
The Effect of Tuberculosis Antimicrobials on the Immunometabolic Profiles of Primary Human Macrophages Stimulated with Mycobacterium tuberculosis

 ,
,  , , ,
, , ,
Abstract
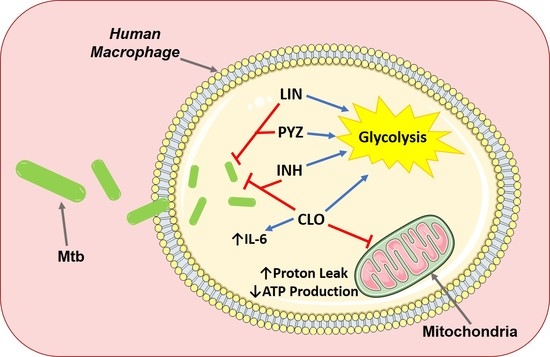
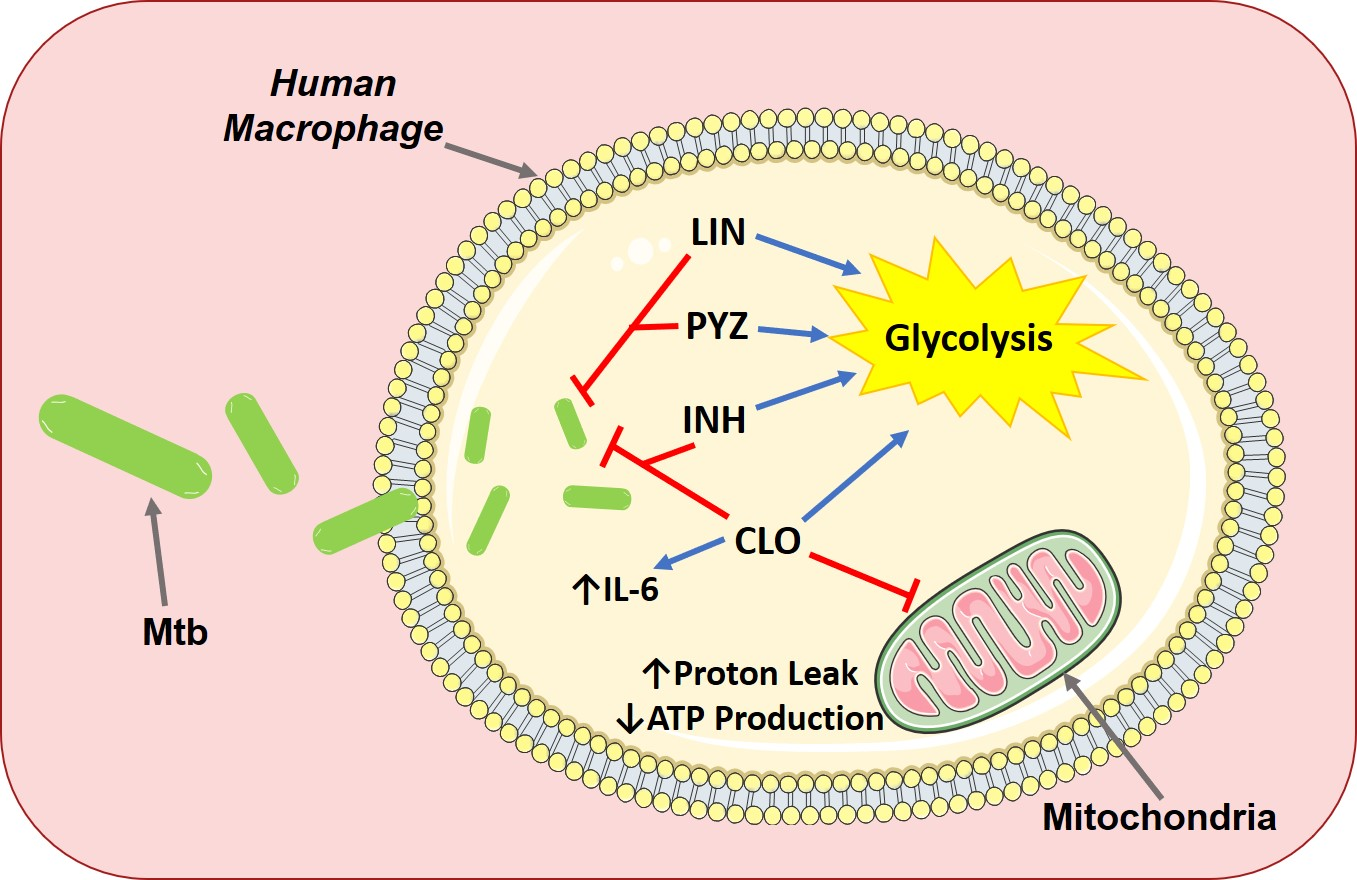
:
1. Introduction
2. Results
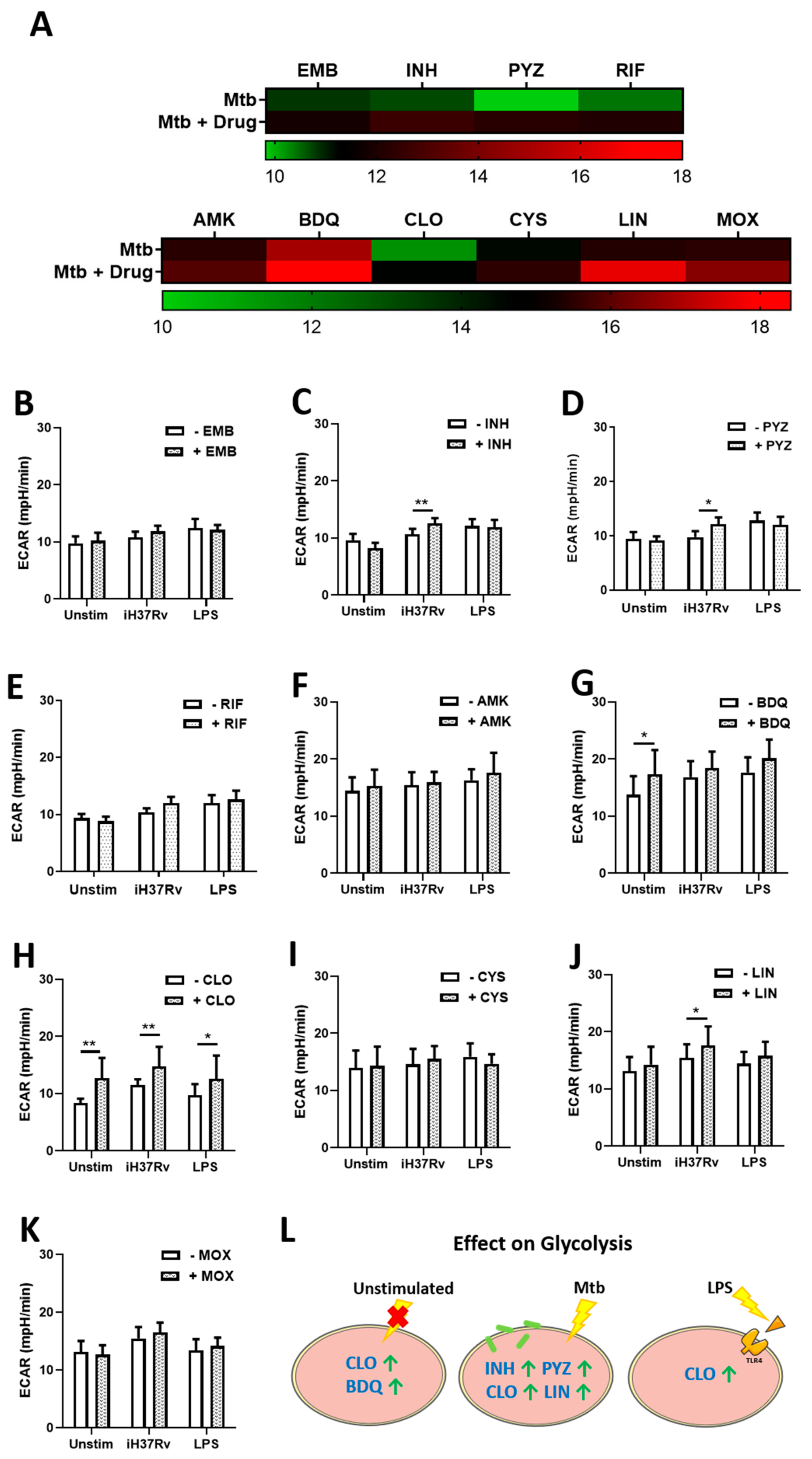
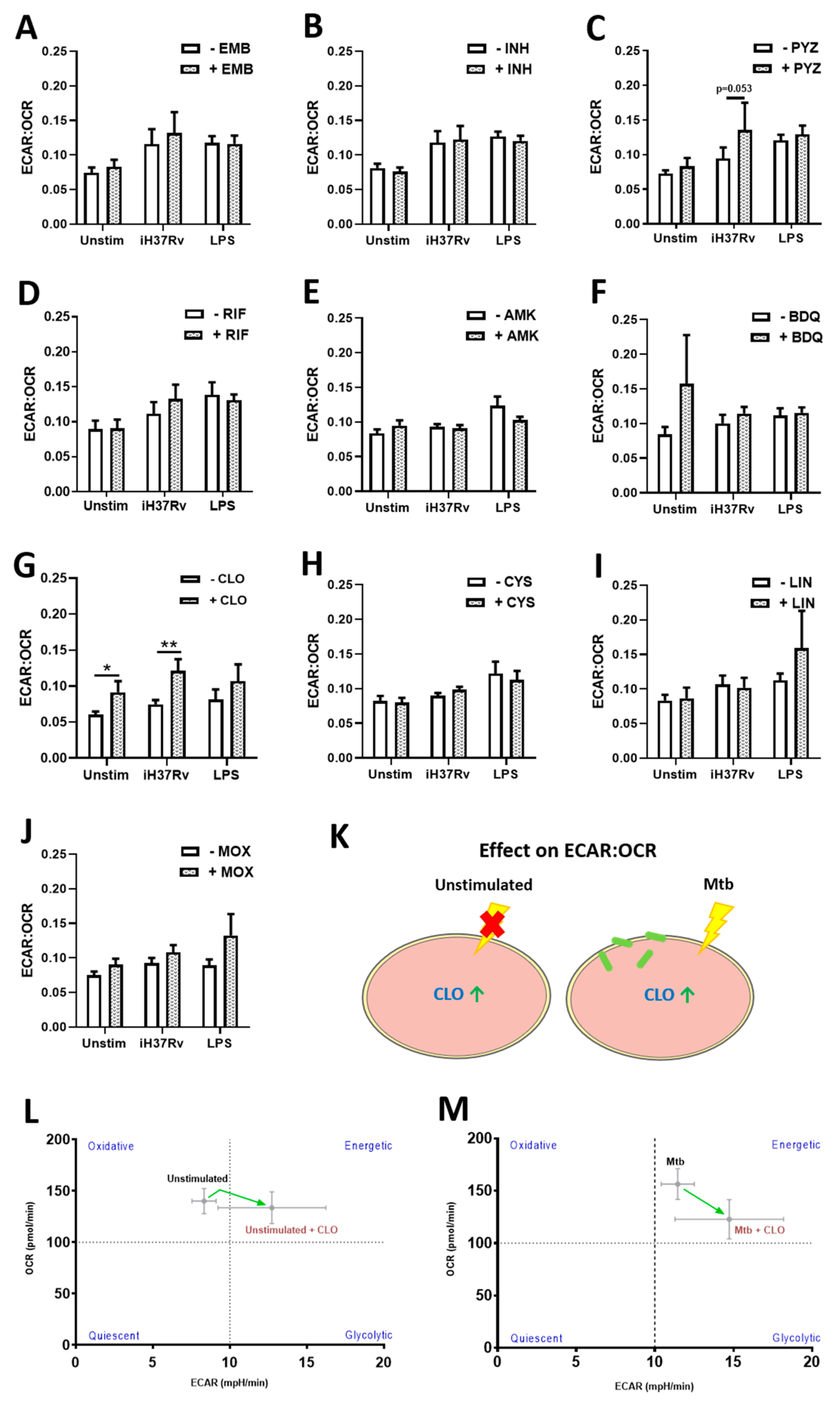
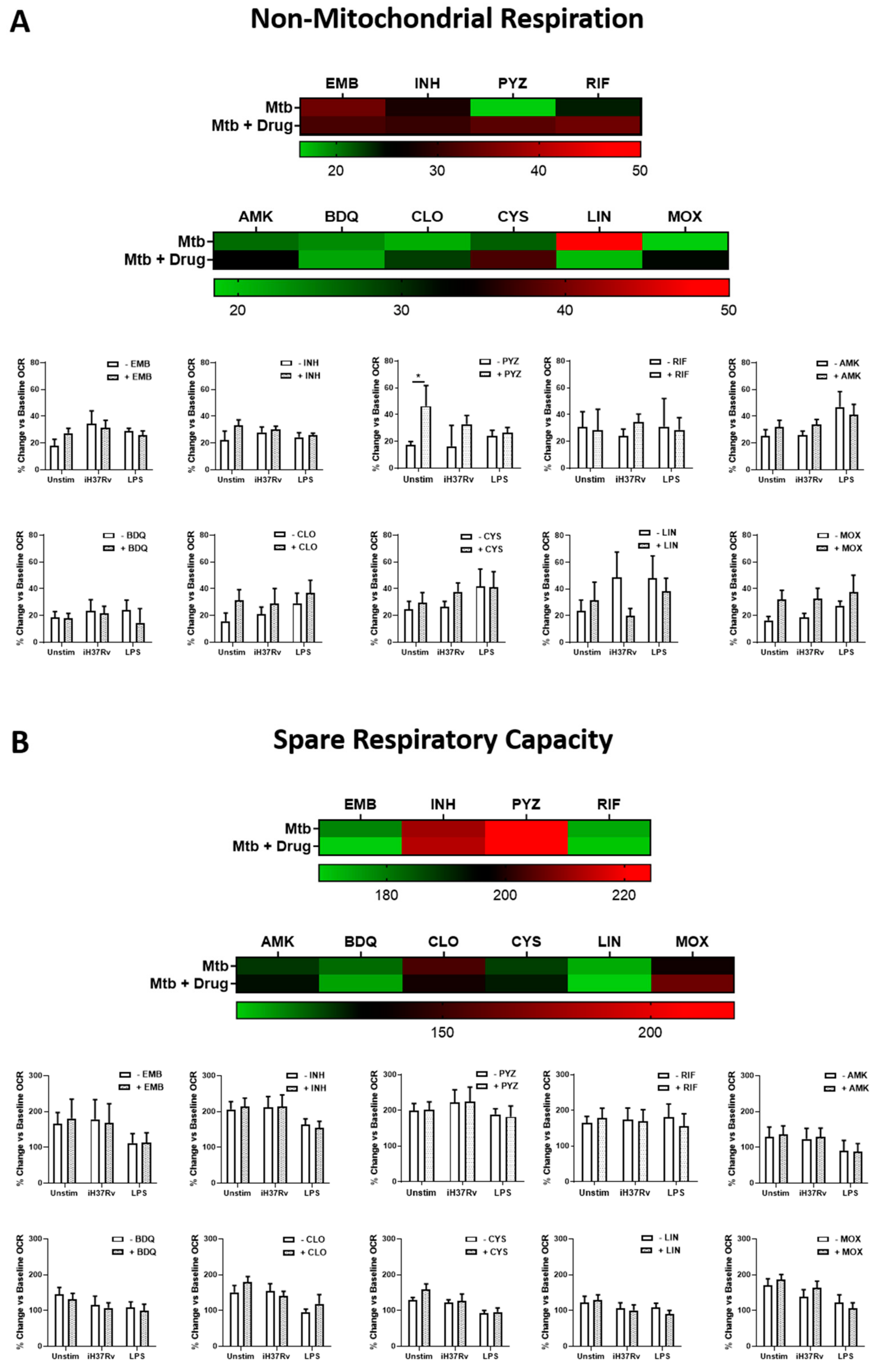
2.1. TB Antimicrobials Differentially Affect Glycolysis and Oxidative Phosphorylation Levels in Unstimulated, iH37Rv-Mtb- and LPS-Stimulated hMDMs
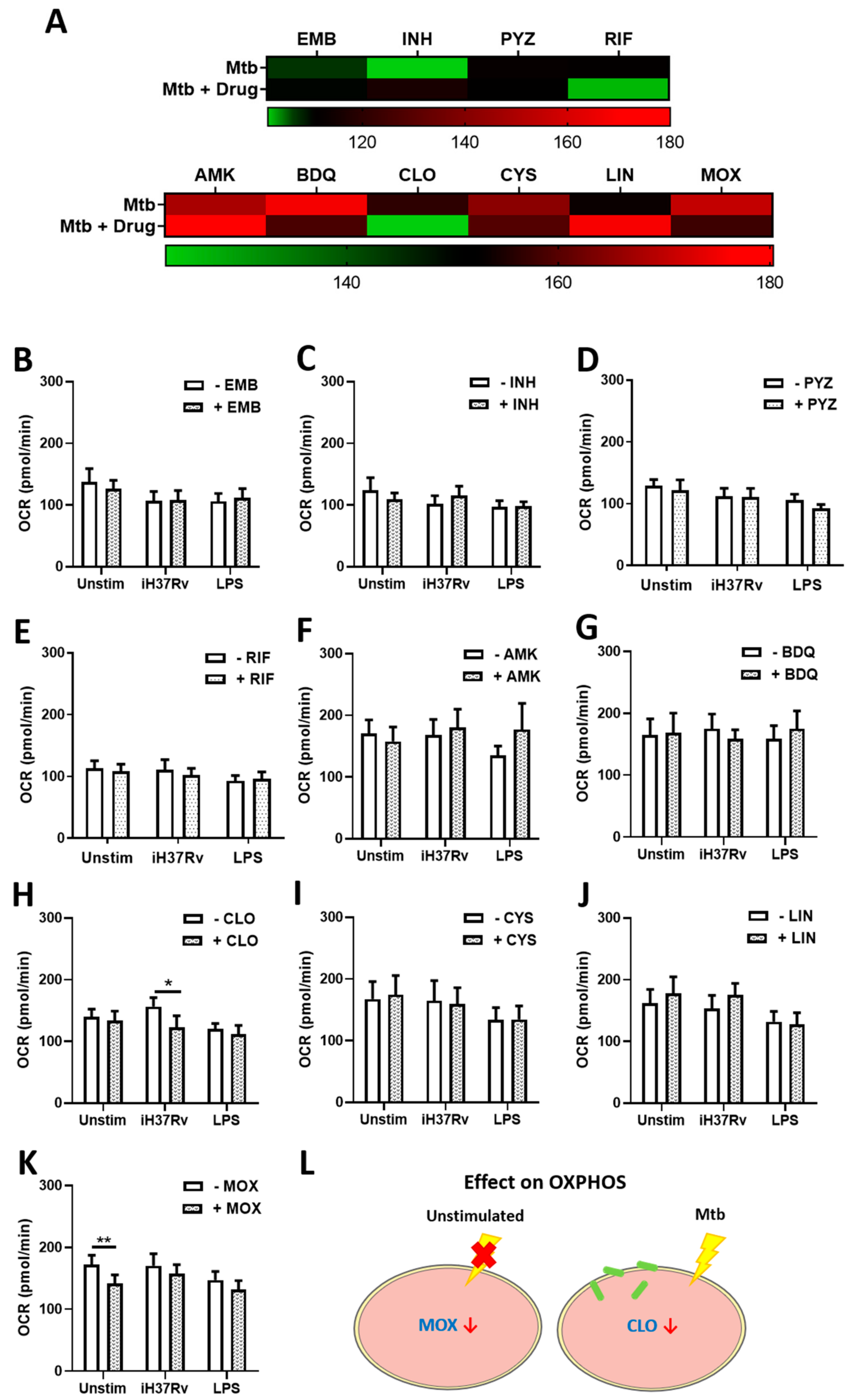
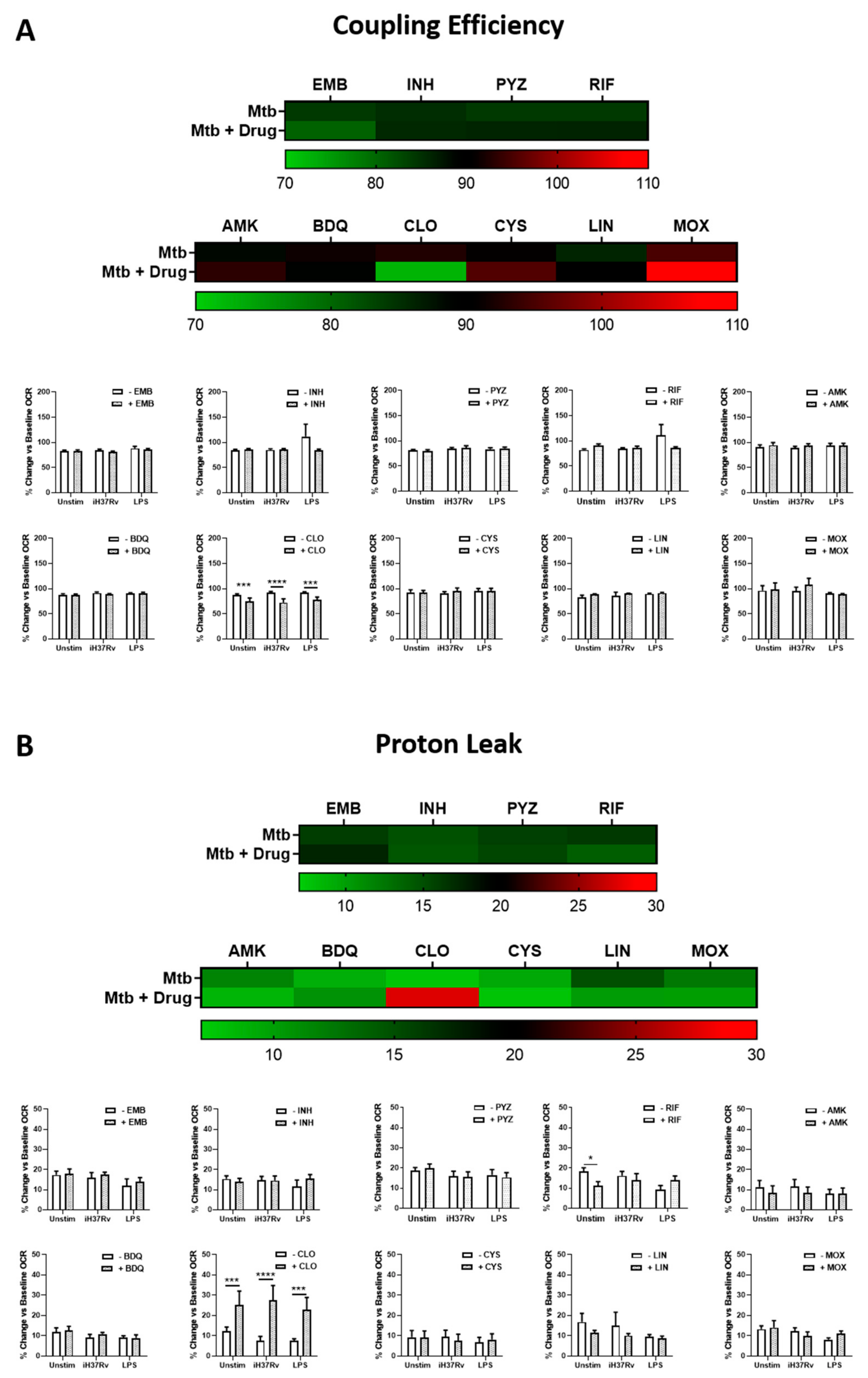
2.2. Clofazimine Alters Mitochondrial Coupling Efficacy and Protein Leak in Primary hMDMs Stimulated with Mtb
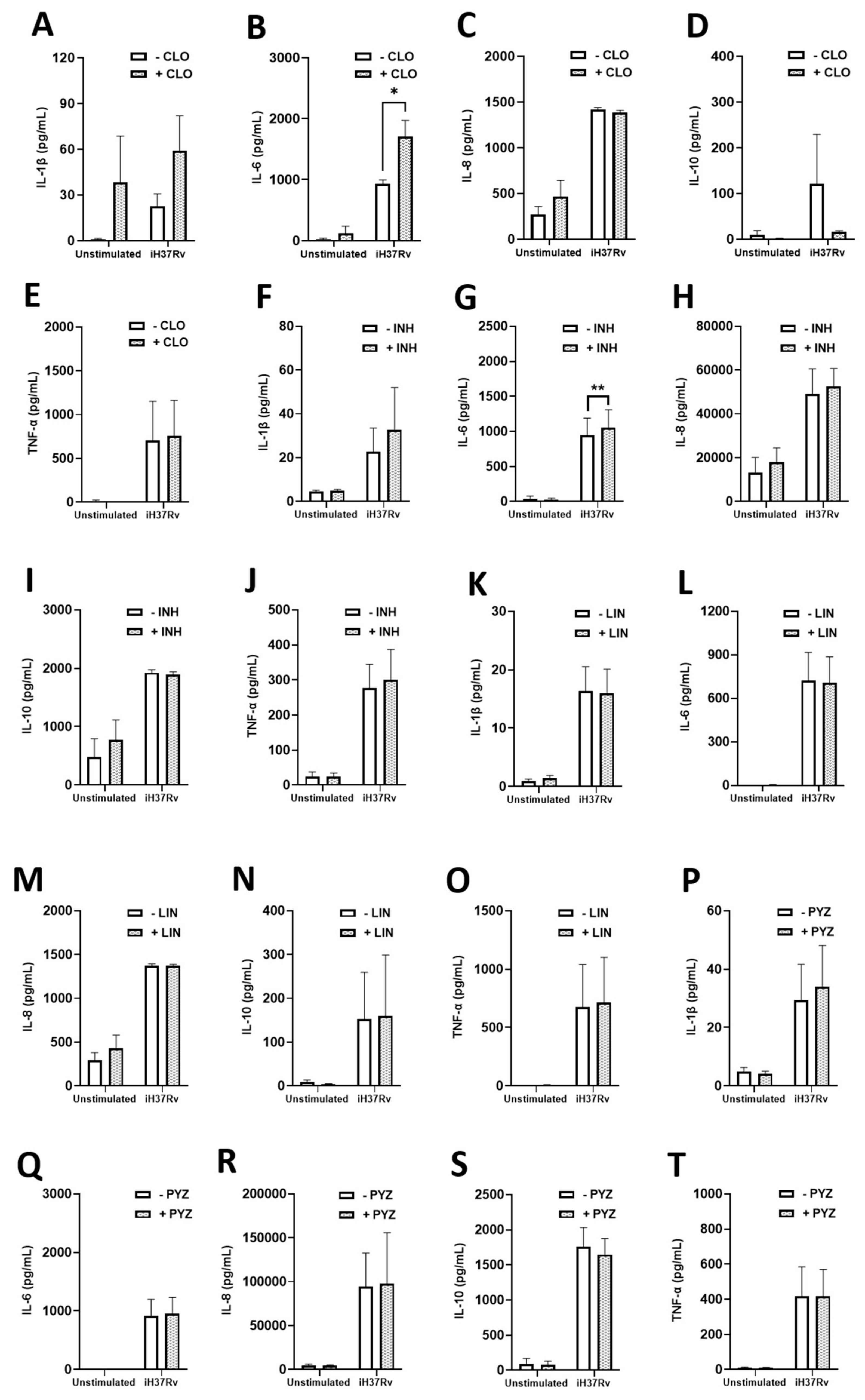
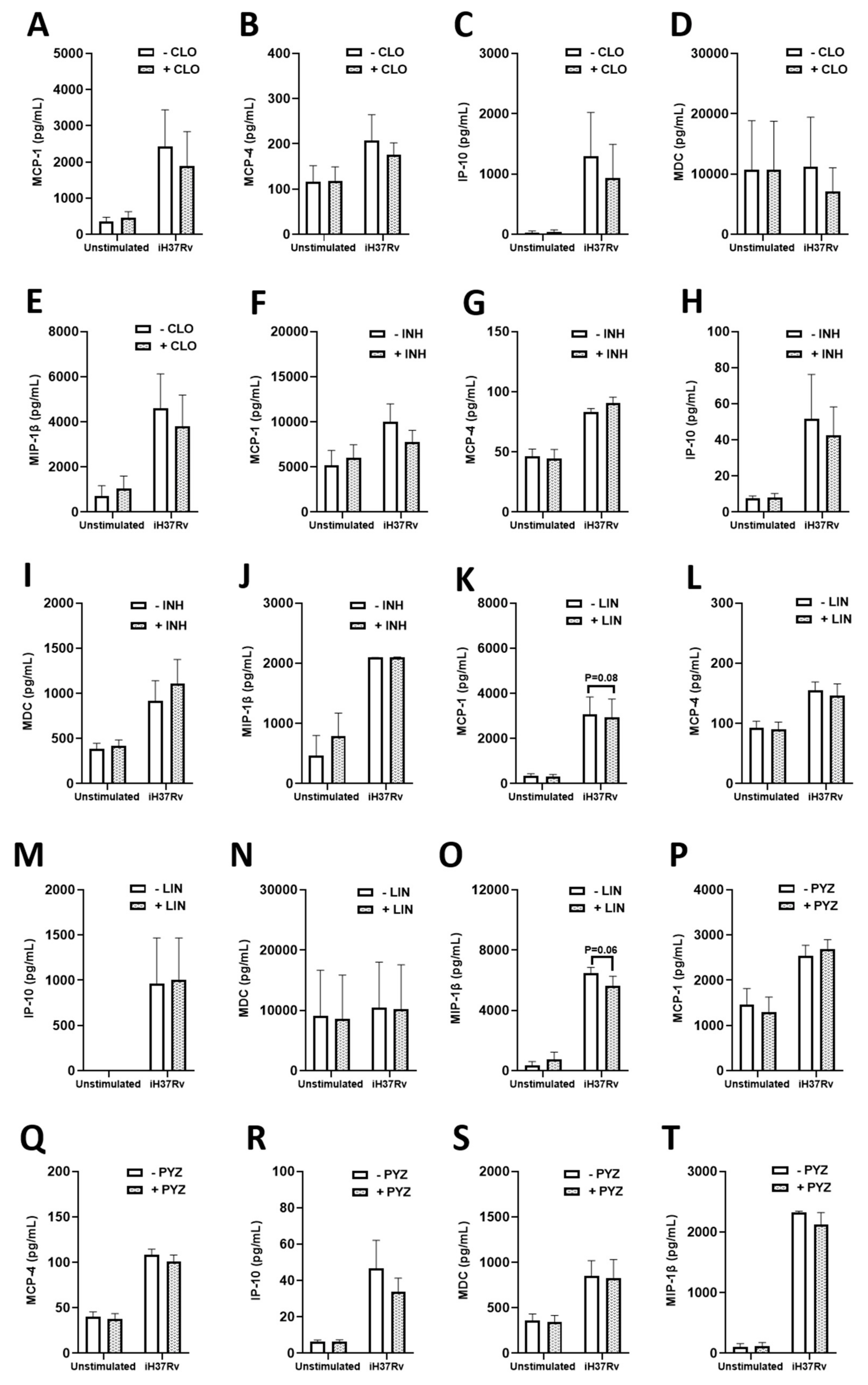
2.3. TB Antimicrobials Differentially Affect Levels of Secreted Cytokines and Chemokines in Unstimulated, iH37Rv-Mtb Stimulated and LPS-Stimulated hMDMs
3. Discussion
4. Materials and Methods
4.1. hMDM Cell Culture
4.2. Stimulation of hMDMs with iH37Rv-Mtb
4.3. Estimating Cell Number and Cell Viability Using Crystal Violet and Propidium Iodide (PI) Based Cell Exclusion Assays
4.4. Characterizing the Effect of Tuberculosis Antimicrobials on Metabolism Profiles and Mitochondrial Function Utilizing the Seahorse XFe24 Analyzer
4.5. Ex Vivo MSD Multiplex ELISA Analysis
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mitini-Nkhoma, S.C.; Chimbayo, E.T.; Mzinza, D.T.; Mhango, D.V.; Chirambo, A.P.; Mandalasi, C.; Lakudzala, A.E.; Tembo, D.L.; Jambo, K.C.; Mwandumba, H.C. Something Old, Something New: Ion Channel Blockers as Potential Anti-Tuberculosis Agents. Front. Immunol. 2021, 12, 2203. [Google Scholar] [CrossRef] [PubMed]
- Krug, S.; Parveen, S.; Bishai, W.R. Host-Directed Therapies: Modulating Inflammation to Treat Tuberculosis. Front. Immunol. 2021, 12, 1247. [Google Scholar] [CrossRef] [PubMed]
- Hayford, F.E.A.; Dolman, R.C.; Blaauw, R.; Nienaber, A.; Smuts, C.M.; Malan, L.; Ricci, C. The effects of anti-inflammatory agents as host-directed adjunct treatment of tuberculosis in humans: A systematic review and meta-analysis. Respir. Res. 2020, 21, 223. [Google Scholar] [CrossRef] [PubMed]
- Cox, D.J.; Coleman, A.M.; Gogan, K.M.; Phelan, J.J.; Maoldomhnaigh, C.Ó.; Dunne, P.J.; Basdeo, S.A.; Keane, J. Inhibiting Histone Deacetylases in Human Macrophages Promotes Glycolysis, IL-1β, and T Helper Cell Responses to Mycobacterium tuberculosis. Front. Immunol. 2020, 11, 1609. [Google Scholar] [CrossRef]
- Cahill, C.; O’Connell, F.; Gogan, K.M.; Cox, D.J.; Basdeo, S.A.; O’Sullivan, J.; Gordon, S.V.; Keane, J.; Phelan, J.J. The Iron Chelator Desferrioxamine Increases the Efficacy of Bedaquiline in Primary Human Macrophages Infected with BCG. Int. J. Mol. Sci. 2021, 22, 2938. [Google Scholar] [CrossRef]
- Phelan, J.J.; McQuaid, K.; Kenny, C.; Gogan, K.M.; Cox, D.J.; Basdeo, S.A.; O’Leary, S.; Tazoll, S.C.; Maoldomhnaigh, C.Ó.; O’Sullivan, M.P.; et al. Desferrioxamine Supports Metabolic Function in Primary Human Macrophages Infected With Mycobacterium tuberculosis. Front. Immunol. 2020, 11, 836. [Google Scholar] [CrossRef]
- Phelan, J.J.; Basdeo, S.A.; Tazoll, S.C.; McGivern, S.; Saborido, J.R.; Keane, J. Modulating Iron for Metabolic Support of TB Host Defense. Front. Immunol. 2018, 9, 2296. [Google Scholar] [CrossRef]
- Kim, J.-S.; Kim, Y.-R.; Yang, C.-S. Host-Directed Therapy in Tuberculosis: Targeting Host Metabolism. Front. Immunol. 2020, 11, 1790. [Google Scholar] [CrossRef]
- Cahill, C.; Phelan, J.J.; Keane, J. Understanding and Exploiting the Effect of Tuberculosis Antimicrobials on Host Mitochondrial Function and Bioenergetics. Front. Cell. Infect. Microbiol. 2020, 10, 493. [Google Scholar] [CrossRef]
- Gleeson, L.E.; Sheedy, F.J.; Palsson-McDermott, E.M.; Triglia, D.; O’Leary, S.M.; O’Sullivan, M.P.; O’Neill, L.A.; Keane, J. Cutting Edge: Mycobacterium tuberculosis Induces Aerobic Glycolysis in Human Alveolar Macrophages That Is Required for Control of Intracellular Bacillary Replication. J. Immunol. 2016, 196, 2444–2449. [Google Scholar] [CrossRef] [Green Version]
- Gleeson, L.E.; Ryan, D.; O’Leary, S.M.; McLaughlin, A.M.; Sheedy, F.J.; Keane, J.M. Cigarette Smoking Impairs the Bioenergetic Immune Response to Mycobacterium tuberculosis Infection. Am. J. Respir. Cell Mol. Biol. 2018, 59, 572–579. [Google Scholar] [CrossRef]
- Guillet, V.; Chevrollier, A.; Cassereau, J.; Letournel, F.; Gueguen, N.; Richard, L.; Desquiret, V.; Verny, C.; Procaccio, V.; Amati-Bonneau, P.; et al. Ethambutol-induced optic neuropathy linked to OPA1 mutation and mitochondrial toxicity. Mitochondrion 2010, 10, 115–124. [Google Scholar] [CrossRef] [Green Version]
- Ahadpour, M.; Eskandari, M.R.; Mashayekhi, V.; Haj Mohammad Ebrahim Tehrani, K.; Jafarian, I.; Naserzadeh, P.; Hosseini, M.J. Mitochondrial oxidative stress and dysfunction induced by isoniazid: Study on isolated rat liver and brain mitochondria. Drug Chem. Toxicol. 2016, 39, 224–232. [Google Scholar] [CrossRef]
- Chowdhury, A.; Santra, A.; Bhattacharjee, K.; Ghatak, S.; Saha, D.R.; Dhali, G.K. Mitochondrial oxidative stress and permeability transition in isoniazid and rifampicin induced liver injury in mice. J. Hepatol. 2006, 45, 117–126. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, K.; Hassan, H.M.; Guo, H.; Ding, P.; Han, L.; He, Q.; Chen, W.; Hsiao, C.D.; Zhang, L.; et al. Liver Fatty Acid Binding Protein Deficiency Provokes Oxidative Stress, Inflammation, and Apoptosis-Mediated Hepatotoxicity Induced by Pyrazinamide in Zebrafish Larvae. Antimicrob. Agents Chemother. 2016, 60, 7347–7356. [Google Scholar] [CrossRef] [Green Version]
- Erokhina, M.V.; Kurynina, A.V.; Onishchenko, G.E. Mitochondria are targets for the antituberculosis drug rifampicin in cultured epithelial cells. Biochemistry 2013, 78, 1155–1163. [Google Scholar] [CrossRef]
- Belosludtsev, K.N.; Belosludtseva, N.V.; Talanov, E.Y.; Tenkov, K.S.; Starinets, V.S.; Agafonov, A.V.; Pavlik, L.L.; Dubinin, M.V. Effect of bedaquiline on the functions of rat liver mitochondria. Biochim. Biophys. Acta Biomembr. 2019, 1861, 288–297. [Google Scholar] [CrossRef]
- Fiorillo, M.; Lamb, R.; Tanowitz, H.B.; Cappello, A.R.; Martinez-Outschoorn, U.E.; Sotgia, F.; Lisanti, M.P. Bedaquiline, an FDA-approved antibiotic, inhibits mitochondrial function and potently blocks the proliferative expansion of stem-like cancer cells (CSCs). Aging 2016, 8, 1593–1607. [Google Scholar] [CrossRef] [Green Version]
- Sri-Pathmanathan, R.M.; Plumb, J.A.; Fearon, K.C. Clofazimine alters the energy metabolism and inhibits the growth rate of a human lung-cancer cell line in vitro and in vivo. Int. J. Cancer 1994, 56, 900–905. [Google Scholar] [CrossRef]
- De Vriese, A.S.; Coster, R.V.; Smet, J.; Seneca, S.; Lovering, A.; Van Haute, L.L.; Vanopdenbosch, L.J.; Martin, J.J.; Groote, C.C.; Vandecasteele, S.; et al. Linezolid-induced inhibition of mitochondrial protein synthesis. Clin. Infect. Dis. 2006, 42, 1111–1117. [Google Scholar] [CrossRef]
- Weiss, T.; Shalit, I.; Blau, H.; Werber, S.; Halperin, D.; Levitov, A.; Fabian, I. Anti-inflammatory effects of moxifloxacin on activated human monocytic cells: Inhibition of NF-kappaB and mitogen-activated protein kinase activation and of synthesis of proinflammatory cytokines. Antimicrob. Agents Chemother. 2004, 48, 1974–1982. [Google Scholar] [CrossRef] [Green Version]
- Cavanaugh, J.S.; Jou, R.; Wu, M.H.; Dalton, T.; Kurbatova, E.; Ershova, J.; Cegielski, J.P. Susceptibilities of MDR Mycobacterium tuberculosis isolates to unconventional drugs compared with their reported pharmacokinetic/pharmacodynamic parameters. J. Antimicrob. Chemother. 2017, 72, 1678–1687. [Google Scholar] [CrossRef]
- Forsman, L.D.; Jonsson, J.; Wagrell, C.; Werngren, J.; Mansjö, M.; Wijkander, M.; Groenheit, R.; Hammar, U.; Giske, C.G.; Schön, T.; et al. Minimum Inhibitory Concentrations of Fluoroquinolones and Pyrazinamide Susceptibility Correlate to Clinical Improvement in Multidrug-resistant Tuberculosis Patients: A Nationwide Swedish Cohort Study Over 2 Decades. Clin. Infect. Dis. 2019, 69, 1394–1402. [Google Scholar] [CrossRef]
- Gillespie, S.H.; Billington, O. Activity of moxifloxacin against mycobacteria. J. Antimicrob. Chemother. 1999, 44, 393–395. [Google Scholar] [CrossRef] [Green Version]
- Pina, R.Z.; Caleffi-Ferracioli, K.R.; Campanerut-Sá, P.A.; Ghiraldi-Lopez, L.D.; Pavan, F.R.; Siqueira, V.L.; Scodro, R.B.; Cardoso, R.F. Pyrazinamide susceptibility testing in Mycobacterium tuberculosis using the fast resazurin microtiter assay plate. Int. J. Tuberc. Lung Dis. 2016, 20, 1535–1538. [Google Scholar] [CrossRef]
- Rastogi, N.; Goh, K.S.; Horgen, L.; Barrow, W.W. Synergistic activities of antituberculous drugs with cerulenin and trans-cinnamic acid against Mycobacterium tuberculosis. FEMS Immunol. Med. Microbiol. 1998, 21, 149–157. [Google Scholar] [CrossRef]
- Shetty, A.; Dick, T. Mycobacterial Cell Wall Synthesis Inhibitors Cause Lethal ATP Burst. Front. Microbiol. 2018, 9, 1898. [Google Scholar] [CrossRef] [Green Version]
- Carmona, J.; Cruz, A.; Moreira-Teixeira, L.; Sousa, C.; Sousa, J.; Osorio, N.S.; Saraiva, A.L.; Svenson, S.; Kallenius, G.; Pedrosa, J.; et al. Mycobacterium tuberculosis Strains Are Differentially Recognized by TLRs with an Impact on the Immune Response. PLoS ONE 2013, 8, e67277. [Google Scholar] [CrossRef] [Green Version]
- Phelan, J.J.; MacCarthy, F.; Feighery, R.; O’Farrell, N.J.; Lynam-Lennon, N.; Doyle, B.; O’Toole, D.; Ravi, N.; Reynolds, J.V.; O’Sullivan, J. Differential expression of mitochondrial energy metabolism profiles across the metaplasia-dysplasia-adenocarcinoma disease sequence in Barrett’s oesophagus. Cancer Lett. 2014, 354, 122–131. [Google Scholar] [CrossRef]
- Reddy, V.P.; Chinta, K.C.; Saini, V.; Glasgow, J.N.; Hull, T.D.; Traylor, A.; Rey-Stolle, F.; Soares, M.P.; Madansein, R.; Rahman, M.A.; et al. Ferritin H Deficiency in Myeloid Compartments Dysregulates Host Energy Metabolism and Increases Susceptibility to Mycobacterium tuberculosis Infection. Front. Immunol. 2018, 9, 860. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Roca, P.; Mancilla-Ramirez, J.; Santos-Segura, A.; Fernández-Avilés, M.; Calderon-Jaimes, E. Linezolid diminishes inflammatory cytokine production from human peripheral blood mononuclear cells. Arch. Med. Res. 2006, 37, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Giraud-Gatineau, A.; Coya, J.M.; Maure, A.; Biton, A.; Thomson, M.; Bernard, E.M.; Marrec, J.; Gutierrez, M.G.; Larrouy-Maumus, G.; Brosch, R.; et al. The antibiotic bedaquiline activates host macrophage innate immune resistance to bacterial infection. Elife 2020, 9, e55692. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.J.; Lee, H.M.; Shin, D.M.; Kim, W.; Yuk, J.M.; Jin, H.S.; Lee, S.H.; Cha, G.H.; Kim, J.M.; Lee, Z.W.; et al. Host cell autophagy activated by antibiotics is required for their effective antimycobacterial drug action. Cell Host Microbe 2012, 11, 457–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, G.S.; Keswani, R.K.; Sud, S.; Rzeczycki, P.M.; Murashov, M.D.; Koehn, T.A.; Standiford, T.J.; Stringer, K.A.; Rosania, G.R. Clofazimine Biocrystal Accumulation in Macrophages Upregulates Interleukin 1 Receptor Antagonist Production To Induce a Systemic Anti-Inflammatory State. Antimicrob. Agents Chemother. 2016, 60, 3470–3479. [Google Scholar] [CrossRef] [Green Version]
- Ziglam, H.M.; Daniels, I.; Finch, R.G. Immunomodulating activity of rifampicin. J. Chemother. 2004, 16, 357–361. [Google Scholar] [CrossRef]
- Cumming, B.M.; Addicott, K.W.; Adamson, J.H.; Steyn, A.J. Mycobacterium tuberculosis induces decelerated bioenergetic metabolism in human macrophages. Elife 2018, 7, e39169. [Google Scholar] [CrossRef]
- Santini, A.; Ronchi, D.; Garbellini, M.; Piga, D.; Protti, A. Linezolid-induced lactic acidosis: The thin line between bacterial and mitochondrial ribosomes. Expert Opin. Drug Saf. 2017, 16, 833–843. [Google Scholar] [CrossRef]
- Trexel, J.; Yoon, G.S.; Keswani, R.K.; McHugh, C.; Yeomans, L.; Vitvitsky, V.; Banerjee, R.; Sud, S.; Sun, Y.; Rosania, G.R.; et al. Macrophage-Mediated Clofazimine Sequestration Is Accompanied by a Shift in Host Energy Metabolism. J. Pharm. Sci. 2017, 106, 1162–1174. [Google Scholar] [CrossRef] [Green Version]
- Poltorak, A.; He, X.; Smirnova, I.; Liu, M.Y.; Van Huffel, C.; Du, X.; Birdwell, D.; Alejos, E.; Silva, M.; Galanos, C.; et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: Mutations in Tlr4 gene. Science 1998, 282, 2085–2088. [Google Scholar] [CrossRef] [Green Version]
- Dubey, R.K. Assuming the role of mitochondria in mycobacterial infection. Int. J. Mycobacteriol. 2016, 5, 379–383. [Google Scholar] [CrossRef] [Green Version]
- Feng, X.; Zhu, W.; Schurig-Briccio, L.A.; Lindert, S.; Shoen, C.; Hitchings, R.; Li, J.; Wang, Y.; Baig, N.; Zhou, T.; et al. Antiinfectives targeting enzymes and the proton motive force. Proc. Natl. Acad. Sci. USA 2015, 112, E7073–E7082. [Google Scholar] [CrossRef] [Green Version]
- Baik, J.; Rosania, G.R. Molecular imaging of intracellular drug-membrane aggregate formation. Mol. Pharm. 2011, 8, 1742–1749. [Google Scholar] [CrossRef] [Green Version]
- Ladel, C.H.; Blum, C.; Dreher, A.; Reifenberg, K.; Kopf, M.; Kaufmann, S.H. Lethal tuberculosis in interleukin-6-deficient mutant mice. Infect. Immun. 1997, 65, 4843–4849. [Google Scholar] [CrossRef] [Green Version]
- Romani, L.; Mencacci, A.; Cenci, E.; Spaccapelo, R.; Toniatti, C.; Puccetti, P.; Bistoni, F.; Poli, V. Impaired neutrophil response and CD4+ T helper cell 1 development in interleukin 6-deficient mice infected with Candida albicans. J. Exp. Med. 1996, 183, 1345–1355. [Google Scholar] [CrossRef]
- Takai, Y.; Wong, G.G.; Clark, S.C.; Burakoff, S.J.; Herrmann, S.H. B cell stimulatory factor-2 is involved in the differentiation of cytotoxic T lymphocytes. J. Immunol. 1988, 140, 508–512. [Google Scholar]
- Uyttenhove, C.; Coulie, P.G.; Van Snick, J. T cell growth and differentiation induced by interleukin-HP1/IL-6, the murine hybridoma/plasmacytoma growth factor. J. Exp. Med. 1988, 167, 1417–1427. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Meng, Q.; Xi, Q.; Zhang, Y.; Zhuang, Q.; Han, Y.; Jiang, Y.; Ding, Q.; Wu, G. Interleukin-6 stimulates aerobic glycolysis by regulating PFKFB3 at early stage of colorectal cancer. Int. J. Oncol. 2016, 48, 215–224. [Google Scholar] [CrossRef]
- Ando, M.; Uehara, I.; Kogure, K.; Asano, Y.; Nakajima, W.; Abe, Y.; Kawauchi, K.; Tanaka, N. Interleukin 6 enhances glycolysis through expression of the glycolytic enzymes hexokinase 2 and 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3. J. Nippon Med. Sch. 2010, 77, 97–105. [Google Scholar] [CrossRef] [Green Version]
- Sigal, G.B.; Segal, M.R.; Mathew, A.; Jarlsberg, L.; Wang, M.; Barbero, S.; Small, N.; Haynesworth, K.; Davis, J.L.; Weiner, M.; et al. Biomarkers of Tuberculosis Severity and Treatment Effect: A Directed Screen of 70 Host Markers in a Randomized Clinical Trial. EBioMedicine 2017, 25, 112–121. [Google Scholar] [CrossRef] [Green Version]
- Richter, J.R.; Sutton, J.M.; Belizaire, R.M.; Friend, L.A.; Schuster, R.M.; Johannigman, T.A.; Miller, S.G.; Lentsch, A.B.; Pritts, T.A. Macrophage-derived chemokine (CCL22) is a novel mediator of lung inflammation following hemorrhage and resuscitation. Shock 2014, 42, 525–531. [Google Scholar] [CrossRef] [Green Version]
- Imai, T.; Nagira, M.; Takagi, S.; Kakizaki, M.; Nishimura, M.; Wang, J.; Gray, P.W.; Matsushima, K.; Yoshie, O. Selective recruitment of CCR4-bearing Th2 cells toward antigen-presenting cells by the CC chemokines thymus and activation-regulated chemokine and macrophage-derived chemokine. Int. Immunol. 1999, 11, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Maoldomhnaigh, C.Ó.; Cox, D.J.; Phelan, J.J.; Malone, F.D.; Keane, J.; Basdeo, S.A. The Warburg Effect Occurs Rapidly in Stimulated Human Adult but Not Umbilical Cord Blood Derived Macrophages. Front. Immunol. 2021, 12, 1115. [Google Scholar] [CrossRef] [PubMed]
- Maoldomhnaigh, C.Ó.; Cox, D.J.; Phelan, J.J.; Mitermite, M.; Murphy, D.M.; Leisching, G.; Thong, L.; O’Leary, S.M.; Gogan, K.M.; McQuaid, K.; et al. Lactate Alters Metabolism in Human Macrophages and Improves Their Ability to Kill Mycobacterium tuberculosis. Front. Immunol. 2021, 12, 4104. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.L.; Lamprecht, D.A.; Mandizvo, T.; Jones, T.T.; Naidoo, V.; Addicott, K.W.; Moodley, C.; Ngcobo, B.; Crossman, D.K.; Wells, G.; et al. Compromised Metabolic Reprogramming Is an Early Indicator of CD8(+) T Cell Dysfunction during Chronic Mycobacterium tuberculosis Infection. Cell Rep. 2019, 29, 3564–3579.e3565. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Name | Abbreviation | Clinical Use | In Vitro Solvent | Mechanism of Action | Known Effects on Eukaryotic Cells |
|---|---|---|---|---|---|
| Ethambutol | EMB | First-line | dH2O | Functions by obstructing the formation of cell wall | Inhibits complex IV and OXPHOS in human fibroblasts [12] |
| Isoniazid | INH | First-line | dH2O | Inhibits mycolic acid synthesis; interferes with cell wall synthesis | Reduces complex I-III activity & increases ROS and lipid peroxidation in rat mitochondria [13] and alters murine mitochondria [14] |
| Pyrazinamide | PYZ | First-line | dH2O | Interferes with the bacteriums’ ability to synthesize new fatty acids (required for growth and replication) | Associated with increased ROS production in zebrafish larvae [15] |
| Rifampicin | RIF | First-line | dH2O | Inhibits bacterial DNA-dependent RNA synthesis | Alters structure of mitochondria resulting in excessive ROS and release of cyto-chrome c [16] and alters murine mitpochondria [14] |
| Amikacin | AMK | Second-line | dH2O | Interferes with mRNA binding (interferes with bacterial growth) | Unknown effects on eukaryotic cells [9] |
| Bedaquiline | BDQ | Second-line | DMSO | Inhibits the proton pump of mycobacterial ATP synthase (essential for ATP production) | Inhibits the mitochondrial transport chain in rat models [17], reduces mitochondrial membrane potential and increases ROS production in human breast cancer cells [18] |
| Clofazimine | CLO | Second-line | DMSO | Blocks the template function of DNA, inhibiting proliferation | CLO increases glycolysis in human non-small-cell bronchial-carcinoma cells [19] |
| Cycloserine | CYS | Second-line | dH2O | Inhibits cell-wall biosynthesis | Unknown effects on eukaryotic cells [9] |
| Linezolid | LIN | Second-line | DMSO | Inhibits bacterial protein synthesis | Decreases protein levels and the enzymatic activity of various mitochondrial complexes [20] |
| Moxifloxacin | MOX | Second-line | dH2O | Blocks bacterial DNA replication | Exhibits anti-inflammatory effects in human THP-1 cells [21] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cahill, C.; Cox, D.J.; O’Connell, F.; Basdeo, S.A.; Gogan, K.M.; Ó’Maoldomhnaigh, C.; O’Sullivan, J.; Keane, J.; Phelan, J.J. The Effect of Tuberculosis Antimicrobials on the Immunometabolic Profiles of Primary Human Macrophages Stimulated with Mycobacterium tuberculosis. Int. J. Mol. Sci. 2021, 22, 12189. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222212189
Cahill C, Cox DJ, O’Connell F, Basdeo SA, Gogan KM, Ó’Maoldomhnaigh C, O’Sullivan J, Keane J, Phelan JJ. The Effect of Tuberculosis Antimicrobials on the Immunometabolic Profiles of Primary Human Macrophages Stimulated with Mycobacterium tuberculosis. International Journal of Molecular Sciences. 2021; 22(22):12189. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222212189
Chicago/Turabian StyleCahill, Christina, Dónal J. Cox, Fiona O’Connell, Sharee A. Basdeo, Karl M. Gogan, Cilian Ó’Maoldomhnaigh, Jacintha O’Sullivan, Joseph Keane, and James J. Phelan. 2021. "The Effect of Tuberculosis Antimicrobials on the Immunometabolic Profiles of Primary Human Macrophages Stimulated with Mycobacterium tuberculosis" International Journal of Molecular Sciences 22, no. 22: 12189. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222212189