky, M.E.; Gokce, N.; Keaney, J.F., Jr.; Vita, J.A. The clinical implications of endothelial dysfunction. J. Am. Coll. Cardiol. 2003, 42, 1149–1160. [Google Scholar] [CrossRef] [Green Version]
- Lerman, A.; Zeiher, A.M. Endothelial function: Cardiac events. Circulation 2005, 111, 363–368. [Google Scholar] [CrossRef]
- Csiszar, A.; Ungvari, Z.; Koller, A.; Edwards, J.G.; Kaley, G. Proinflammatory phenotype of coronary arteries promotes endothelial apoptosis in aging. Physiol. Genom. 2004, 17, 21–30. [Google Scholar] [CrossRef] [Green Version]
- Asai, K.; Kudej, R.K.; Shen, Y.T.; Yang, G.P.; Takagi, G.; Kudej, A.B.; Geng, Y.J.; Sato, N.; Nazareno, J.B.; Vatner, D.E.; et al. Peripheral vascular endothelial dysfunction and apoptosis in old monkeys. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1493–1499. [Google Scholar] [CrossRef] [Green Version]
- Crackower, M.A.; Sarao, R.; Oudit, G.Y.; Yagil, C.; Kozieradzki, I.; Scanga, S.E.; Oliveira-dos-Santos, A.J.; da Costa, J.; Zhang, L.; Pei, Y.; et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature 2002, 417, 822–828. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Koibuchi, N.; Nishimatsu, H.; Higashikuni, Y.; Hirata, Y.; Kugiyama, K.; Nagai, R.; Sata, M. Candesartan ameliorates cardiac dysfunction observed in angiotensin-converting enzyme 2-deficient mice. Hypertens. Res. 2008, 31, 1953–1961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrario, C.M.; Jessup, J.; Chappell, M.C.; Averill, D.B.; Brosnihan, K.B.; Tallant, E.A.; Diz, D.I.; Gallagher, P.E. Effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockers on cardiac angiotensin-converting enzyme 2. Circulation 2005, 111, 2605–2610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishiyama, Y.; Gallagher, P.E.; Averill, D.B.; Tallant, E.A.; Brosnihan, K.B.; Ferrario, C.M. Upregulation of angiotensin-converting enzyme 2 after myocardial infarction by blockade of angiotensin II receptors. Hypertension 2004, 43, 970–976. [Google Scholar] [CrossRef] [Green Version]
- Furuhashi, M.; Moniwa, N.; Mita, T.; Fuseya, T.; Ishimura, S.; Ohno, K.; Shibata, S.; Tanaka, M.; Watanabe, Y.; Akasaka, H.; et al. Urinary angiotensin-converting enzyme 2 in hypertensive patients may be increased by olmesartan, an angiotensin II receptor blocker. Am. J. Hypertens. 2015, 28, 15–21. [Google Scholar] [CrossRef] [Green Version]
- Epelman, S.; Shrestha, K.; Troughton, R.W.; Francis, G.S.; Sen, S.; Klein, A.L.; Tang, W.H. Soluble angiotensin-converting enzyme 2 in human heart failure: Relation with myocardial function and clinical outcomes. J. Card. Fail. 2009, 15, 565–571. [Google Scholar] [CrossRef] [Green Version]
- Lopes, R.D.; Macedo, A.V.S.; de Barros, E.S.P.G.M.; Moll-Bernardes, R.J.; Dos Santos, T.M.; Mazza, L.; Feldman, A.; D’Andrea Saba Arruda, G.; de Albuquerque, D.C.; Camiletti, A.S.; et al. Effect of Discontinuing vs Continuing Angiotensin-Converting Enzyme Inhibitors and Angiotensin II Receptor Blockers on Days Alive and Out of the Hospital in Patients Admitted With COVID-19: A Randomized Clinical Trial. J. Am. Med. Assoc. 2021, 325, 254–264. [Google Scholar] [CrossRef]
- Cohen, J.B.; Hanff, T.C.; William, P.; Sweitzer, N.; Rosado-Santander, N.R.; Medina, C.; Rodriguez-Mori, J.E.; Renna, N.; Chang, T.I.; Corrales-Medina, V.; et al. Continuation versus discontinuation of renin-angiotensin system inhibitors in patients admitted to hospital with COVID-19: A prospective, randomised, open-label trial. Lancet Respir. Med. 2021, 9, 275–284. [Google Scholar] [CrossRef]
- Bauer, A.; Schreinlechner, M.; Sappler, N.; Dolejsi, T.; Tilg, H.; Aulinger, B.A.; Weiss, G.; Bellmann-Weiler, R.; Adolf, C.; Wolf, D.; et al. Discontinuation versus continuation of renin-angiotensin-system inhibitors in COVID-19 (ACEI-COVID): A prospective, parallel group, randomised, controlled, open-label trial. Lancet Respir. Med. 2021, 9, 863–872. [Google Scholar] [CrossRef]
- Jara, A.; Undurraga, E.A.; Gonzalez, C.; Paredes, F.; Fontecilla, T.; Jara, G.; Pizarro, A.; Acevedo, J.; Leo, K.; Leon, F.; et al. Effectiveness of an Inactivated SARS-CoV-2 Vaccine in Chile. N. Engl. J. Med. 2021, 385, 875–884. [Google Scholar] [CrossRef]
- Lopez Bernal, J.; Andrews, N.; Gower, C.; Gallagher, E.; Simmons, R.; Thelwall, S.; Stowe, J.; Tessier, E.; Groves, N.; Dabrera, G.; et al. Effectiveness of Covid-19 Vaccines against the B.1.617.2 (Delta) Variant. N. Engl. J. Med. 2021, 385, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Tartof, S.Y.; Slezak, J.M.; Fischer, H.; Hong, V.; Ackerson, B.K.; Ranasinghe, O.N.; Frankland, T.B.; Ogun, O.A.; Zamparo, J.M.; Gray, S.; et al. Effectiveness of mRNA BNT162b2 COVID-19 vaccine up to 6 months in a large integrated health system in the USA: A retrospective cohort study. Lancet 2021, 398, 1407–1416. [Google Scholar] [CrossRef]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Bar-On, Y.M.; Goldberg, Y.; Mandel, M.; Bodenheimer, O.; Freedman, L.; Kalkstein, N.; Mizrahi, B.; Alroy-Preis, S.; Ash, N.; Milo, R.; et al. Protection of BNT162b2 Vaccine Booster against Covid-19 in Israel. N. Engl. J. Med. 2021, 385, 1393–1400. [Google Scholar] [CrossRef] [PubMed]
- Chemaitelly, H.; Yassine, H.M.; Benslimane, F.M.; Al Khatib, H.A.; Tang, P.; Hasan, M.R.; Malek, J.A.; Coyle, P.; Ayoub, H.H.; Al Kanaani, Z.; et al. mRNA-1273 COVID-19 vaccine effectiveness against the B.1.1.7 and B.1.351 variants and severe COVID-19 disease in Qatar. Nat. Med. 2021, 27, 1614–1621. [Google Scholar] [CrossRef]
- Chemaitelly, H.; Tang, P.; Hasan, M.R.; AlMukdad, S.; Yassine, H.M.; Benslimane, F.M.; Al Khatib, H.A.; Coyle, P.; Ayoub, H.H.; Al Kanaani, Z.; et al. Waning of BNT162b2 Vaccine Protection against SARS-CoV-2 Infection in Qatar. N. Engl. J. Med. 2021, NEJMoa2114114. [Google Scholar] [CrossRef]
- Beigel, J.H.; Tomashek, K.M.; Dodd, L.E.; Mehta, A.K.; Zingman, B.S.; Kalil, A.C.; Hohmann, E.; Chu, H.Y.; Luetkemeyer, A.; Kline, S.; et al. Remdesivir for the Treatment of Covid-19-Final Report. N. Engl. J. Med. 2020, 383, 1813–1826. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, D.; Du, G.; Du, R.; Zhao, J.; Jin, Y.; Fu, S.; Gao, L.; Cheng, Z.; Lu, Q.; et al. Remdesivir in adults with severe COVID-19: A randomised, double-blind, placebo-controlled, multicentre trial. Lancet 2020, 395, 1569–1578. [Google Scholar] [CrossRef]
- Cao, B.; Wang, Y.; Wen, D.; Liu, W.; Wang, J.; Fan, G.; Ruan, L.; Song, B.; Cai, Y.; Wei, M.; et al. A Trial of Lopinavir-Ritonavir in Adults Hospitalized with Severe Covid-19. N. Engl. J. Med. 2020, 382, 1787–1799. [Google Scholar] [CrossRef]
- Cruz-Topete, D.; Cidlowski, J.A. One hormone, two actions: Anti- and pro-inflammatory effects of glucocorticoids. Neuroimmunomodulation 2015, 22, 20–32. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; Chen, X.; Cai, Y.; Xia, J.; Zhou, X.; Xu, S.; Huang, H.; Zhang, L.; Zhou, X.; Du, C.; et al. Risk Factors Associated with Acute Respiratory Distress Syndrome and Death in Patients With Coronavirus Disease 2019 Pneumonia in Wuhan, China. JAMA Intern. Med. 2020, 180, 934–943. [Google Scholar] [CrossRef] [Green Version]
- Fadel, R.; Morrison, A.R.; Vahia, A.; Smith, Z.R.; Chaudhry, Z.; Bhargava, P.; Miller, J.; Kenney, R.M.; Alangaden, G.; Ramesh, M.S.; et al. Early Short-Course Corticosteroids in Hospitalized Patients With COVID-19. Clin. Infect. Dis. 2020, 71, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Sterne, J.A.C.; Murthy, S.; Diaz, J.V.; Slutsky, A.S.; Villar, J.; Angus, D.C.; Annane, D.; Azevedo, L.C.P.; Berwanger, O.; Cavalcanti, A.B.; et al. Association between Administration of Systemic Corticosteroids and Mortality among Critically Ill Patients with COVID-19: A Meta-analysis. J. Am. Med. Assoc. 2020, 324, 1330–1341. [Google Scholar]
- Horby, P.; Lim, W.S.; Emberson, J.R.; Mafham, M.; Bell, J.L.; Linsell, L.; Staplin, N.; Brightling, C.; Ustianowski, A.; Elmahi, E.; et al. Dexamethasone in Hospitalized Patients with Covid-19. N. Engl. J. Med. 2021, 384, 693–704. [Google Scholar] [PubMed]
- Kalil, A.C.; Patterson, T.F.; Mehta, A.K.; Tomashek, K.M.; Wolfe, C.R.; Ghazaryan, V.; Marconi, V.C.; Ruiz-Palacios, G.M.; Hsieh, L.; Kline, S.; et al. Baricitinib plus Remdesivir for Hospitalized Adults with Covid-19. N. Engl. J. Med. 2021, 384, 795–807. [Google Scholar] [CrossRef]
- Marconi, V.C.; Ramanan, A.V.; de Bono, S.; Kartman, C.E.; Krishnan, V.; Liao, R.; Piruzeli, M.L.B.; Goldman, J.D.; Alatorre-Alexander, J.; de Cassia Pellegrini, R.; et al. Efficacy and safety of baricitinib for the treatment of hospitalised adults with COVID-19 (COV-BARRIER): A randomised, double-blind, parallel-group, placebo-controlled phase 3 trial. Lancet Respir. Med. 2021. [Google Scholar] [CrossRef]
- Guimaraes, P.O.; Quirk, D.; Furtado, R.H.; Maia, L.N.; Saraiva, J.F.; Antunes, M.O.; Kalil Filho, R.; Junior, V.M.; Soeiro, A.M.; Tognon, A.P.; et al. Tofacitinib in Patients Hospitalized with Covid-19 Pneumonia. N. Engl. J. Med. 2021, 385, 406–415. [Google Scholar] [CrossRef]
- Del Valle, D.M.; Kim-Schulze, S.; Huang, H.H.; Beckmann, N.D.; Nirenberg, S.; Wang, B.; Lavin, Y.; Swartz, T.H.; Madduri, D.; Stock, A.; et al. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat. Med. 2020, 26, 1636–1643. [Google Scholar] [CrossRef]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.C.; Uhl, S.; Hoagland, D.; Moller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 1036–1045.e9. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, T.S.; de Sa, K.S.G.; Ishimoto, A.Y.; Becerra, A.; Oliveira, S.; Almeida, L.; Goncalves, A.V.; Perucello, D.B.; Andrade, W.A.; Castro, R.; et al. Inflammasomes are activated in response to SARS-CoV-2 infection and are associated with COVID-19 severity in patients. J. Exp. Med. 2021, 218, e20201707. [Google Scholar] [CrossRef]
- Alattar, R.; Ibrahim, T.B.H.; Shaar, S.H.; Abdalla, S.; Shukri, K.; Daghfal, J.N.; Khatib, M.Y.; Aboukamar, M.; Abukhattab, M.; Alsoub, H.A.; et al. Tocilizumab for the treatment of severe coronavirus disease 2019. J. Med. Virol. 2020, 92, 2042–2049. [Google Scholar] [CrossRef] [PubMed]
- Guaraldi, G.; Meschiari, M.; Cozzi-Lepri, A.; Milic, J.; Tonelli, R.; Menozzi, M.; Franceschini, E.; Cuomo, G.; Orlando, G.; Borghi, V.; et al. Tocilizumab in patients with severe COVID-19: A retrospective cohort study. Lancet Rheumatol. 2020, 2, e474–e484. [Google Scholar] [CrossRef]
- Dimopoulos, G.; de Mast, Q.; Markou, N.; Theodorakopoulou, M.; Komnos, A.; Mouktaroudi, M.; Netea, M.G.; Spyridopoulos, T.; Verheggen, R.J.; Hoogerwerf, J.; et al. Favorable Anakinra Responses in Severe Covid-19 Patients with Secondary Hemophagocytic Lymphohistiocytosis. Cell Host Microbe 2020, 28, 117–123.e1. [Google Scholar] [CrossRef] [PubMed]
- Gordon, A.C.; Mouncey, P.R.; Al-Beidh, F.; Rowan, K.M.; Nichol, A.D.; Arabi, Y.M.; Annane, D.; Beane, A.; Van Bentum-Puijk, W.; Berry, L.R.; et al. Interleukin-6 Receptor Antagonists in Critically Ill Patients with Covid-19. N. Engl. J. Med. 2021, 384, 1491–1502. [Google Scholar] [PubMed]
- RECOVERY Collaborative Group. Tocilizumab in patients admitted to hospital with COVID-19 (RECOVERY): A randomised, controlled, open-label, platform trial. Lancet 2021, 397, 1637–1645. [Google Scholar] [CrossRef]
- Kyriazopoulou, E.; Poulakou, G.; Milionis, H.; Metallidis, S.; Adamis, G.; Tsiakos, K.; Fragkou, A.; Rapti, A.; Damoulari, C.; Fantoni, M.; et al. Early treatment of COVID-19 with anakinra guided by soluble urokinase plasminogen receptor plasma levels: A double-blind, randomized controlled phase 3 trial. Nat. Med. 2021, 27, 1752–1760. [Google Scholar] [CrossRef]
- The CORIMUNO-19 Collaborative group. Effect of anakinra versus usual care in adults in hospital with COVID-19 and mild-to-moderate pneumonia (CORIMUNO-ANA-1): A randomised controlled trial. Lancet Respir. Med. 2021, 9, 295–304. [Google Scholar] [CrossRef]
- Caricchio, R.; Abbate, A.; Gordeev, I.; Meng, J.; Hsue, P.Y.; Neogi, T.; Arduino, R.; Fomina, D.; Bogdanov, R.; Stepanenko, T.; et al. Effect of Canakinumab vs Placebo on Survival Without Invasive Mechanical Ventilation in Patients Hospitalized with Severe COVID-19: A Randomized Clinical Trial. J. Am. Med. Assoc. 2021, 326, 230–239. [Google Scholar] [CrossRef]
- Park, H.H.; Park, W.; Lee, Y.Y.; Kim, H.; Seo, H.S.; Choi, D.W.; Kwon, H.K.; Na, D.H.; Kim, T.H.; Choy, Y.B.; et al. Bioinspired DNase-I-Coated Melanin-Like Nanospheres for Modulation of Infection-Associated NETosis Dysregulation. Adv. Sci. 2021, 8, e2103748. [Google Scholar] [CrossRef]
- Lasky, J.A.; Fuloria, J.; Morrison, M.E.; Lanier, R.; Naderer, O.; Brundage, T.; Melemed, A. Design and Rationale of a Randomized, Double-Blind, Placebo-Controlled, Phase 2/3 Study Evaluating Dociparstat in Acute Lung Injury Associated with Severe COVID-19. Adv. Ther. 2021, 38, 782–791. [Google Scholar] [CrossRef]
- Liu, Y.; Pan, Y.; Hu, Z.; Wu, M.; Wang, C.; Feng, Z.; Mao, C.; Tan, Y.; Liu, Y.; Chen, L.; et al. Thymosin Alpha 1 Reduces the Mortality of Severe Coronavirus Disease 2019 by Restoration of Lymphocytopenia and Reversion of Exhausted T Cells. Clin. Infect. Dis. 2020, 71, 2150–2157. [Google Scholar] [CrossRef] [PubMed]
- Ahn, E.; Araki, K.; Hashimoto, M.; Li, W.; Riley, J.L.; Cheung, J.; Sharpe, A.H.; Freeman, G.J.; Irving, B.A.; Ahmed, R. Role of PD-1 during effector CD8 T cell differentiation. Proc. Natl. Acad. Sci. USA 2018, 115, 4749–4754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, H.T.; Anderson, A.C.; Tan, W.G.; West, E.E.; Ha, S.J.; Araki, K.; Freeman, G.J.; Kuchroo, V.K.; Ahmed, R. Cooperation of Tim-3 and PD-1 in CD8 T-cell exhaustion during chronic viral infection. Proc. Natl. Acad. Sci. USA 2010, 107, 14733–14738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Li, Z.; Liu, S.; Sun, J.; Chen, Z.; Jiang, M.; Zhang, Q.; Wei, Y.; Wang, X.; Huang, Y.Y.; et al. Potential therapeutic effects of dipyridamole in the severely ill patients with COVID-19. Acta Pharm. Sin. B 2020, 10, 1205–1215. [Google Scholar] [CrossRef]
- Savarapu, P.; Baral, N.; Adhikari, G.; Akanbi, M.; Abdelazeem, B.; Isa, S.O.; Khan, A.; Ali, M.; Jenumula, S.; Kesari, K.; et al. Aspirin Use is Associated with Decreased Mortality in Patients with COVID-19: A Systematic Review and Meta-analysis. medRxiv 2021, medRxiv:2021.07.08.21260236. [Google Scholar]
- Horby, P.W.; Pessoa-Amorim, G.; Staplin, N.; Emberson, J.R.; Campbell, M.; Spata, E.; Peto, L.; Brunskill, N.J.; Tiberi, S.; Chew, V.; et al. Aspirin in patients admitted to hospital with COVID-19 (RECOVERY): A randomised, controlled, open-label, platform trial. medRxiv 2021, medRxiv: 2021.06.08.21258132. [Google Scholar]
- Connors, J.M.; Brooks, M.M.; Sciurba, F.C.; Krishnan, J.A.; Bledsoe, J.R.; Kindzelski, A.; Baucom, A.L.; Kirwan, B.A.; Eng, H.; Martin, D.; et al. Effect of Antithrombotic Therapy on Clinical Outcomes in Outpatients With Clinically Stable Symptomatic COVID-19: The ACTIV-4B Randomized Clinical Trial. J. Am. Med. Assoc. 2021, 326, 1703–1712. [Google Scholar] [CrossRef]
- Goligher, E.C.; Bradbury, C.A.; McVerry, B.J.; Lawler, P.R.; Berger, J.S.; Gong, M.N.; Carrier, M.; Reynolds, H.R.; Kumar, A.; Turgeon, A.F.; et al. Therapeutic Anticoagulation with Heparin in Critically Ill Patients with Covid-19. N. Engl. J. Med. 2021, 385, 777–789. [Google Scholar]
- Spyropoulos, A.C.; Goldin, M.; Giannis, D.; Diab, W.; Wang, J.; Khanijo, S.; Mignatti, A.; Gianos, E.; Cohen, M.; Sharifova, G.; et al. Efficacy and Safety of Therapeutic-Dose Heparin vs Standard Prophylactic or Intermediate-Dose Heparins for Thromboprophylaxis in High-risk Hospitalized Patients With COVID-19: The HEP-COVID Randomized Clinical Trial. JAMA Intern. Med. 2021, e216203. [Google Scholar]
- Lawler, P.R.; Goligher, E.C.; Berger, J.S.; Neal, M.D.; McVerry, B.J.; Nicolau, J.C.; Gong, M.N.; Carrier, M.; Rosenson, R.S.; Reynolds, H.R.; et al. Therapeutic Anticoagulation with Heparin in Noncritically Ill Patients with Covid-19. N. Engl. J. Med. 2021, 385, 790–802. [Google Scholar]
- Yamamoto, M.; Matsuyama, S.; Li, X.; Takeda, M.; Kawaguchi, Y.; Inoue, J.I.; Matsuda, Z. Identification of Nafamostat as a Potent Inhibitor of Middle East Respiratory Syndrome Coronavirus S Protein-Mediated Membrane Fusion Using the Split-Protein-Based Cell-Cell Fusion Assay. Antimicrob. Agents Chemother. 2016, 60, 6532–6539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mastaglio, S.; Ruggeri, A.; Risitano, A.M.; Angelillo, P.; Yancopoulou, D.; Mastellos, D.C.; Huber-Lang, M.; Piemontese, S.; Assanelli, A.; Garlanda, C.; et al. The first case of COVID-19 treated with the complement C3 inhibitor AMY-101. Clin. Immunol. 2020, 215, 108450. [Google Scholar] [CrossRef] [PubMed]
- Mastellos, D.C.; Pires da Silva, B.G.P.; Fonseca, B.A.L.; Fonseca, N.P.; Auxiliadora-Martins, M.; Mastaglio, S.; Ruggeri, A.; Sironi, M.; Radermacher, P.; Chrysanthopoulou, A.; et al. Complement C3 vs C5 inhibition in severe COVID-19: Early clinical findings reveal differential biological efficacy. Clin. Immunol. 2020, 220, 108598. [Google Scholar] [CrossRef] [PubMed]
- Neri, T.; Nieri, D.; Celi, A. P-selectin blockade in COVID-19-related ARDS. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 318, L1237–L1238. [Google Scholar] [CrossRef] [PubMed]
- Kow, C.S.; Hasan, S.S. Meta-analysis of Effect of Statins in Patients with COVID-19. Am. J. Cardiol. 2020, 134, 153–155. [Google Scholar] [CrossRef]
- Diaz-Arocutipa, C.; Melgar-Talavera, B.; Alvarado-Yarasca, A.; Saravia-Bartra, M.M.; Cazorla, P.; Belzusarri, I.; Hernandez, A.V. Statins reduce mortality in patients with COVID-19: An updated meta-analysis of 147 824 patients. Int. J. Infect. Dis. 2021, 110, 374–381. [Google Scholar] [CrossRef]
- Forstermann, U.; Munzel, T. Endothelial nitric oxide synthase in vascular disease: From marvel to menace. Circulation 2006, 113, 1708–1714. [Google Scholar] [CrossRef] [Green Version]
- Lei, C.; Su, B.; Dong, H.; Bellavia, A.; Di Fenza, R.; Safaee Fakhr, B.; Gianni, S.; Grassi, L.G.; Kacmarek, R.; Araujo Morais, C.C.; et al. Protocol of a randomized controlled trial testing inhaled Nitric Oxide in mechanically ventilated patients with severe acute respiratory syndrome in COVID-19 (SARS-CoV-2). medRxiv 2020, medRxiv:2020.03.09.20033530. [Google Scholar]
- Van de Veerdonk, F.L.; Kouijzer, I.J.E.; De Nooijer, A.H.; Van der Hoeven, H.G.; Maas, C.; Netea, M.G.; Bruggemann, R.J.M. Outcomes Associated With Use of a Kinin B2 Receptor Antagonist Among Patients With COVID-19. JAMA Net. Open 2020, 3, e2017708. [Google Scholar] [CrossRef]
- Tanaka, S.; Nelson, G.; Olson, C.A.; Buzko, O.; Higashide, W.; Shin, A.; Gonzalez, M.; Taft, J.; Patel, R.; Buta, S.; et al. An ACE2 Triple Decoy that neutralizes SARS-CoV-2 shows enhanced affinity for virus variants. Sci. Rep. 2021, 11, 12740. [Google Scholar] [CrossRef]
- Channappanavar, R.; Perlman, S. Age-related susceptibility to coronavirus infections: Role of impaired and dysregulated host immunity. J. Clin. Investig. 2020, 130, 6204–6213. [Google Scholar] [CrossRef] [PubMed]
- Abdelrahman, Z.; Li, M.; Wang, X. Comparative Review of SARS-CoV-2, SARS-CoV, MERS-CoV, and Influenza A Respiratory Viruses. Front. Immunol. 2020, 11, 552909. [Google Scholar] [CrossRef]
- Zhang, Y.Y.; Li, B.R.; Ning, B.T. The Comparative Immunological Characteristics of SARS-CoV, MERS-CoV, and SARS-CoV-2 Coronavirus Infections. Front. Immunol. 2020, 11, 2033. [Google Scholar] [CrossRef] [PubMed]
- Arons, M.M.; Hatfield, K.M.; Reddy, S.C.; Kimball, A.; James, A.; Jacobs, J.R.; Taylor, J.; Spicer, K.; Bardossy, A.C.; Oakley, L.P.; et al. Presymptomatic SARS-CoV-2 Infections and Transmission in a Skilled Nursing Facility. N. Engl. J. Med. 2020, 382, 2081–2090. [Google Scholar] [CrossRef] [PubMed]
- Fogarty, H.; Townsend, L.; Morrin, H.; Ahmad, A.; Comerford, C.; Karampini, E.; Englert, H.; Byrne, M.; Bergin, C.; O’Sullivan, J.M.; et al. Persistent endotheliopathy in the pathogenesis of long COVID syndrome. J. Thromb. Haemost. 2021, 19, 2546–2553. [Google Scholar] [CrossRef]
- Nalbandian, A.; Sehgal, K.; Gupta, A.; Madhavan, M.V.; McGroder, C.; Stevens, J.S.; Cook, J.R.; Nordvig, A.S.; Shalev, D.; Sehrawat, T.S.; et al. Post-acute COVID-19 syndrome. Nat. Med. 2021, 27, 601–615. [Google Scholar] [CrossRef]
- Wolfel, R.; Corman, V.M.; Guggemos, W.; Seilmaier, M.; Zange, S.; Muller, M.A.; Niemeyer, D.; Jones, T.C.; Vollmar, P.; Rothe, C.; et al. Virological assessment of hospitalized patients with COVID-2019. Nature 2020, 581, 465–469. [Google Scholar] [CrossRef] [Green Version]
- Bikdeli, B.; Madhavan, M.V.; Gupta, A.; Jimenez, D.; Burton, J.R.; Der Nigoghossian, C.; Chuich, T.; Nouri, S.N.; Dreyfus, I.; Driggin, E.; et al. Pharmacological Agents Targeting Thromboinflammation in COVID-19: Review and Implications for Future Research. Thromb. Haemost. 2020, 120, 1004–1024. [Google Scholar]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Higashikuni, Y.; Liu, W.; Obana, T.; Sata, M. Pathogenic Basis of Thromboinflammation and Endothelial Injury in COVID-19: Current Findings and Therapeutic Implications. Int. J. Mol. Sci. 2021, 22, 12081. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222112081
Higashikuni Y, Liu W, Obana T, Sata M. Pathogenic Basis of Thromboinflammation and Endothelial Injury in COVID-19: Current Findings and Therapeutic Implications. International Journal of Molecular Sciences. 2021; 22(21):12081. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222112081
Chicago/Turabian StyleHigashikuni, Yasutomi, Wenhao Liu, Takumi Obana, and Masataka Sata. 2021. "Pathogenic Basis of Thromboinflammation and Endothelial Injury in COVID-19: Current Findings and Therapeutic Implications" International Journal of Molecular Sciences 22, no. 21: 12081. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222112081