
The Use of Inhibitors of Tyrosine Kinase in Paediatric Haemato-Oncology—When and Why?


Abstract
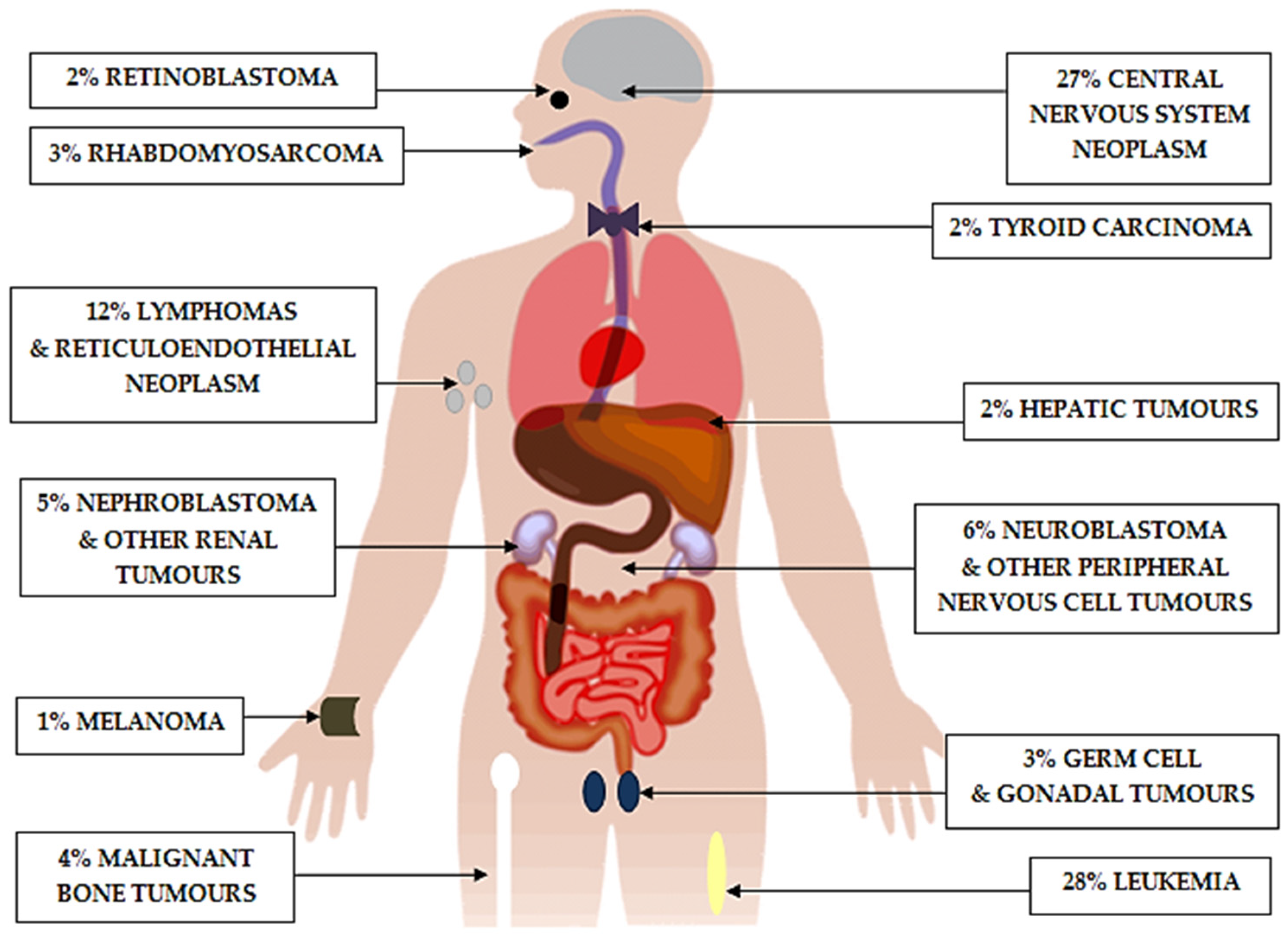
:1. Introduction
2. Historical Perspective and Current Treatment Challenges
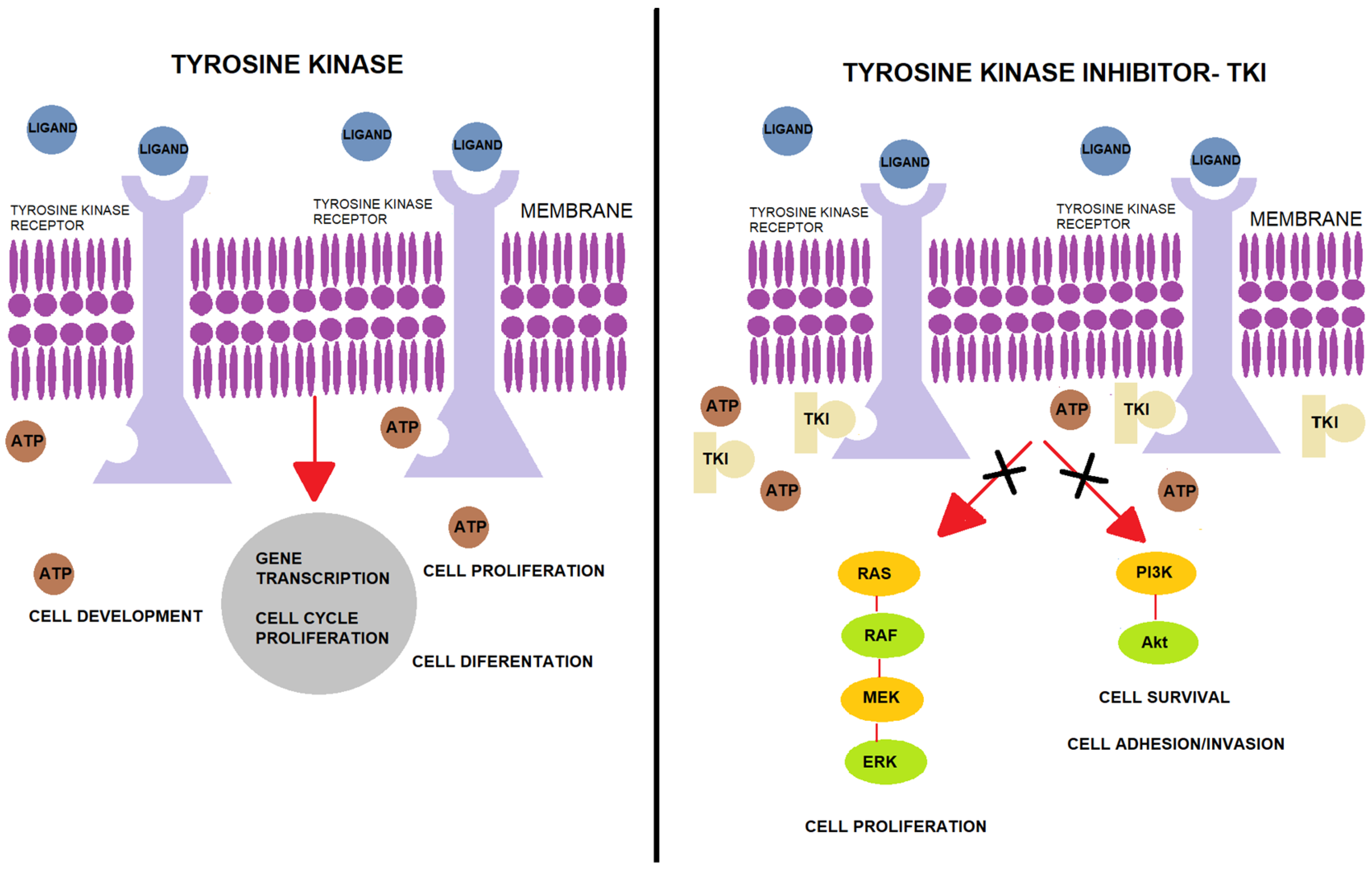
3. Protein Tyrosine Kinase
4. Tyrosine Kinase Inhibitors
4.1. First-Generation TKIs: Imatinib
4.2. Second-Generation TKIs: Dasatinib
4.3. Third-Generation TKIs: Ponatinib
5. The Use of TKIs in Paediatric Haemato-Oncology
5.1. CML
5.2. Ph+ ALL and Ph-Like ALL
6. TKIs in Solid Tumours
6.1. GIST
6.2. Central Nervous System (CNS) Tumours
6.3. Neuroblastoma
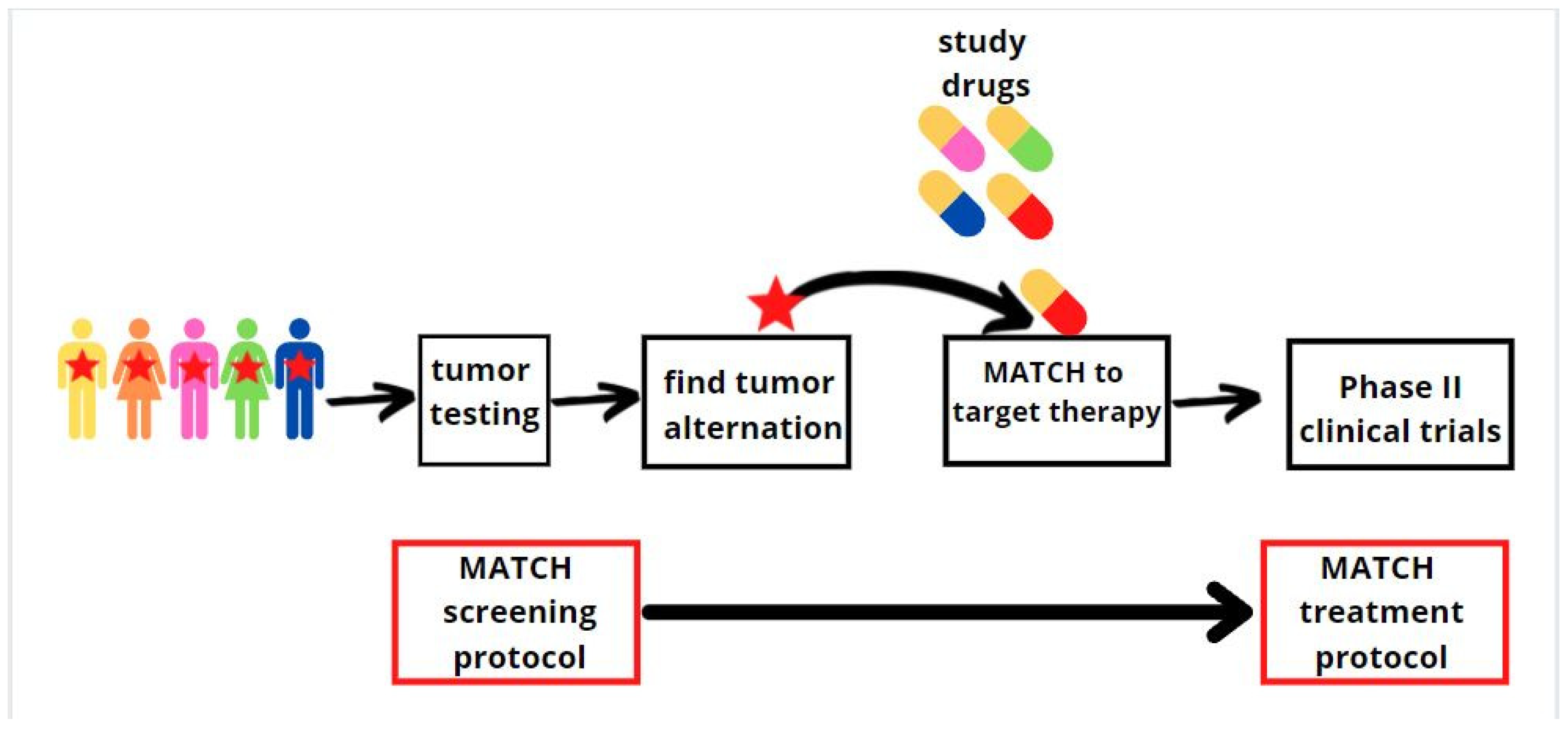
7. Paediatric Molecular Analysis for Therapeutic Choice (Paediatric MATCH)
8. Future Use of New TKIs in Cancer Treatment
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ABL | V-abl Abelson murine leukaemia viral oncogene homolog 1 |
| Akt | serine/threonine kinase 1 |
| ALK | anaplastic lymphoma kinase |
| ALL | acute lymphoblastic leukaemia |
| AML | acute myeloid leukaemia |
| ATM | ATM serine/threonine kinase |
| ATP | adenosine triphosphate |
| BCR | ABL1-breakpoint cluster region and V-abl Abelson murine leukaemia viral oncogene homolog |
| BRAF | gene encoding protein B-Raf |
| BRCA1 | breast cancer 1 gene |
| BRCA2 | breast cancer 2 gene |
| CCyR | complete cytogenetic response |
| CDK4/6 | cyclin-dependent kinase |
| CHR | complete haematologic response |
| c-KIT | tyrosine-protein kinase KIT |
| c-MET | hepatocyte growth factor receptor |
| CML-BP | chronic myeloid leukaemia plastic phase |
| CML | chronic myeloid leukaemia |
| CNS | central nervous system |
| COG | Children’s Oncology Group |
| CSF-1R | Colony stimulating factor 1 receptor |
| EGFR | epidermal growth factor receptor |
| ELN | European Leukaemia Net |
| ERK1/2 | extracellular signal-regulated kinases |
| EsPhALL | European Study of Postinduction Treatment of Ph-Positive ALL |
| EZH2 | enhancer of zeste homolog 2 |
| FDA | Food and Drug Administration |
| FGFR1 | fibroblast growth factor receptor 1 |
| FGFR2 | fibroblast growth factor receptor 2 |
| FGFR3 | fibroblast growth factor receptor 3 |
| FGFR4 | fibroblast growth factor receptor 4 |
| FLT3 | cluster of differentiation antigen 135 |
| GCT | germ cell tumour |
| GIST | gastrointestinal stromal tumour |
| HER2 | human epidermal growth factor receptor 2 |
| HRAS | v-Ha-ras Harvey rat sarcoma viral oncogene homolog |
| HSCT | haematopoietic stem cell transplantation |
| IRIS | International Randomised Study of Interferon and STI571 |
| KIT | tyrosine-protein kinase KIT cluster of differentiation 117 |
| MATCH | Molecular Analysis for Therapeutic Choice |
| MAPK | mitogen-activated protein kinase |
| MET | MET proto-oncogene, tyrosine kinase receptor |
| MM-GBMV | molecular mechanics—generalised Born molecular volume |
| MMR | major molecular response |
| NCI | National Cancer Institute |
| NCI-MATCH | National Cancer Institute—Molecular Analysis for Therapeutic Choice |
| NRTK | non-receptor protein tyrosine kinase |
| NSCLC | non-small-cell lung cancer |
| OS | overall survival |
| P loop | phosphate binding loop |
| P13K/mTOR | intracellular signalling pathway |
| pan-TRK | family of receptor tyrosine kinases |
| PARP | poly (ADP-ribose) polymerase |
| PDGFRα | platelet-derived growth factor receptor alpha |
| PDGFRβ | platelet-derived growth factor receptor beta |
| Ph-like ALL | Philadelphia chromosome-like acute lymphoblastic leukaemia |
| Ph+ ALL | Philadelphia chromosome acute lymphoblastic leukaemia |
| PI3K | phosphoinositide 3-kinase |
| PI3K/mTOR | phosphatidylinositol 3-kinase/mammalian target of rapamycin kinase |
| PTK | protein tyrosine kinase |
| Rb | retinoblastoma |
| RAF | proto-oncogene |
| RAS | rat sarcoma virus protein family |
| Rb | retinoblastoma |
| RET | “rearranged during transfection” proto-oncogene |
| RR | relative resistance |
| ROS1 | ROS proto-oncogene 1, tyrosine kinase receptor |
| RTK | receptor protein tyrosine kinase |
| SMARCB1 | SWI/SNF-related, matrix-associated, actin-dependent regulator of chromatin, Subfamily B, Member 1 |
| SMARCA4 | SWI/SNF-related, matrix-associated, actin-dependent regulator of chromatin, Subfamily A, Member 4 |
| SRC | Src proto-oncogene, non-receptor tyrosine kinase |
| STAMP | specifically targeting the myristoyl pocket |
| STI571 | imatinib |
| TKI | tyrosine kinase inhibitor |
| TRKA | tropomyosin receptor kinase A |
| TRKB | tropomyosin receptor kinase B |
| TRKC | tropomyosin receptor kinase C |
| TSC1 | Tuberous sclerosis 1 |
| TSC2 | Tuberous sclerosis 2 |
| UK | United Kingdom |
| USA | United States of America |
| VEGFR | vascular endothelial growth factor |
| VEGFR2 | vascular endothelial growth factor 2 |
| VEGFR3 | vascular endothelial growth factor 3 |
References
- O’Leary, M.; Krailo, M.; Anderson, J.R.; Reaman, G.H. Progress in Childhood Cancer: 50 Years of Research Collaboration, a Report from the Children’s Oncology Group. Semin. Oncol. 2008, 35, 484–493. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, R.M.; Walton, M.A.; Carter, P.M. The Major Causes of Death in Children and Adolescents in the United States. N. Engl. J. Med. 2018, 379, 2468–2475. [Google Scholar] [CrossRef]
- Smith, M.A.; Seibel, N.L.; Altekruse, S.F.; Ries, L.A.G.; Melbert, D.L.; O’Leary, M.; Smith, F.O.; Reaman, G.H. Outcomes for Children and Adolescents with Cancer: Challenges for the Twenty-First Century. J. Clin. Oncol. 2010, 28, 2625–2634. [Google Scholar] [CrossRef]
- Gupta, S.; Howard, S.C.; Hunger, S.P.; Antillon, F.G.; Metzger, M.L.; Israels, T.; Harif, M.; Rodriguez-Galindo, C. Cancer: Disease Control Priorities, 3rd ed.; The International Bank for Reconstruction and Development / The World Bank: Washington, DC, USA, 2015; Volume 3, pp. 121–146. ISBN 978-1-4648-0349-9. [Google Scholar]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Joseph, P.D.; Craig, J.C.; Caldwell, P.H.Y. Clinical Trials in Children. Br. J. Clin. Pharmacol. 2015, 79, 357–369. [Google Scholar] [CrossRef] [Green Version]
- Baudino, T.A. Targeted Cancer Therapy: The Next Generation of Cancer Treatment. Curr. Drug Discov. Technol. 2015, 12, 3–20. [Google Scholar] [CrossRef]
- Farber, S. Some Observations on the Effect of Folic Acid Antagonists on Acute Leukemia and Other Forms of Incurable Cancer. Blood 1949, 4, 160–167. [Google Scholar] [CrossRef] [Green Version]
- Farber, S.; Diamond, L.K.; Mercer, R.D.; Sylvester, R.F.; Wolff, J.A. Temporary Remissions in Acute Leukemia in Children Produced by Folic Acid Antagonist, 4-Aminopteroyl-Glutamic Acid (Aminopterin). N. Engl. J. Med. 1948, 238, 787–793. [Google Scholar] [CrossRef]
- Farber, S. Chemotherapy in the Treatment of Leukemia and Wilms’ Tumor. JAMA 1966, 198, 826–836. [Google Scholar] [CrossRef]
- Cantrell, M.A.; Ruble, K. Multidisciplinary care in pediatric oncology. J Multidiscip. Healthc. 2011, 4, 171–181. [Google Scholar] [CrossRef] [Green Version]
- Koren, G.; Schechter, T. Cancer Chemotherapy in Young Children: Challenges and Solutions. Pediatr. Blood Cancer 2007, 49 (Suppl. S7), 1091–1092. [Google Scholar] [CrossRef]
- Frattarelli, D.A.; Galinkin, J.L.; Green, T.P.; Johnson, T.D.; Neville, K.A.; Paul, I.M.; Van Den Anker, J.N. Off-Label Use of Drugs in Children. Pediatrics 2014, 133, 563–567. [Google Scholar] [CrossRef] [Green Version]
- Decamp, M.; Joffe, S.; Fernandez, C.V.; Faden, R.R.; Unguru, Y. Chemotherapy Drug Shortages in Pediatric Oncology: A Consensus Statement. Pediatrics 2014, 133, e716–e724. [Google Scholar] [CrossRef] [Green Version]
- Call, R.J.; Burlison, J.D.; Robertson, J.J.; Scott, J.R.; Baker, D.K.; Rossi, M.G.; Howard, S.C.; Hoffman, J.M. Adverse Drug Event Detection in Pediatric Oncology and Hematology Patients: Using Medication Triggers to Identify Patient Harm in a Specialized Pediatric Patient Population. J. Pediatr. 2014, 165, 447–452.e4. [Google Scholar] [CrossRef] [Green Version]
- Aleksakhina, S.N.; Kashyap, A.; Imyanitov, E.N. Mechanisms of Acquired Tumor Drug Resistance. Biochim. Biophys. Acta Rev. Cancer 2019, 1872, 188310. [Google Scholar] [CrossRef]
- Winkler, G.C.; Barle, E.L.; Galati, G.; Kluwe, W.M. Functional Differentiation of Cytotoxic Cancer Drugs and Targeted Cancer Therapeutics. Regul. Toxicol. Pharmacol. 2014, 70, 46–53. [Google Scholar] [CrossRef]
- Filbin, M.; Monje, M. Developmental Origins and Emerging Therapeutic Opportunities for Childhood Cancer. Nat. Med. 2019, 25, 367–376. [Google Scholar] [CrossRef]
- Wang, Z.; Cole, P.A. Catalytic Mechanisms and Regulation of Protein Kinases. Methods Enzymol. 2014, 548, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Jiao, Q.; Bi, L.; Ren, Y.; Song, S.; Wang, Q.; Wang, Y.-S. Advances in Studies of Tyrosine Kinase Inhibitors and Their Acquired Resistance. Mol. Cancer 2018, 17, 36. [Google Scholar] [CrossRef]
- Drake, J.M.; Lee, J.K.; Witte, O.N. Clinical Targeting of Mutated and Wild-Type Protein Tyrosine Kinases in Cancer. Mol. Cell. Biol. 2014, 34, 1722–1732. [Google Scholar] [CrossRef] [Green Version]
- Madhusudan, S.; Ganesan, T.S. Tyrosine Kinase Inhibitors in Cancer Therapy. Clin. Biochem. 2004, 37, 618–635. [Google Scholar] [CrossRef]
- Sulzbacher, I.; Birner, P.; Trieb, K.; Träxler, M.; Lang, S.; Chott, A. Expression of Platelet-Derived Growth Factor-AA Is Associated with Tumor Progression in Osteosarcoma. Mod. Pathol. 2003, 16, 66–71. [Google Scholar] [CrossRef] [Green Version]
- Tamborini, E.; Bonadiman, L.; Greco, A.; Gronchi, A.; Riva, C.; Bertulli, R.; Casali, P.G.; Pierotti, M.A.; Pilotti, S. Expression of Ligand-Activated KIT and Platelet-Derived Growth Factor Receptor β Tyrosine Kinase Receptors in Synovial Sarcoma. Clin. Cancer Res. 2004, 10, 938–943. [Google Scholar] [CrossRef] [Green Version]
- Smithey, B.E.; Pappo, A.S.; Hill, D.A. C-Kit Expression in Pediatric Solid Tumors: A Comparative Immunohistochemical Study. Am. J. Surg. Pathol. 2002, 26, 486–492. [Google Scholar] [CrossRef]
- Robinson, D.R.; Wu, Y.M.; Lin, S.F. The Protein Tyrosine Kinase Family of the Human Genome. Oncogene 2000, 19, 5548–5557. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, J.T.; Haap, M.; Kopp, H.-G.; Lipp, H.-P. Tyrosine Kinase Inhibitors—A Review on Pharmacology, Metabolism and Side Effects. Curr. Drug Metab. 2009, 10, 470–481. [Google Scholar] [CrossRef]
- Yaghmaie, M.; Yeung, C. Molecular mechanisms of resistance to tyrosine kinase inhibitors. Curr. Hematol. Malig. Rep. 2019, 14, 395–404. [Google Scholar] [CrossRef]
- Redaelli, S.; Piazza, R.; Rostagno, R.; Magistroni, V.; Perini, P.; Marega, M.; Gambacorti-Passerini, C.; Boschelli, F. Activity of bosutinib, dasatinib, and nilotinib against 18 imatinib-resistant BCR/ABL mutants. J. Clin. Oncol. 2009, 27, 469–471. [Google Scholar] [CrossRef]
- Cohen, M.H.; Williams, G.; Johnson, J.R.; Duan, J.; Gobburu, J.; Rahman, A.; Benson, K.; Leighton, J.; Kim, S.K.; Wood, R.; et al. Approval Summary for Imatinib Mesylate Capsules in the Treatment of Chronic Myelogenous Leukemia. Clin. Cancer Res. 2002, 8, 935–942. [Google Scholar]
- O’Hare, T.; Shakespeare, W.C.; Zhu, X.; Eide, C.A.; Rivera, V.M.; Wang, F.; Adrian, L.T.; Zhou, T.; Huang, W.S. AP24534, a Pan-BCR-ABL Inhibitor for Chronic Myeloid Leukemia, Potently Inhibits the T315I Mutant and Overcomes Mutation-Based Resistance. Cancer Cell 2009, 16, 401–412. [Google Scholar] [CrossRef] [Green Version]
- Alloo, A.; Sheu, J.; Butrynski, J.E.; DeAngelo, D.J.; George, S.; Murphy, G.F.; LeBoeuf, N.R. Ponatinib-Induced Pityriasiform, Folliculocentric and Ichthyosiform Cutaneous Toxicities. Br. J. Dermatol. 2015, 173, 574–577. [Google Scholar] [CrossRef]
- Millot, F.; Guilhot, J.; Baruchel, A.; Petit, A.; Bertrand, Y.; Mazingue, F.; Lutz, P.; Vérité, C.; Berthou, C.; Galambrun, C.; et al. Impact of Early Molecular Response in Children with Chronic Myeloid Leukemia Treated in the French Glivec Phase 4 Study. Blood 2014, 124, 2408–2410. [Google Scholar] [CrossRef] [Green Version]
- Suttorp, M.; Schulze, P.; Glauche, I.; Göhring, G.; von Neuhoff, N.; Metzler, M.; Sedlacek, P.; de Bont, E.S.J.M.; Balduzzi, A.; Lausen, B. Front-Line Imatinib Treatment in Children and Adolescents with Chronic Myeloid Leukemia: Results from a Phase III Trial. Leukemia 2018, 32, 1657–1669. [Google Scholar] [CrossRef]
- Druker, B.J.; Guilhot, F.; O’Brien, S.G.; Gathmann, I.; Kantarjian, H.; Gattermann, N.; Deininger, M.W.N.; Silver, R.T.; Goldman, J.M.; Stone, R.M.A.; et al. Five-Year Follow-up of Patients Receiving Imatinib for Chronic Myeloid Leukemia. N. Engl. J. Med. 2006, 355, 2408–2417. [Google Scholar] [CrossRef]
- Hochhaus, A.; Larson, R.A.; Guilhot, F.; Radich, J.P.; Branford, S.; Hughes, T.P.; Baccarani, M.; Deininger, M.W.; Cervantes, F.; Fujihara, S.; et al. Long-Term Outcomes of Imatinib Treatment for Chronic Myeloid Leukemia. N. Engl. J. Med. 2017, 376, 917–927. [Google Scholar] [CrossRef]
- Biondi, A.; Cario, G.; De Lorenzo, P.; Castor, A.; Conter, V.; Leoni, V.; Gandemer, V.; Pieters, R.; Stary, J.; Escherich, G.; et al. Long-Term Follow up of Pediatric Philadelphia Positive Acute Lymphoblastic Leukemia Treated with the EsPhALL2004 Study: High White Blood Cell Count at Diagnosis Is the Strongest Prognostic Factor. Haematologica 2019, 104, e13–e16. [Google Scholar] [CrossRef] [Green Version]
- Fielding, A.K.; Rowe, J.M.; Buck, G.; Foroni, L.; Gerrard, G.; Litzow, M.R.; Lazarus, H.; Luger, S.M.; Marks, D.I.; McMillan, A.K.; et al. UKALLXII/ECOG2993: Addition of Imatinib to a Standard Treatment Regimen Enhances Long-Term Outcomes in Philadelphia Positive Acute Lymphoblastic Leukemia. Blood 2014, 123, 843–850. [Google Scholar] [CrossRef]
- Agaram, N.P.; Laquaglia, M.P.; Ustun, B.; Guo, T.; Wong, G.C.; Socci, N.D.; Maki, R.G.; DeMatteo, R.P.; Besmer, P.; Antonescu, C.R. Molecular Characterization of Pediatric Gastrointestinal Stromal Tumors. Clin. Cancer Res. 2008, 14, 3204–3215. [Google Scholar] [CrossRef] [Green Version]
- Joensuu, H.; Eriksson, M.; Sundby Hall, K.; Hartmann, J.T.; Pink, D.; Schütte, J.; Ramadori, G.; Hohenberger, P.; Duyster, J.; Al-Batran, S.-E.; et al. One vs Three Years of Adjuvant Imatinib for Operable Gastrointestinal Stromal Tumor: A Randomized Trial. JAMA 2012, 307, 1265–1272. [Google Scholar] [CrossRef] [Green Version]
- Raut, C.P.; Espat, N.J.; Maki, R.G.; Araujo, D.M.; Trent, J.; Williams, T.F.; Purkayastha, D.D.; DeMatteo, R.P. Efficacy and Tolerability of 5-Year Adjuvant Imatinib Treatment for Patients with Resected Intermediate- or High-Risk Primary Gastrointestinal Stromal Tumor: The PERSIST-5 Clinical Trial. JAMA Oncol. 2018, 4, e184060. [Google Scholar] [CrossRef]
- Pollack, I.F.; Jakacki, R.I.; Blaney, S.M.; Hancock, M.L.; Kieran, M.W.; Phillips, P.; Kun, L.E.; Friedman, H.; Packer, R.; Banerjee, A.; et al. Phase I Trial of Imatinib in Children with Newly Diagnosed Brainstem and Recurrent Malignant Gliomas: A Pediatric Brain Tumor Consortium Report. Neuro Oncol. 2007, 9, 145–160. [Google Scholar] [CrossRef] [Green Version]
- Raymond, E.; Brandes, A.A.; Dittrich, C.; Fumoleau, P.; Coudert, B.; Clement, P.M.J.; Frenay, M.; Rampling, R.; Stupp, R.; Kros, J.M.; et al. Phase II Study of Imatinib in Patients with Recurrent Gliomas of Various Histologies: A European Organisation for Research and Treatment of Cancer Brain Tumor Group Study. J. Clin. Oncol. 2008, 26, 4659–4665. [Google Scholar] [CrossRef] [Green Version]
- Shaw, A.T.; Kim, D.-W.; Nakagawa, K.; Seto, T.; Crinó, L.; Ahn, M.-J.; De Pas, T.; Besse, B.; Solomon, B.J.; Blackhall, F.A.; et al. Crizotinib versus Chemotherapy in Advanced ALK-Positive Lung Cancer. N. Engl. J. Med. 2013, 368, 2385–2394. [Google Scholar] [CrossRef] [Green Version]
- Broniscer, A.; Jia, S.; Mandrell, B.; Hamideh, D.; Huang, J.; Onar-Thomas, A.; Gajjar, A.; Raimondi, S.C.; Tatevossian, R.G.; Stewart, C.F. Phase 1 Trial, Pharmacokinetics, and Pharmacodynamics of Dasatinib Combined with Crizotinib in Children with Recurrent or Progressive High-Grade and Diffuse Intrinsic Pontine Glioma. Pediatr. Blood Cancer 2018, 65, e27035. [Google Scholar] [CrossRef]
- Greengard, E.; Mosse, Y.P.; Liu, X.; Minard, C.G.; Reid, J.M.; Voss, S.; Wilner, K.; Fox, E.; Balis, F.; Blaney, S.M.; et al. Safety, Tolerability and Pharmacokinetics of Crizotinib in Combination with Cytotoxic Chemotherapy for Pediatric Patients with Refractory Solid Tumors or Anaplastic Large Cell Lymphoma (ALCL): A Children’s Oncology Group Phase 1 Consortium Study (ADVL121. Cancer Chemother. Pharmacol. 2020, 86, 829–840. [Google Scholar] [CrossRef]
- Junca, A.; Villalva, C.; Tachon, G.; Rivet, P.; Cortes, U.; Guilloteau, K.; Balbous, A.; Godet, J.; Wager, M.; Karayan-Tapon, L. Crizotinib Targets in Glioblastoma Stem Cells. Cancer Med. 2017, 6, 2625–2634. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.-L.; Yang, J.C.-H.; Kim, D.-W.; Lu, S.; Zhou, J.; Seto, T.; Yang, J.-J.; Yamamoto, N.; Ahn, M.-J.; Takahashi, T.; et al. Phase II Study of Crizotinib in East Asian Patients with ROS1-Positive Advanced Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2018, 36, 1405–1411. [Google Scholar] [CrossRef]
- Chowdhury, T.; Prichard-Jones, K.; Sebire, N.J.; Bier, N.; Cherian, A.; O’Sullivan, M.; O’Meara, A.; Anderson, J. Persistent Complete Response after Single-Agent Sunitinib Treatment in a Case of TFE Translocation Positive Relapsed Metastatic Pediatric Renal Cell Carcinoma. J. Pediatr. Hematol. Oncol. 2013, 35, e1–e3. [Google Scholar] [CrossRef]
- Motzer, R.J.; Rini, B.I.; Bukowski, R.M.; Curti, B.D.; George, D.J.; Hudes, G.R.; Redman, B.G.; Margolin, K.A.; Merchan, J.R.; Wilding, G.; et al. Sunitinib in Patients with Metastatic Renal Cell Carcinoma. JAMA 2006, 295, 2516–2524. [Google Scholar] [CrossRef] [Green Version]
- Janeway, K.A.; Albritton, K.H.; Van Den Abbeele, A.D.; D’Amato, G.Z.; Pedrazzoli, P.; Siena, S.; Picus, J.; Butrynski, J.E.; Schlemmer, M.; Heinrich, M.C.; et al. Sunitinib Treatment in Pediatric Patients with Advanced GIST Following Failure of Imatinib. Pediatr. Blood Cancer 2009, 52, 767–771. [Google Scholar] [CrossRef]
- Riedel, R.F.; Ballman, K.V.; Lu, Y.; Attia, S.; Loggers, E.T.; Ganjoo, K.N.; Livingston, M.B.; Chow, W.; Wright, J.; Ward, J.H.; et al. A Randomized, Double-Blind, Placebo-Controlled, Phase II Study of Regorafenib Versus Placebo in Advanced/Metastatic, Treatment-Refractory Liposarcoma: Results from the SARC024 Study. Oncologist 2020, 25, e1655–e1662. [Google Scholar] [CrossRef]
- Demetri, G.D.; Reichardt, P.; Kang, Y.-K.; Blay, J.-Y.; Rutkowski, P.; Gelderblom, H.; Hohenberger, P.; Leahy, M.; von Mehren, M.; Joensuu, H.; et al. Efficacy and Safety of Regorafenib for Advanced Gastrointestinal Stromal Tumours after Failure of Imatinib and Sunitinib (GRID): An International, Multicentre, Randomised, Placebo-Controlled, Phase 3 Trial. Lancet 2013, 381, 295–302. [Google Scholar] [CrossRef] [Green Version]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.-H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V. Regorafenib for Patients with Hepatocellular Carcinoma Who Progressed on Sorafenib Treatment (RESORCE): A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef] [Green Version]
- Zwaan, C.M.; Rizzari, C.; Mechinaud, F.; Lancaster, D.L.; Lehrnbecher, T.; van der Velden, V.H.J.; Beverloo, B.B.; den Boer, M.L.; Pieters, R.; Reinhardt, D.; et al. Dasatinib in Children and Adolescents with Relapsed or Refractory Leukemia: Results of the CA180-018 Phase I Dose-Escalation Study of the Innovative Therapies for Children with Cancer Consortium. J. Clin. Oncol. 2013, 31, 2460–2468. [Google Scholar] [CrossRef]
- Gore, L.; Kearns, P.; de Martino, M.L.; Lee; De Souza, C.A.; Bertrand, Y.; Hijiya, N.; Stork, L.C.; Chung, N.-G.; Cardos, R.C.; et al. Dasatinib in Pediatric Patients with Chronic Myeloid Leukemia in Chronic Phase: Results from a Phase II Trial. J. Clin. Oncol. 2018, 36, 1330–1338. [Google Scholar] [CrossRef]
- Foà, R.; Vitale, A.; Vignetti, M.; Meloni, G.; Guarini, A.; De Propris, M.S.; Elia, L.; Paoloni, F.; Fazi, P.; Cimino, G.; et al. Dasatinib as First-Line Treatment for Adult Patients with Philadelphia Chromosome-Positive Acute Lymphoblastic Leukemia. Blood 2011, 118, 6521–6528. [Google Scholar] [CrossRef] [Green Version]
- Kantarjian, H.M.; Hochhaus, A.; Saglio, G.; De Souza, C.; Flinn, I.W.; Stenke, L.; Goh, Y.-T.; Rosti, G.; Nakamae, H.; Gallagher, N.J.; et al. Nilotinib versus Imatinib for the Treatment of Patients with Newly Diagnosed Chronic Phase, Philadelphia Chromosome-Positive, Chronic Myeloid Leukaemia: 24-Month Minimum Follow-up of the Phase 3 Randomised ENESTnd Trial. Lancet Oncol. 2011, 12, 841–851. [Google Scholar] [CrossRef]
- Cortes, J.E.; Khoury, H.J.; Kantarjian, H.M.; Lipton, J.H.; Kim, D.-W.; Schafhausen, P.; Matczak, E.; Leip, E.; Noonan, K.; Brümmendorf, T.H.; et al. Long-Term Bosutinib for Chronic Phase Chronic Myeloid Leukemia after Failure of Imatinib plus Dasatinib and/or Nilotinib. Am. J. Hematol. 2016, 91, 1206–1214. [Google Scholar] [CrossRef] [Green Version]
- Taylor, J.W.; Dietrich, J.; Gerstner, E.R.; Norden, A.D.; Rinne, M.L.; Cahill, D.P.; Stemmer-Rachamimov, A.; Wen, P.Y.; Betensky, R.A.; Giorgio, D.H.; et al. Phase 2 Study of Bosutinib, a Src Inhibitor, in Adults with Recurrent Glioblastoma. J. Neurooncol. 2015, 121, 557–563. [Google Scholar] [CrossRef] [Green Version]
- Hijiya, N.; Maschan, A.; Rizzari, C.; Shimada, H.; Dufour, C.; Goto, H.; Kang, H.J.; Guinipero, T.; Karakas, Z.; Bautista, F.; et al. A Phase 2 Study of Nilotinib in Pediatric Patients with CML: Long-Term Update on Growth Retardation and Safety. Blood Adv. 2021, 5, 2925–2934. [Google Scholar] [CrossRef]
- Gómez-Almaguer, D.; Saldaña-Vázquez, R.; Tarín-Arzaga, L.; Herrera-Rojas, M.A.; Vázquez-Mellado de Larracoechea, A.; Cantú-Rodríguez, O.G.; Gutiérrez-Aguirre, C.H.; Jaime-Pérez, J.C. Combination of Low-Dose Imatinib plus Nilotinib for the Treatment of Chronic-Phase Chronic Myeloid Leukaemia after Imatinib Failure. Hematology 2016, 21, 411–414. [Google Scholar] [CrossRef] [Green Version]
- Blay, J.-Y.; Shen, L.; Kang, Y.-K.; Rutkowski, P.; Qin, S.; Nosov, D.; Wan, D.; Trent, J.; Srimuninnimit, V.; Pápai, Z.; et al. Nilotinib versus Imatinib as First-Line Therapy for Patients with Unresectable or Metastatic Gastrointestinal Stromal Tumours (ENESTg1): A Randomised Phase 3 Trial. Lancet. Oncol. 2015, 16, 550–560. [Google Scholar] [CrossRef] [Green Version]
- Vairy, S.; Le Teuff, G.; Bautista, F.; De Carli, E.; Bertozzi, A.-I.; Pagnier, A.; Fouyssac, F.; Nysom, K.; Aerts, I.; Leblond, P.; et al. Phase I Study of Vinblastine in Combination with Nilotinib in Children, Adolescents, and Young Adults with Refractory or Recurrent Low-Grade Glioma. Neurooncol. Adv. 2020, 2, vdaa075. [Google Scholar] [CrossRef]
- Lee, E.Q.; Muzikansky, A.; Duda, D.G.; Gaffey, S.; Dietrich, J.; Nayak, L.; Chukwueke, U.N.; Beroukhim, R.; Doherty, L.; Laub, C.K.; et al. Phase II Trial of Ponatinib in Patients with Bevacizumab-Refractory Glioblastoma. Cancer Med. 2019, 8, 5988–5994. [Google Scholar] [CrossRef]
- Jabbour, E.; Short, N.J.; Ravandi, F.; Huang, X.; Daver, N.; DiNardo, C.D.; Konopleva, M.; Pemmaraju, N.; Wierda, W.; Garcia-Manero, G.; et al. Combination of Hyper-CVAD with Ponatinib as First-Line Therapy for Patients with Philadelphia Chromosome-Positive Acute Lymphoblastic Leukaemia: Long-Term Follow-up of a Single-Centre, Phase 2 Study. Lancet Haematol. 2018, 5, e618–e627. [Google Scholar] [CrossRef]
- Shah, N.P.; Talpaz, M.; Deininger, M.W.N.; Mauro, M.J.; Flinn, I.W.; Bixby, D.; Lustgarten, S.; Gozgit, J.M.; Clackson, T.; Turner, C.D.; et al. Ponatinib in Patients with Refractory Acute Myeloid Leukaemia: Findings from a Phase 1 Study. Br. J. Haematol. 2013, 367, 2075–2088. [Google Scholar] [CrossRef] [Green Version]
- Millot, F.; Suttorp, M.; Versluys, A.B.; Kalwak, K.; Nelken, B.; Ducassou, S.; Bertrand, Y.; Baruchel, A. Ponatinib in Childhood Philadelphia Chromosome—Positive Leukaemias: An International Registry of Childhood Chronic Myeloid Leukaemia Study. Eur. J. Cancer 2020, 136, 107–112. [Google Scholar] [CrossRef]
- Lipton, J.H.; Chuah, C.; Guerci-Bresler, A.; Rosti, G.; Simpson, D.; Assouline, S.; Etienne, G.; Nicolini, F.E.; le Coutre, P.; Clark, R.E.; et al. Ponatinib versus Imatinib for Newly Diagnosed Chronic Myeloid Leukaemia: An International, Randomised, Open-Label, Phase 3 Trial. Lancet Oncol. 2016, 17, 612–621. [Google Scholar] [CrossRef]
- Soria, J.-C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in Untreated EGFR -Mutated Advanced Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 113–125. [Google Scholar] [CrossRef]
- Kim, D.-W.; Lee, D.H.; Han, J.-Y.; Lee, J.; Cho, B.C.; Kang, J.H.; Lee, K.H.; Cho, E.K.; Kim, J.-S.; Min, Y.J.; et al. Safety, Tolerability, and Anti-Tumor Activity of Olmutinib in Non-Small Cell Lung Cancer with T790M Mutation: A Single Arm, Open Label, Phase 1/2 Trial. Lung Cancer 2019, 135, 66–72. [Google Scholar] [CrossRef]
- Tan, D.S.-W.; Leighl, N.B.; Riely, G.J.; Yang, J.C.-H.; Sequist, L.V.; Wolf, J.; Seto, T.; Felip, E.; Aix, S.P.; Jonnaert, M.; et al. Safety and Efficacy of Nazartinib (EGF816) in Adults with EGFR-Mutant Non-Small-Cell Lung Carcinoma: A Multicentre, Open-Label, Phase 1 Study. Lancet Respir. Med. 2020, 8, 561–572. [Google Scholar] [CrossRef]
- Kaczmarska, A.; Śliwa, P.; Zawitkowska, J.; Lejman, M. Genomic Analyses of Pediatric Acute Lymphoblastic Leukemia Ph+ and Ph−like—Recent Progress in Treatment. Int. J. Mol. Sci. 2021, 22, 6411. [Google Scholar] [CrossRef]
- Kalmanti, L.; Saussele, S.; Lauseker, M.; Müller, M.C.; Dietz, C.T.; Heinrich, L.; Hanfstein, B.; Proetel, U.; Fabarius, A.; Krause, S.W.; et al. Safety and efficacy of imatinib in CML over a period of 10 years: Data from the randomized CML-study IV. Leukemia. 2015, 29, 1123–1132. [Google Scholar] [CrossRef]
- O’Brien, S.G.; Guilhot, F.; Larson, R.A.; Gathmann, I.; Baccarani, M.; Cervantes, F.; Cornelissen, J.J.; Fischer, T.; Hochhaus, A.; Hughes, T.; et al. Imatinib Compared with Interferon and Low-Dose Cytarabine for Newly Diagnosed Chronic-Phase Chronic Myeloid Leukemia. N. Engl. J. Med. 2003, 348, 994–1004. [Google Scholar] [CrossRef] [Green Version]
- Millot, F.; Baruchel, A.; Guilhot, J.; Petit, A.; Leblanc, T.; Bertrand, Y.; Mazingue, F.; Lutz, P.; Vérité, C.; Berthou, C.; et al. Imatinib Is Effective in Children with Previously Untreated Chronic Myelogenous Leukemia in Early Chronic Phase: Results of the French National Phase IV Trial. J. Clin. Oncol. 2011, 29, 2827–2832. [Google Scholar] [CrossRef] [Green Version]
- Hijiya, N.; Zwaan, C.M.; Rizzari, C.; Foà, R.; Abbink, F.; Lancaster, D.; Landman-Parker, J.; Millot, F.; Moppett, J.; Nelken, B.; et al. Pharmacokinetics of Nilotinib in Pediatric Patients with Philadelphia Chromosome-Positive Chronic Myeloid Leukemia or Acute Lymphoblastic Leukemia. Clin. Cancer Res. 2020, 26, 812–820. [Google Scholar] [CrossRef]
- Ward, E.; DeSantis, C.; Robbins, A.; Kohler, B.; Jemal, A. Childhood and Adolescent Cancer Statistics, 2014. CA Cancer J. Clin. 2014, 64, 83–103. [Google Scholar] [CrossRef]
- Pui, C.H.; Pei, D.; Campana, D.; Cheng, C.; Sandlund, J.T.; Bowman, W.P.; Hudson, M.M.; Ribeiro, R.C.; Raimondi, S.C.; Jeha, S.; et al. A Revised Definition for Cure of Childhood Acute Lymphoblastic Leukemia. Leukemia 2014, 28, 2336–2343. [Google Scholar] [CrossRef] [Green Version]
- Kebriaei, P.; Saliba, R.; Rondon, G.; Chiattone, A.; Luthra, R.; Anderlini, P.; Andersson, B.; Shpall, E.; Popat, U.; Jones, R.; et al. Long-Term Follow-up of Allogeneic Hematopoietic Stem Cell Transplantation for Patients with Philadelphia Chromosome-Positive Acute Lymphoblastic Leukemia: Impact of Tyrosine Kinase Inhibitors on Treatment Outcomes. Biol. Blood Marrow Transplant. 2012, 18, 584–592. [Google Scholar] [CrossRef] [Green Version]
- Biondi, A.; Gandemer, V.; De Lorenzo, P.; Cario, G.; Campbell, M.; Castor, A.; Pieters, R.; Baruchel, A.; Vora, A.; Leoni, V.; et al. Imatinib Treatment of Paediatric Philadelphia Chromosome-Positive Acute Lymphoblastic Leukaemia (EsPhALL2010): A Prospective, Intergroup, Open-Label, Single-Arm Clinical Trial. Lancet Haematol. 2018, 5, e641–e652. [Google Scholar] [CrossRef] [Green Version]
- Biondi, A.; Schrappe, M.; De Lorenzo, P.; Castor, A.; Lucchini, G.; Gandemer, V.; Pieters, R.; Stary, J.; Escherich, G.; Campbell, M.; et al. Imatinib after Induction for Treatment of Children and Adolescents with Philadelphia-Chromosome-Positive Acute Lymphoblastic Leukaemia (EsPhALL): A Randomised, Open-Label, Intergroup Study. Lancet Oncol. 2012, 13, 936–945. [Google Scholar] [CrossRef]
- Ribera, J.-M.; Oriol, A.; González, M.; Vidriales, B.; Brunet, S.; Esteve, J.; Del Potro, E.; Rivas, C.; Moreno, M.J.; Tormo, M.; et al. Concurrent Intensive Chemotherapy and Imatinib before and after Stem Cell Transplantation in Newly Diagnosed Philadelphia Chromosome-Positive Acute Lymphoblastic Leukemia. Final Results of the CSTIBES02 Trial. Haematologica 2010, 95, 87–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Den Boer, M.L.; van Slegtenhorst, M.; De Menezes, R.X.; Cheok, M.H.; Buijs-Gladdines, J.G.; Peters, S.T.; Van Zutven, L.J.; Beverloo, H.B.; Van der Spek, P.J.; Escherich, G.; et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: A genome-wide classification study. Lancet Oncol. 2009, 10, 125–134. [Google Scholar] [CrossRef] [Green Version]
- Tasian, S.K.; Loh, M.L.; Hunger, S.P. Philadelphia Chromosome–like Acute Lymphoblastic Leukemia. Blood 2017, 130, 2064–2072. [Google Scholar] [CrossRef] [PubMed]
- Tanasi, I.; Ba, I.; Sirvent, N.; Braun, T.; Cuccuini, W.; Ballerini, P.; Duployez, N.; Tanguy-Schmidt, A.; Tamburini, J.; Maury, S.; et al. Efficacy of tyrosine kinase inhibitors in Ph-like acute lymphoblastic leukemia harboring ABL-class rearrangements. Blood 2019, 134, 1351–1355. [Google Scholar] [CrossRef] [PubMed]
- Bond, M.; Bernstein, M.L.; Pappo, A.; Schultz, K.R.; Krailo, M.; Blaney, S.M.; Adamson, P.C. A Phase II Study of Imatinib Mesylate in Children with Refractory or Relapsed Solid Tumors: A Children’s Oncology Group Study. Pediatr. Blood Cancer 2008, 50, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Siehl, J.; Thiel, E. C-Kit, GIST, and Imatinib. In Targeted Theraphies in Cancer; Dietel, M., Ed.; Recent Results Cancer Research Series; Springer: Berlin/Heidelberg, Germany, 2007; Volume 176, pp. 145–151. [Google Scholar] [CrossRef]
- Demetri, G.D.; von Mehren, M.; Blanke, C.D.; Van den Abbeele, A.D.; Eisenberg, B.; Roberts, P.J.; Heinrich, M.C.; Tuveson, D.A.; Singer, S.; Janicek, M.; et al. Efficacy and Safety of Imatinib Mesylate in Advanced Gastrointestinal Stromal Tumors. N. Engl. J. Med. 2002, 347, 472–480. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.Y.; Janeway, K.; Pappo, A. Pediatric and Wild-Type Gastrointestinal Stromal Tumor: New Therapeutic Approaches. Curr. Opin. Oncol. 2010, 22, 347–350. [Google Scholar] [CrossRef] [Green Version]
- Hawkins, M.; Lancashire, E.; Winter, D.; Frobisher, C.; Reulen, R.; Taylor, A.; Stevens, M.; Jenney, M. The British Childhood Cancer Survivor Study: Objectives, Methods, Population Structure, Response Rates and Initial Descriptive Information. Pediatr. Blood Cancer 2008, 50, 1018–1025. [Google Scholar] [CrossRef]
- Son, M.K.; Ryu, M.-H.; Park, J.O.; Im, S.-A.; Kim, T.-Y.; Lee, S.J.; Ryoo, B.-Y.; Park, S.R.; Kang, Y.-K. Efficacy and Safety of Regorafenib in Korean Patients with Advanced Gastrointestinal Stromal Tumor after Failure of Imatinib and Sunitinib: A Multicenter Study Based on the Management Access Program. Cancer Res. Treat. 2017, 49, 350–357. [Google Scholar] [CrossRef] [Green Version]
- Cavnar, M.J.; Seier, K.; Curtin, C.; Balachandran, V.P.; Coit, D.G.; Yoon, S.S.; Crago, A.M.; Strong, V.E.; Tap, W.D.; Gönen, M.; et al. Outcome of 1000 Patients with Gastrointestinal Stromal Tumor (GIST) Treated by Surgery in the Pre- and Post-Imatinib Eras. Ann. Surg. 2021, 273, 128–138. [Google Scholar] [CrossRef]
- Takeshima, H.; Kuratsu, J.-I. A Review of Soluble C-Kit (s-Kit) as a Novel Tumor Marker and Possible Molecular Target for the Treatment of CNS Germinoma. Surg. Neurol. 2003, 60, 321–325. [Google Scholar] [CrossRef]
- Osorio, D.S.; Finlay, J.L.; Dhall, G.; Goldman, S.; Eisenstat, D.; Brown, R.J. Feasibility of Dasatinib in Children and Adolescents with New or Recurrent Central Nervous System Germinoma. Pediatr. Blood Cancer 2013, 60, E100–E102. [Google Scholar] [CrossRef] [PubMed]
- Pollack, I.F.; Stewart, C.F.; Kocak, M.; Poussaint, T.Y.; Broniscer, A.; Banerjee, A.; Douglas, J.G.; Kun, L.E.; Boyett, J.M.; Geyer, J.R. A Phase II Study of Gefitinib and Irradiation in Children with Newly Diagnosed Brainstem Gliomas: A Report from the Pediatric Brain Tumor Consortium. Neuro-Oncology 2011, 13, 290–297. [Google Scholar] [CrossRef] [Green Version]
- Daw, N.C.; Furman, W.L.; Stewart, C.F.; Iacono, L.C.; Krailo, M.; Bernstein, M.L.; Dancey, J.E.; Speights, R.A.; Blaney, S.M.; Croop, J.M.; et al. Phase I and Pharmacokinetic Study of Gefitinib in Children with Refractory Solid Tumors: A Children’s Oncology Group Study. J. Clin. Oncol. 2005, 23, 6172–6180. [Google Scholar] [CrossRef] [PubMed]
- Furman, W.L.; McGregor, L.M.; McCarville, M.B.; Onciu, M.; Davidoff, A.M.; Kovach, S.; Hawkins, D.; McPherson, V.; Houghton, P.J.; Billups, C.A.; et al. A Single-Arm Pilot Phase II Study of Gefitinib and Irinotecan in Children with Newly Diagnosed High-Risk Neuroblastoma. Investig. New Drugs 2012, 30, 1660–1670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freeman, B.B.; Daw, N.C.; Geyer, J.R.; Furman, W.L.; Stewart, C.F. Evaluation of Gefitinib for Treatment of Refractory Solid Tumors and Central Nervous System Malignancies in Pediatric Patients. Cancer Investig. 2006, 24, 310–317. [Google Scholar] [CrossRef]
- Stucklin, A.S.G.; Ryall, S.; Fukuoka, K.; Zapotocky, M.; Lassaletta, A.; Li, C.; Bridge, T.; Kim, B.; Arnoldo, A.; Kowalski, P.E. Alterations in ALK/ROS1/NTRK/MET Drive a Group of Infantile Hemispheric Gliomas. Nat. Commun. 2019, 10, 4343. [Google Scholar] [CrossRef] [Green Version]
- Mossé, Y.P.; Voss, S.D.; Lim, M.S.; Rolland, D.; Minard, C.G.; Fox, E.; Adamson, P.; Wilner, K.; Blaney, S.M.; Weigel, B.J. Targeting ALK With Crizotinib in Pediatric Anaplastic Large Cell Lymphoma and Inflammatory Myofibroblastic Tumor: A Children’s Oncology Group Study. J. Clin. Oncol. 2017, 35, 3215–3221. [Google Scholar] [CrossRef]
- Mossé, Y.P.; Laudenslager, M.; Longo, L.; Cole, K.A.; Wood, A.; Attiyeh, E.F.; Laquaglia, M.J.; Sennett, R.; Lynch, J.E.; Perri, P.; et al. Identification of ALK as a Major Familial Neuroblastoma Predisposition Gene. Nature 2008, 455, 930–935. [Google Scholar] [CrossRef] [Green Version]
- Henderson, T.O.; Bhatia, S.; Pinto, N.; London, W.B.; McGrady, P.; Crotty, C.; Sun, C.-L.; Cohn, S.L. Racial and Ethnic Disparities in Risk and Survival in Children with Neuroblastoma: A Children’s Oncology Group Study. J. Clin. Oncol. 2011, 29, 76–82. [Google Scholar] [CrossRef] [Green Version]
- Whittle, S.B.; Smith, V.; Doherty, E.; Zhao, S.; McCarty, S.; Zage, P.E. Overview and Recent Advances in the Treatment of Neuroblastoma. Expert Rev. Anticancer Ther. 2017, 17, 369–386. [Google Scholar] [CrossRef] [Green Version]
- Irwin, M.S.; Park, J.R. Neuroblastoma: Paradigm for Precision Medicine. Pediatr. Clin. North Am. 2015, 62, 225–256. [Google Scholar] [CrossRef]
- Guan, J.; Fransson, S.; Siaw, J.T.; Treis, D.; Van Den Eynden, J.; Chand, D.; Umapathy, G.; Ruuth, K.; Svenberg, P.; Wessman, S.; et al. Clinical Response of the Novel Activating ALK-I1171T Mutation in Neuroblastoma to the ALK Inhibitor Ceritinib. Mol. Case Stud. 2018, 4, a002550. [Google Scholar] [CrossRef]
- Mossé, Y.P.; Lim, M.S.; Voss, S.D.; Wilner, K.; Ruffner, K.; Laliberte, J.; Rolland, D.; Balis, F.M.; Maris, J.M.; Weigel, B.J.; et al. Safety and Activity of Crizotinib for Paediatric Patients with Refractory Solid Tumours or Anaplastic Large-Cell Lymphoma. Lancet Oncol. 2013, 14, 472–480. [Google Scholar] [CrossRef] [Green Version]
- Savary, C.; Kim, A.; Lespagnol, A.; Gandemer, V.; Pellier, I.; Andrieu, C.; Pagès, G.; Galibert, M.-D.; Blum, Y.; de Tayrac, M. Depicting the Genetic Architecture of Pediatric Cancers through an Integrative Gene Network Approach. Sci. Rep. 2020, 10, 1224. [Google Scholar] [CrossRef] [Green Version]
- Allen, C.E.; Laetsch, T.W.; Mody, R.; Irwin, M.S.; Lim, M.S.; Adamson, P.C.; Seibel, N.L.; Parsons, D.W.; Cho, Y.J.; Janeway, K. Target and Agent Prioritization for the Children’s Oncology Group—National Cancer Institute Pediatric MATCH Trial. J. Natl. Cancer Inst. 2017, 109, djw274. [Google Scholar] [CrossRef] [Green Version]
- Chen, A.P.; Eljanne, M.; Harris, L.; Malik, S.; Seibel, N.L. National Cancer Institute Basket/Umbrella Clinical Trials: MATCH, LungMAP, and Beyond. Cancer J. 2019, 25, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Parsons, D.W.; Janeway, K.A.; Patton, D.; Coffey, B.; Williams, P.M.; Hamilton, S.R.; Purkayastha, A.; Tsongalis, G.J.; Routbort, M.; Gastier-Foster, J.M.; et al. Identification of Targetable Molecular Alterations in the NCI-COG Pediatric MATCH Trial. J. Clin. Oncol. 2019, 37, 10011. [Google Scholar] [CrossRef]
- Drilon, A.; Laetsch, T.W.; Kummar, S.; DuBois, S.G.; Lassen, U.N.; Demetri, G.D.; Nathenson, M.; Doebele, R.C.; Farago, A.F.; Pappo, A.S.; et al. Efficacy of Larotrectinib in TRK Fusion-Positive Cancers in Adults and Children. N. Engl. J. Med. 2018, 378, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.S.; DuBois, S.G.; Kummar, S.; Farago, A.F.; Albert, C.M.; Rohrberg, K.S.; van Tilburg, C.M.; Nagasubramanian, R.; Berlin, J.D.; Federman, N.; et al. Larotrectinib in Patients with TRK Fusion-Positive Solid Tumours: A Pooled Analysis of Three Phase 1/2 Clinical Trials. Lancet. Oncol. 2020, 21, 531–540. [Google Scholar] [CrossRef]
- Roth, J.A.; Carlson, J.J.; Xia, F.; Williamson, T.; Sullivan, S.D. The Potential Long-Term Comparative Effectiveness of Larotrectinib and Entrectinib for Second-Line Treatment of TRK Fusion-Positive Metastatic Lung Cancer. J. Manag. Care Spec. Pharm. 2020, 26, 981–986. [Google Scholar] [CrossRef] [PubMed]
- Laetsch, T.W.; DuBois, S.G.; Mascarenhas, L.; Turpin, B.; Federman, N.; Albert, C.M.; Nagasubramanian, R.; Davis, J.L.; Rudzinski, E.; Feraco, A.M.; et al. Larotrectinib for Paediatric Solid Tumours Harbouring NTRK Gene Fusions: Phase 1 Results from a Multicentre, Open-Label, Phase 1/2 Study. Lancet Oncol. 2018, 19, 705–714. [Google Scholar] [CrossRef]
- Bahleda, R.; Italiano, A.; Hierro, C.; Mita, A.; Cervantes, A.; Chan, N.; Awad, M.; Calvo, E.; Moreno, V.; Govindan, R.; et al. Multicenter Phase I Study of Erdafitinib (JNJ-42756493), Oral Pan-Fibroblast Growth Factor Receptor Inhibitor, in Patients with Advanced or Refractory Solid Tumors. Clin. Cancer Res. 2019, 25, 4888–4897. [Google Scholar] [CrossRef]
- Loriot, Y.; Necchi, A.; Park, S.H.; Garcia-Donas, J.; Huddart, R.; Burgess, E.; Fleming, M.; Rezazadeh, A.; Mellado, B.; Varlamov, S.; et al. Erdafitinib in Locally Advanced or Metastatic Urothelial Carcinoma. N. Engl. J. Med. 2019, 381, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Tabernero, J.; Bahleda, R.; Dienstmann, R.; Infante, J.R.; Mita, A.; Italiano, A.; Calvo, E.; Moreno, V.; Adamo, B.; Gazzah, A.; et al. Phase I Dose-Escalation Study of JNJ-42756493, an Oral Pan-Fibroblast Growth Factor Receptor Inhibitor, in Patients with Advanced Solid Tumors. J. Clin. Oncol. 2015, 33, 3401–3408. [Google Scholar] [CrossRef]
- Van Mater, D.; Gururangan, S.; Becher, O.; Campagne, O.; Leary, S.; Phillips, J.J.; Huang, J.; Lin, T.; Poussaint, T.Y.; Goldman, S.; et al. A Phase I Trial of the CDK 4/6 Inhibitor Palbociclib in Pediatric Patients with Progressive Brain Tumors: A Pediatric Brain Tumor Consortium Study (PBTC-042). Pediatr. Blood Cancer 2021, 68, e28879. [Google Scholar] [CrossRef]
- Finn, R.S.; Martin, M.; Rugo, H.S.; Jones, S.; Im, S.-A.; Gelmon, K.; Harbeck, N.; Lipatov, O.N.; Walshe, J.M.; Moulder, S.; et al. Palbociclib and Letrozole in Advanced Breast Cancer. N. Engl. J. Med. 2016, 375, 1925–1936. [Google Scholar] [CrossRef]
- Edelman, M.J.; Redman, M.W.; Albain, K.S.; McGary, E.C.; Rafique, N.M.; Petro, D.; Waqar, S.N.; Minichiello, K.; Miao, J.; Papadimitrakopoulou, V.A.; et al. SWOG S1400C (NCT02154490)—A Phase II Study of Palbociclib for Previously Treated Cell Cycle Gene Alteration—Positive Patients with Stage IV Squamous Cell Lung Cancer (Lung-MAP Substudy). J. Thorac. Oncol. 2019, 14, 1853–1859. [Google Scholar] [CrossRef]
- Adkins, D.; Ley, J.; Neupane, P.; Worden, F.; Sacco, A.G.; Palka, K.; Grilley-Olson, J.E.; Maggiore, R.; Salama, N.N.; Trinkaus, K. Palbociclib and Cetuximab in Platinum-Resistant and in Cetuximab-Resistant Human Papillomavirus-Unrelated Head and Neck Cancer: A Multicentre, Multigroup, Phase 2 Trial. Lancet Oncol. 2019, 20, 1295–1305. [Google Scholar] [CrossRef]
- Taylor, J.W.; Parikh, M.; Phillips, J.J.; James, C.D.; Molinaro, A.M.; Butowski, N.A.; Clarke, J.L.; Oberheim-Bush, N.A.; Chang, S.M.; Berger, M.S.; et al. Phase-2 Trial of Palbociclib in Adult Patients with Recurrent RB1-Positive Glioblastoma. J. Neurooncol. 2018, 140, 477–483. [Google Scholar] [CrossRef]
- Valiakhmetova, A.; Gorelyshev, S.; Konovalov, A.; Trunin, Y.; Savateev, A.; Kram, D.E.; Severson, E.; Hemmerich, A.; Edgerly, C.; Duncan, D.; et al. Treatment of Pediatric Glioblastoma with Combination Olaparib and Temozolomide Demonstrates 2-Year Durable Response. Oncologist 2020, 25, e198–e202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray-Coquard, I.; Pautier, P.; Pignata, S.; Pérol, D.; González-Martín, A.; Berger, R.; Fujiwara, K.; Vergote, I.; Colombo, N.; Mäenpää, J.; et al. Olaparib plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2416–2428. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Survival with Olaparib in Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 383, 2345–2357. [Google Scholar] [CrossRef]
- Robson, M.; Im, S.-A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef]
- Wang, Y.; Yuan, X.; Xiong, J.; Hao, Z.; Peng, X.; Chen, W.; Cui, L.; Li, H.; Wang, X.; He, X. Pharmacology and Clinical Evaluation of Ensartinib Hydrochloride Capsule. Zhongguo Feiai Zazhi 2020, 23, 719–729. [Google Scholar] [CrossRef]
- Yang, Y.; Zhou, J.; Zhou, J.; Feng, J.; Zhuang, W.; Chen, J.; Zhao, J.; Zhong, W.; Zhao, Y.; Zhang, Y.; et al. Efficacy, Safety, and Biomarker Analysis of Ensartinib in Crizotinib-Resistant, ALK-Positive Non-Small-Cell Lung Cancer: A Multicentre, Phase 2 Trial. Lancet Respir. Med. 2020, 8, 45–53. [Google Scholar] [CrossRef]
- Hong, D.S.; Moore, K.N.; Bendell, J.C.; Karp, D.D.; Wang, J.S.; Ulahannan, S.V.; Jones, S.; Wu, W.; Donoho, G.P.; Ding, Y.; et al. Preclinical Evaluation and Phase Ib Study of Prexasertib, a CHK1 Inhibitor, and Samotolisib (LY3023414), a Dual PI3K/MTOR Inhibitor. Clin. Cancer Res. 2021, 27, 1864–1874. [Google Scholar] [CrossRef]
- Sullivan, R.J.; Infante, J.R.; Janku, F.; Wong, D.J.L.; Sosman, J.A.; Keedy, V.; Patel, M.R.; Shapiro, G.I.; Mier, J.W.; Tolcher, A.W.; et al. First-in-Class ERK1/2 Inhibitor Ulixertinib (BVD-523) in Patients with MAPK Mutant Advanced Solid Tumors: Results of a Phase I Dose-Escalation and Expansion Study. Cancer Discov. 2018, 8, 184–195. [Google Scholar] [CrossRef] [Green Version]
- Mendzelevski, B.; Ferber, G.; Janku, F.; Li, B.T.; Sullivan, R.J.; Welsch, D.; Chi, W.; Jackson, J.; Weng, O.; Sager, P.T. Effect of Ulixertinib, a Novel ERK1/2 Inhibitor, on the QT/QTc Interval in Patients with Advanced Solid Tumor Malignancies. Cancer Chemother. Pharmacol. 2018, 81, 1129–1141. [Google Scholar] [CrossRef] [Green Version]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M. Improved Survival with Vemurafenib in Melanoma with BRAF V600E Mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [Green Version]
- Garbe, C.; Eigentler, T.K. Vemurafenib. In Small Molecules in Oncology; Martins, U.W., Ed.; Recent Results Cancer Research Series; Springer: Berlin/Heidelberg, Germany, 2018; Volume 211, pp. 77–89. [Google Scholar] [CrossRef]
- Banerji, U.; Camidge, D.R.; Verheul, H.M.W.; Agarwal, R.; Sarker, D.; Kaye, S.B.; Desar, I.M.E.; Timmer-Bonte, J.N.H.; Eckhardt, S.G.; Lewis, K.D. The First-in-Human Study of the Hydrogen Sulfate (Hyd-Sulfate) Capsule of the MEK1/2 Inhibitor AZD6244 (ARRY-142886): A Phase I Open-Label Multicenter Trial in Patients with Advanced Cancer. Clin. Cancer Res. 2010, 16, 1613–1623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fangusaro, J.; Onar-Thomas, A.; Young Poussaint, T.; Wu, S.; Ligon, A.H.; Lindeman, N.; Banerjee, A.; Packer, R.J.; Kilburn, L.B.; Goldman, S.; et al. Selumetinib in Paediatric Patients with BRAF-Aberrant or Neurofibromatosis Type 1-Associated Recurrent, Refractory, or Progressive Low-Grade Glioma: A Multicentre, Phase 2 Trial. Lancet Oncol. 2019, 20, 1011–1022. [Google Scholar] [CrossRef]
- Carvajal, R.D.; Piperno-Neumann, S.; Kapiteijn, E.; Chapman, P.B.; Frank, S.; Joshua, A.M.; Piulats, J.M.; Wolter, P.; Cocquyt, V.; Chmielowski, B.; et al. Selumetinib in Combination with Dacarbazine in Patients with Metastatic Uveal Melanoma: A Phase III, Multicenter, Randomized Trial (SUMIT). J. Clin. Oncol. 2018, 36, 1232–1239. [Google Scholar] [CrossRef] [Green Version]
- Hoy, S.M. Tazemetostat: First Approval. Drugs 2020, 80, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Morschhauser, F.; Tilly, H.; Chaidos, A.; McKay, P.; Phillips, T.; Assouline, S.; Batlevi, C.L.; Campbell, P.; Ribrag, V.; Damaj, G.L.; et al. Tazemetostat for Patients with Relapsed or Refractory Follicular Lymphoma: An Open-Label, Single-Arm, Multicentre, Phase 2 Trial. Lancet Oncol. 2020, 21, 1433–1442. [Google Scholar] [CrossRef]
- Izutsu, K.; Ando, K.; Nishikori, M.; Shibayama, H.; Teshima, T.; Kuroda, J.; Kato, K.; Imaizumi, Y.; Nosaka, K.; Sakai, R.; et al. Phase II Study of Tazemetostat for Relapsed or Refractory B-Cell Non-Hodgkin Lymphoma with EZH2 Mutation in Japan. Cancer Sci. 2021, 112, 3627–3635. [Google Scholar] [CrossRef]
- Widemann, B.C.; Arceci, R.J.; Jayaprakash, N.; Fox, E.; Zannikos, P.; Goodspeed, W.; Goodwin, A.; Wright, J.J.; Blaney, S.M.; Adamson, P.C.; et al. Phase 1 Trial and Pharmacokinetic Study of the Farnesyl Transferase Inhibitor Tipifarnib in Children and Adolescents with Refractory Leukemias: A Report from the Children’s Oncology Group. Pediatr. Blood Cancer 2011, 56, 226–233. [Google Scholar] [CrossRef] [Green Version]
- Haas-Kogan, D.A.; Banerjee, A.; Kocak, M.; Prados, M.D.; Geyer, J.R.; Fouladi, M.; McKnight, T.; Poussaint, T.Y.; Broniscer, A.; Blaney, S.M.; et al. Phase I Trial of Tipifarnib in Children with Newly Diagnosed Intrinsic Diffuse Brainstem Glioma. Neuro Oncol. 2008, 10, 341–347. [Google Scholar] [CrossRef] [Green Version]
- Yanamandra, N.; Colaco, N.M.; Parquet, N.A.; Buzzeo, R.W.; Boulware, D.; Wright, G.; Perez, L.E.; Dalton, W.S.; Beaupre, D.M. Tipifarnib and Bortezomib Are Synergistic and Overcome Cell Adhesion-Mediated Drug Resistance in Multiple Myeloma and Acute Myeloid Leukemia. Clin. Cancer Res. 2006, 12, 591–599. [Google Scholar] [CrossRef] [Green Version]
- Jabbour, E.; Kantarjian, H.; Ravandi, F.; Garcia-Manero, G.; Estrov, Z.; Verstovsek, S.; O’Brien, S.; Faderl, S.; Thomas, D.A.; Wright, J.J.; et al. A Phase 1-2 Study of a Farnesyltransferase Inhibitor, Tipifarnib, Combined with Idarubicin and Cytarabine for Patients with Newly Diagnosed Acute Myeloid Leukemia and High-Risk Myelodysplastic Syndrome. Cancer 2011, 117, 1236–1244. [Google Scholar] [CrossRef]
- Drilon, A.; Oxnard, G.R.; Tan, D.S.W.; Loong, H.H.F.; Johnson, M.; Gainor, J.; McCoach, C.E.; Gautschi, O.; Besse, B.; Cho, B.C.; et al. Efficacy of Selpercatinib in RET Fusion-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2020, 383, 813–824. [Google Scholar] [CrossRef]
- Wirth, L.J.; Sherman, E.; Robinson, B.; Solomon, B.; Kang, H.; Lorch, J.; Worden, F.; Brose, M.; Patel, J.; Leboulleux, S.; et al. Efficacy of Selpercatinib in RET-Altered Thyroid Cancers. N. Engl. J. Med. 2020, 383, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.; Ma, C.; DuBois, S.G. New approaches to therapeutic drug development for childhood cancers. Curr. Opin. Pediatr. 2020, 32, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Shoukier, M.; Kubiak, M.; Cortes, J. Review of New-Generation Tyrosine Kinase Inhibitors for Chronic Myeloid Leukemia. Curr. Oncol. Rep. 2021, 23, 91. [Google Scholar] [CrossRef] [PubMed]
- Hughes, T.P.; Mauro, M.J.; Cortes, J.E.; Minami, H.; Rea, D.; DeAngelo, D.J.; Breccia, M.; Goh, Y.T.; Talpaz, M.; Hochhaus, A.; et al. Asciminib in Chronic Myeloid Leukemia after ABL Kinase Inhibitor Failure. N. Engl. J. Med. 2019, 381, 2315–2326. [Google Scholar] [CrossRef]
- Rea, D.; Mauro, M.J.; Boquimpani, C.; Minami, Y.; Lomaia, E.; Voloshin, S.; Turkina, A.G.; Kim, D.W.; Apperley, J.F.; Abdo, A.; et al. A Phase 3, Open-Label, Randomized Study of Asciminib, a STAMP Inhibitor, vs Bosutinib in CML After ≥2 Prior TKIs. Blood 2021, in press. [Google Scholar] [CrossRef]
- Zerbit, J.; Tamburini, J.; Goldwirt, L.; Decroocq, J.; Cayuela, J.M.; Chapuis, N.; Contejean, A.; Batista, R.; Bouscary, D.; Willems, L. Asciminib and ponatinib combination in Philadelphia chromosome-positive acute lymphoblastic leukemia. Leuk. Lymphoma 2021, 1–4. [Google Scholar] [CrossRef]
- Zabkiewicz, J.; Lazenby, M.; Edwards, G.; Bygrave, C.A.; Omidvar, N.; Zhuang, L.; Knapper, S.; Guy, C.; Hills, R.K.; Burnett, A.K.; et al. Combination of a mitogen-activated protein kinase inhibitor with the tyrosine kinase inhibitor pacritinib combats cell adhesion-based residual disease and prevents re-expansion of FLT3-ITD acute myeloid leukaemia. Br. J. Haematol. 2020, 191, 231–242. [Google Scholar] [CrossRef]
- Salvaris, R.; Fedele, P.L. Targeted Therapy in Acute Lymphoblastic Leukaemia. J. Pers. Med. 2021, 11, 715. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Generation of TKIs 1 | Name | Chemical Structure | First FDA 2 Approval | Target | Disease | Children | Adults |
|---|---|---|---|---|---|---|---|
| I | Imatinib |  | 2001 | BCR-ABL1 3 | CML 4 | [33,34] | [35,36] |
| Ph+ ALL 5 | [37] | [38] | |||||
| GIST 6 | [39] | [40,41] | |||||
| Brainstem gliomas, intracranial gliomas | [42] | [43] | |||||
| Crizotinib |  | 2011 | ALK 7, ROS1 8, c-MET 9 | ALK-positive lung cancer | No data | [44] | |
| Intrinsic pontine glioma | [45] | No data | |||||
| Solid tumours (hepatocellular carcinoma, adrenal cortical carcinoma, Wilms’ tumour, neuroblastoma ganglioneuroblastoma, rhabdomyosarcoma, synovial sarcoma, epithelial myoepithelial carcinoma, Ewing sarcoma), anaplastic large-cell lymphoma | [46] | [47] | |||||
| Non-small-cell lung carcinoma | No data | [48] | |||||
| Sunitinib |  | 2006 | VEGFR 10, VEGFR2 11, VEGFR3 12, c- KIT 13, FLT3 14, CSF-1R 15, RET 16 | Renal cell carcinoma | [49] | [50] | |
| GIST | [51] | No data | |||||
| II | Regorafenib |  | 2012 | VEGF, EGFR 17 | Liposarcoma | Clinical trial number NCT02085148 | [52] |
| GIST | [53] | ||||||
| Hepatocellular carcinoma | [54] | ||||||
| Dasatinib |  | 2006 | BCR-ABL1, Src 18, c-Kit, ephrin receptors | CML | [55,56] | [57] | |
| Ph+ ALL | [58] | [57] | |||||
| Bosutinib |  | 2012 | BCR-ABL1, Src | CML | Clinical trial number NCT04258943 | [41,59] | |
| Glioblastoma | No data | [60] | |||||
| Nilotinib |  | 2007 | BCR-ABL1 | Ph+ ALL | No data | [58] | |
| CML | [61] | [62] | |||||
| GIST | No data | [63] | |||||
| Glioma | [64] | No data | |||||
| III | Ponatinib |  | 2012 | BCR-ABL1 | Glioblastoma | No data | [65] |
| ALL 19 | Clinical trial number NCT04501614 | [66] | |||||
| AML 20 | No data | [67] | |||||
| CML | [68] | [69] | |||||
| Osimertinib |  | 2020 | EGFR | Non-small-cell lung carcinoma | No data | [70] | |
| Olmutinib |  | No FDA approval, approval in South Korea | No data | [71] | |||
| Nazartinib |  | No approval, clinical trial number: NCT03040973 | Non-small-cell lung carcinoma, advanced solid tumours | No data | [72] |
| Target | Drug | Used in the Treatment | References |
|---|---|---|---|
| NTRK1 1, NTRK2 2, or NTRK3 3 gene fusion | Larotrectinib | Solid tumours, CNS 4 tumours, lung cancer, sarcomas, papillary thyroid cancer | [112,113,114,115] |
| FGFR1 5, FGFR2 6, FGFR3 7, FGFR4 8 | Erdafitinib | Urothelial carcinoma, glioblastoma, endometrial cancer | [116,117,118] |
| Rb 9-positive, alterations in cell cycle genes | Palbociclib | Breast cancer, lung cancer, head and neck cancer, glioblastoma | [119,120,121,122,123] |
| ATM 10, BRCA1 11, BRCA2 12, RAD51C 13, RAD51D 14 | Olaparib | Prostatic cancer, ovarian cancer, breast cancer | [124,125,126,127] |
| ALK 15 or ROS1 16 | Ensartinib | Non-small-cell lung carcinoma | [128,129] |
| TSC1 17, TSC2 18, PI3K/mTOR 19 | Samotolisib | Preclinical studies only | [130] |
| MAPK 20 pathway mutations | Ulixertinib | Melanoma, colorectal cancer, lung cancer, non-small-cell lung carcinoma | [131,132] |
| BRAF 21 V600 | Vemurafenib | Melanoma | [133,134] |
| Activating MAPK 20 pathway | Selumetinib sulphate | Melanoma, solid tumours | [135,136,137] |
| EZH2 22, SMARCB1 23, SMARCA4 24 | Tazemetostat | Follicular lymphoma, CNS tumours, solid tumours, B-cell non-Hodgkin lymphoma | [138,139,140] |
| HRAS 25 gene alterations | Tipifarnib | Salivary gland cancer, breast cancer, AML26, myelodysplastic syndrome, solid tumours | [141,142,143,144] |
| Activating RET 27 mutations | Selpercatinib | Non-small-cell lung cancers, thyroid cancer | [145,146] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaczmarska, A.; Śliwa, P.; Lejman, M.; Zawitkowska, J. The Use of Inhibitors of Tyrosine Kinase in Paediatric Haemato-Oncology—When and Why? Int. J. Mol. Sci. 2021, 22, 12089. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222112089
Kaczmarska A, Śliwa P, Lejman M, Zawitkowska J. The Use of Inhibitors of Tyrosine Kinase in Paediatric Haemato-Oncology—When and Why? International Journal of Molecular Sciences. 2021; 22(21):12089. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222112089
Chicago/Turabian StyleKaczmarska, Agnieszka, Patrycja Śliwa, Monika Lejman, and Joanna Zawitkowska. 2021. "The Use of Inhibitors of Tyrosine Kinase in Paediatric Haemato-Oncology—When and Why?" International Journal of Molecular Sciences 22, no. 21: 12089. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222112089