
Contribution of the Transcription Factors Sp1/Sp3 and AP-1 to Clusterin Gene Expression during Corneal Wound Healing of Tissue-Engineered Human Corneas

 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
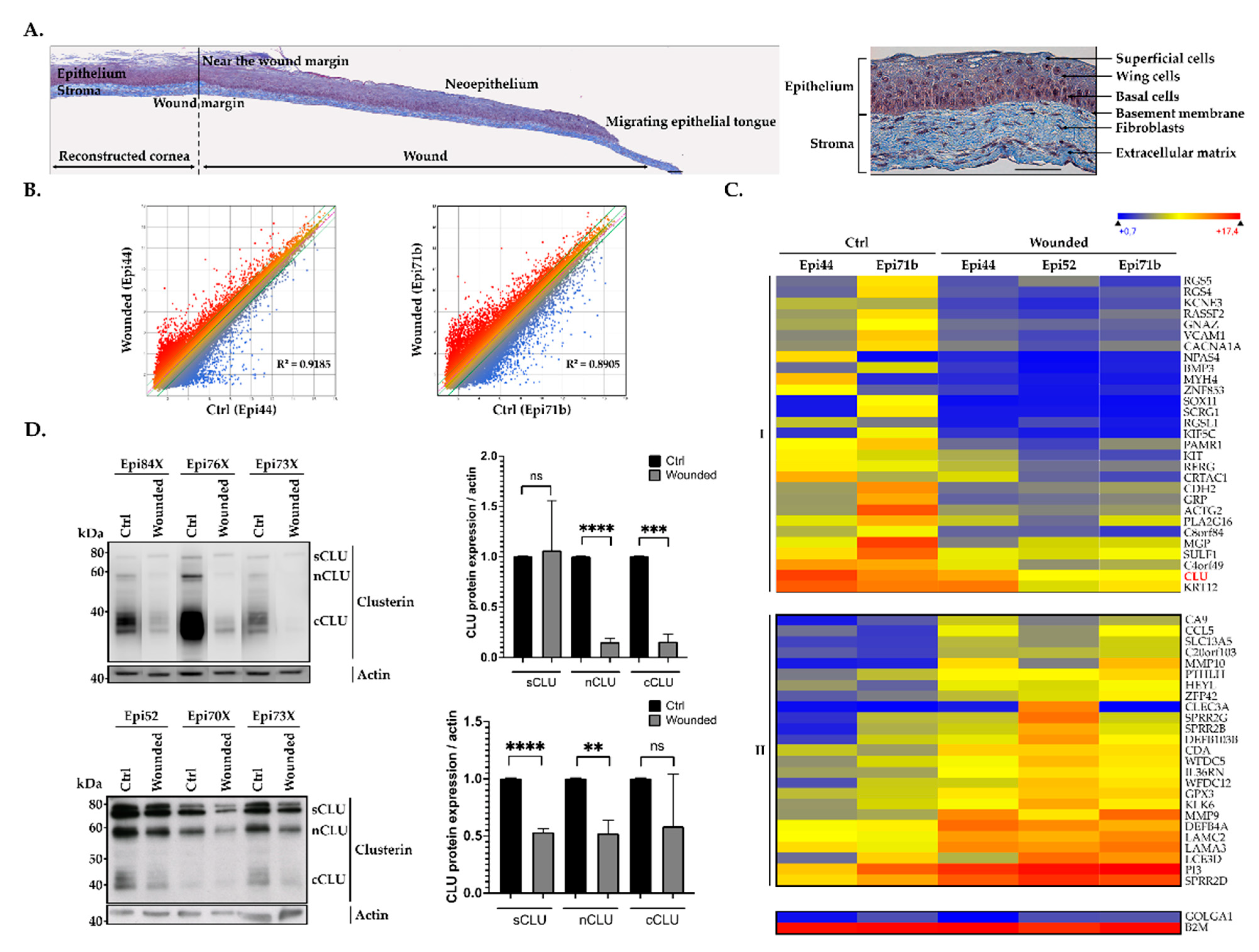
2.1. hTECs Wound Healing Alters CLU Gene Expression in Human Corneal Epithelial Cells
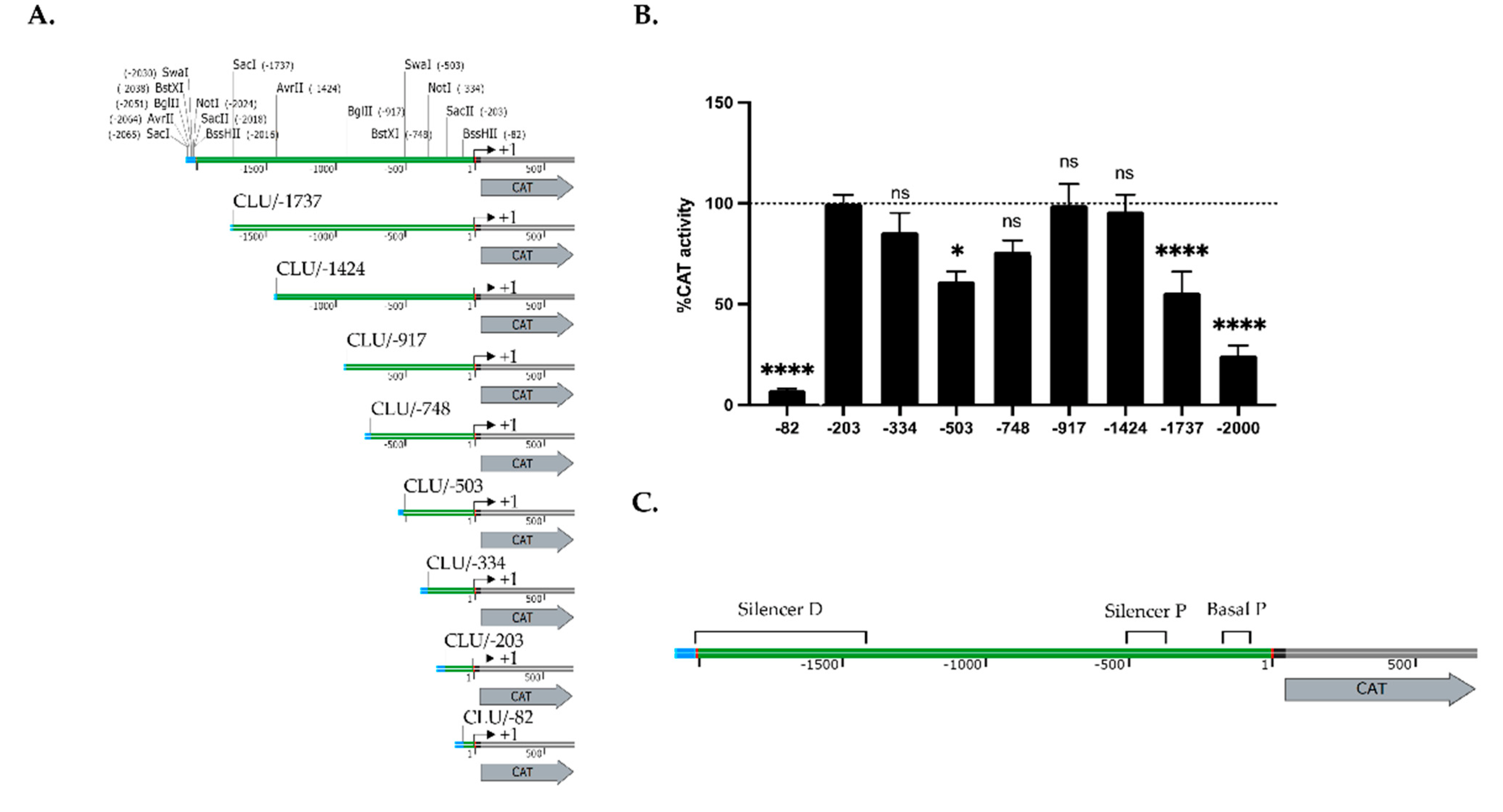
2.2. CLU Gene Transcription Is Modulated by Both Positive and Negative Regulatory Elements
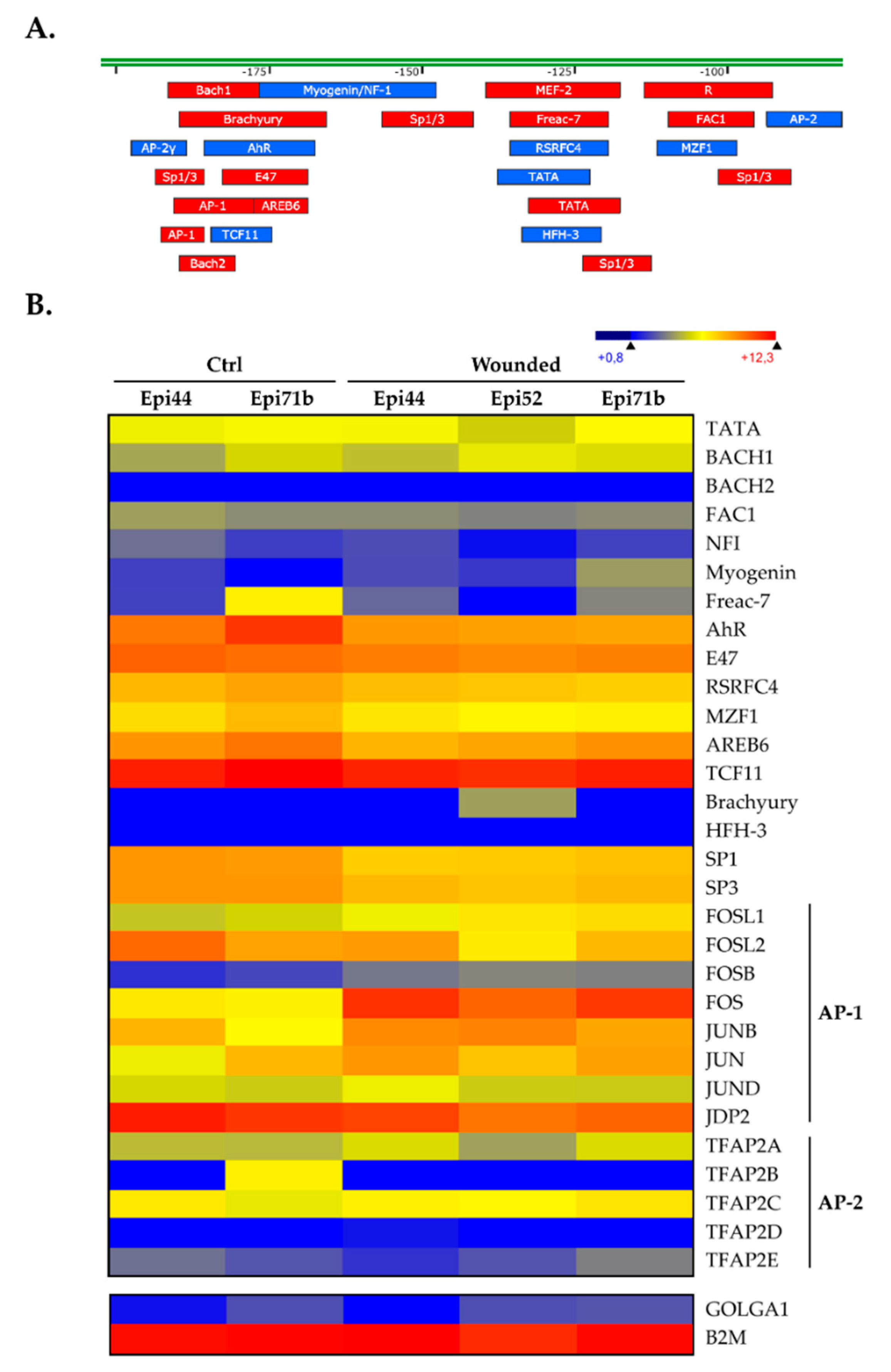
2.3. Analysis of the Transcription Factors Binding to the CLU Basal Promoter in HCECs
2.4. Members from the AP1 and Sp1 Families Bind to the CLU Basal Promoter in hCECs
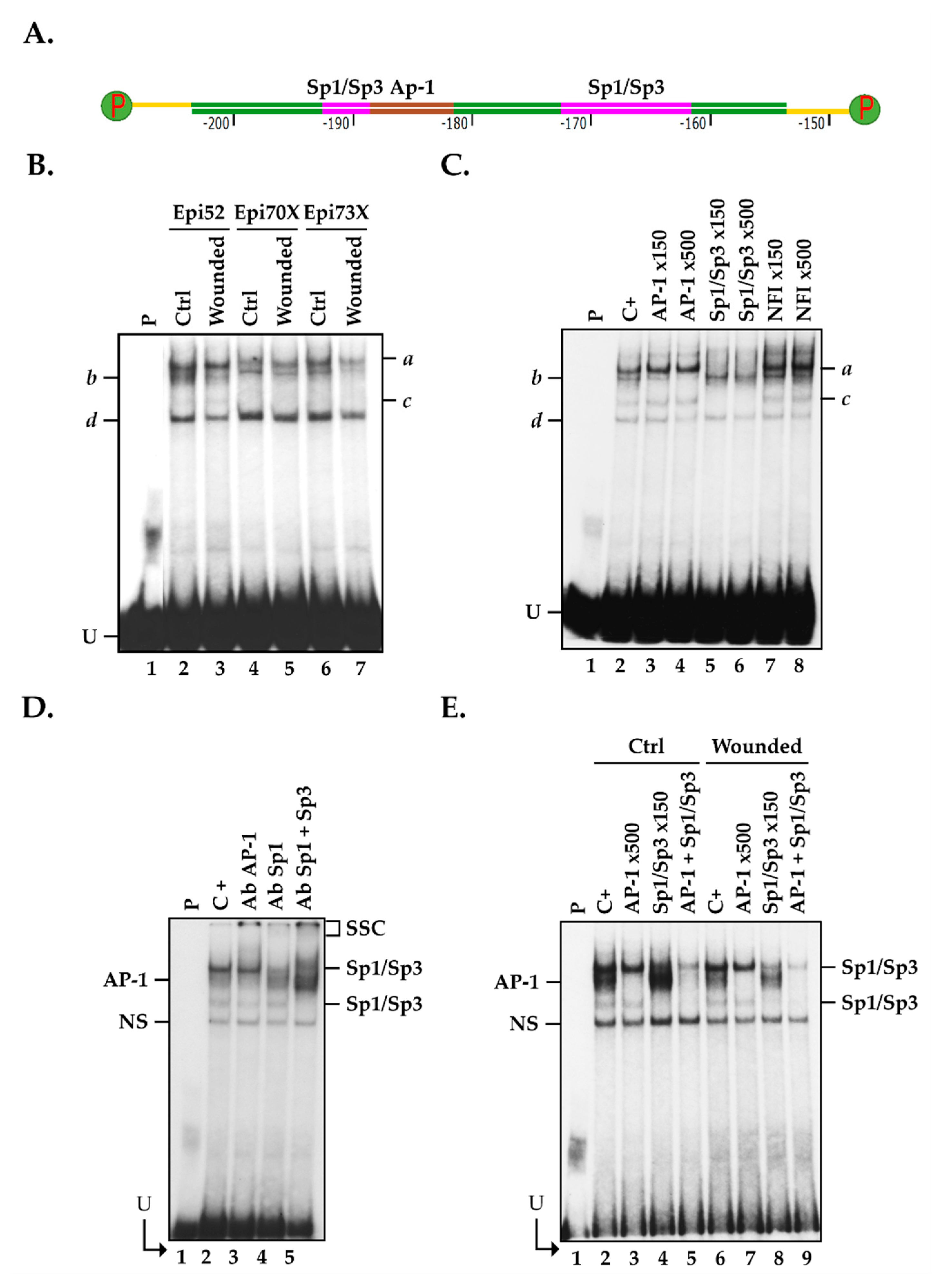
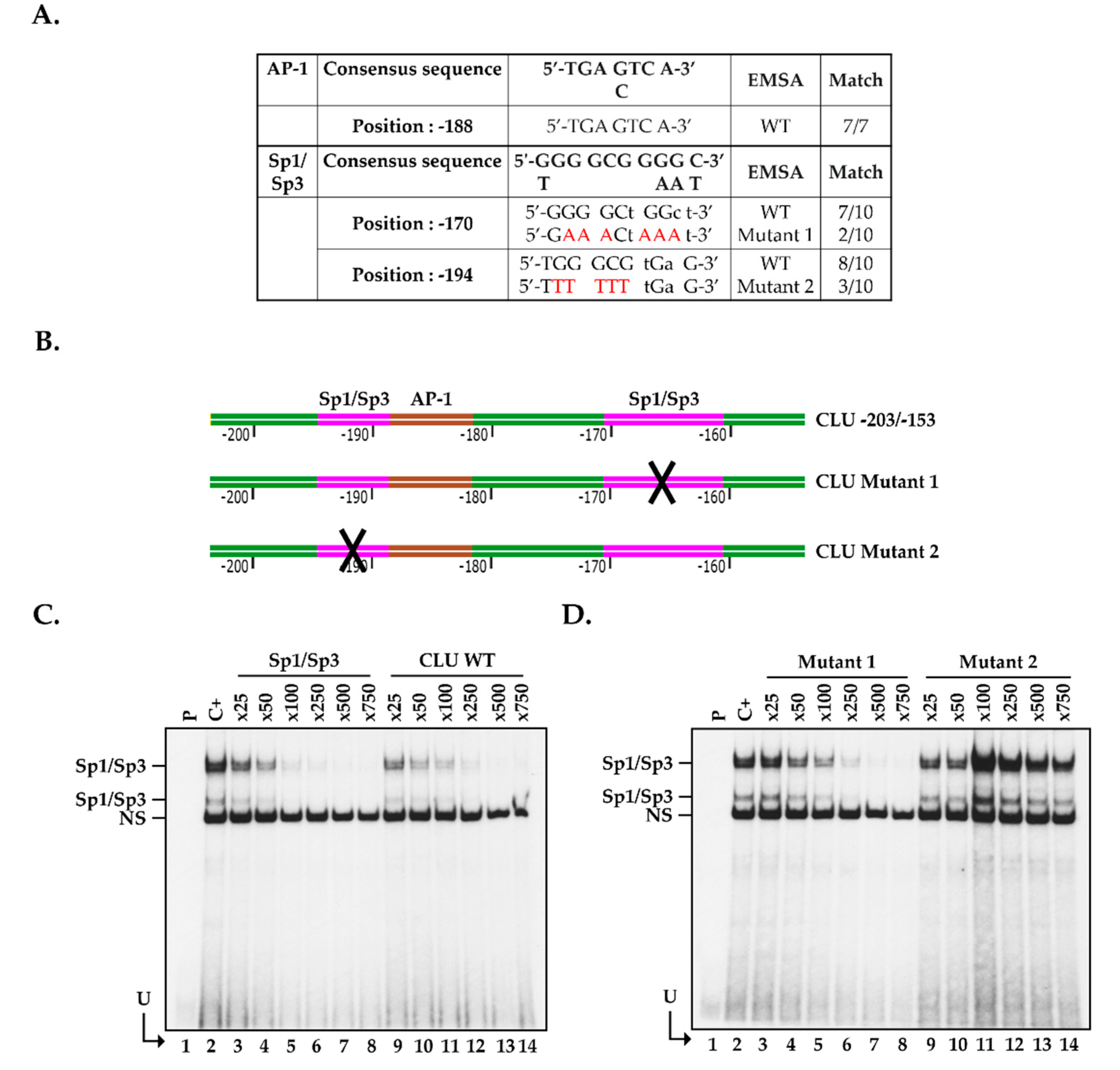
2.5. Positioning of the Functional Sp1/Sp3 Binding Site in the CLU Promoter In Vitro
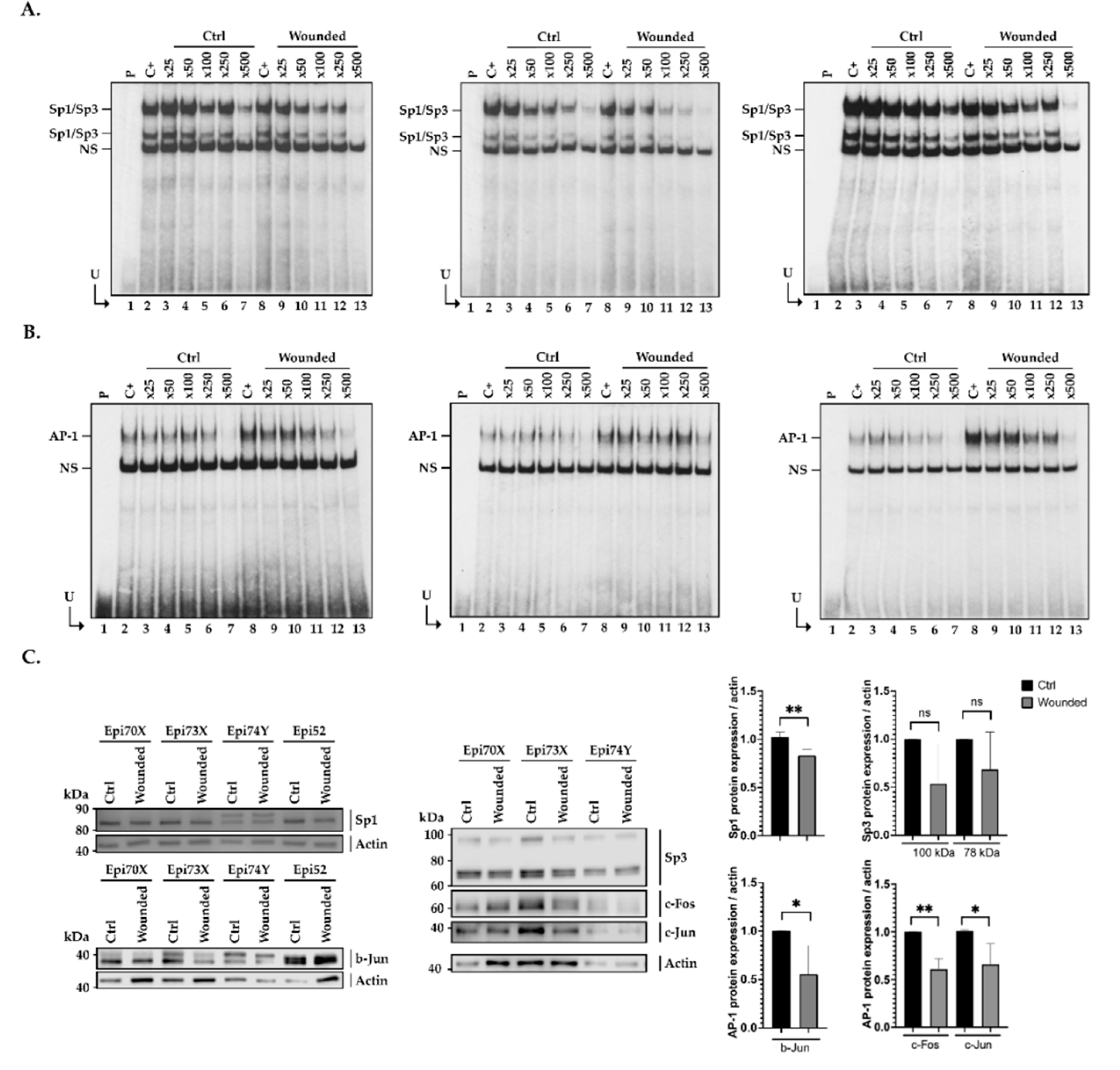
2.6. Influence of Cell Damage on the Binding of Sp1/Sp3 to the CLU Promoter in hCECs
3. Discussion
4. Materials and Methods
4.1. Cell Culture—Human Epithelial Cells (hCECs)
4.2. Human Tissue-Engineered Cornea (hTEC)
4.3. Gene Expression Profiling
4.4. Western Blot Analyses
4.5. Plasmid Constructs and Oligonucleotides
4.6. Transient Transfection and CAT (Chloramphenicol Acetyl Transferase) Assays
4.7. Preparation of Nuclear Extracts and Electrophoretic Mobility Shift Assays (EMSA)
4.8. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Eghrari, A.O.; Riazuddin, S.A.; Gottsch, J.D. Overview of the Cornea: Structure, Function, and Development. Prog. Mol. Biol. Transl. Sci. 2015, 134, 7–23. [Google Scholar] [CrossRef] [PubMed]
- Germain, L.; Carrier, P.; Auger, F.A.; Salesse, C.; Guerin, S.L. Can we produce a human corneal equivalent by tissue engineering? Prog. Retin. Eye Res. 2000, 19, 497–527. [Google Scholar] [CrossRef]
- Gupta, N.; Kalaivani, M.; Tandon, R. Comparison of prognostic value of Roper Hall and Dua classification systems in acute ocular burns. Br. J. Ophthalmol. 2011, 95, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Daniels, J.T.; Dart, J.K.; Tuft, S.J.; Khaw, P.T. Corneal stem cells in review. Wound Repair Regen. 2001, 9, 483–494. [Google Scholar] [CrossRef] [PubMed]
- Vemuganti, G.K.; Murthy, S.I.; Das, S. Update on pathologic diagnosis of corneal infections and inflammations. Middle East Afr. J. Ophthalmol. 2011, 18, 277–284. [Google Scholar] [CrossRef]
- Hong, J.W.; Liu, J.J.; Lee, J.S.; Mohan, R.R.; Mohan, R.R.; Woods, D.J.; He, Y.G.; Wilson, S.E. Proinflammatory chemokine induction in keratocytes and inflammatory cell infiltration into the cornea. Investig. Ophthalmol. Vis. Sci. 2001, 42, 2795–2803. [Google Scholar]
- Dua, H.S.; Azuara-Blanco, A. Autologous limbal transplantation in patients with unilateral corneal stem cell deficiency. Br. J. Ophthalmol. 2000, 84, 273–278. [Google Scholar] [CrossRef] [Green Version]
- Kang, B.S.; Kim, M.K.; Wee, W.R.; Oh, J.Y. Infectious Keratitis in Limbal Stem Cell Deficiency: Stevens-Johnson Syndrome Versus Chemical Burn. Cornea 2016, 35, 51–55. [Google Scholar] [CrossRef]
- Carrier, P.; Deschambeault, A.; Talbot, M.V.; Giasson, C.J.; Auger, F.O.A.; GuéRin, S.L.; Germain, L. Characterization of Wound Reepithelialization Using a New Human Tissue-Engineered Corneal Wound Healing Model. Investig. Opthalmology Vis. Sci. 2008, 49, 1376. [Google Scholar] [CrossRef]
- Couture, C.; Desjardins, P.; Zaniolo, K.; Germain, L.; Guerin, S.L. Enhanced wound healing of tissue-engineered human corneas through altered phosphorylation of the CREB and AKT signal transduction pathways. Acta Biomater. 2018, 73, 312–325. [Google Scholar] [CrossRef]
- Couture, C.; Zaniolo, K.; Carrier, P.; Lake, J.; Patenaude, J.; Germain, L.; Guerin, S.L. The tissue-engineered human cornea as a model to study expression of matrix metalloproteinases during corneal wound healing. Biomaterials 2016, 78, 86–101. [Google Scholar] [CrossRef]
- Guerin, L.P.; Le-Bel, G.; Desjardins, P.; Couture, C.; Gillard, E.; Boisselier, E.; Bazin, R.; Germain, L.; Guerin, S.L. The Human Tissue-Engineered Cornea (hTEC): Recent Progress. Int. J. Mol. Sci. 2021, 22, 1291. [Google Scholar] [CrossRef]
- Proulx, S.; d’Arc Uwamaliya, J.; Carrier, P.; Deschambeault, A.; Audet, C.; Giasson, C.J.; Guerin, S.L.; Auger, F.A.; Germain, L. Reconstruction of a human cornea by the self-assembly approach of tissue engineering using the three native cell types. Mol. Vis. 2010, 16, 2192–2201. [Google Scholar]
- Zaniolo, K.; Carrier, P.; Guerin, S.L.; Auger, F.A.; Germain, L. A tissue-engineered corneal wound healing model for the characterization of reepithelialization. Methods Mol. Biol. 2013, 1037, 59–78. [Google Scholar] [CrossRef]
- Carrier, P.; Deschambeault, A.; Audet, C.; Talbot, M.V.; Gauvin, R.; Giasson, C.J.; Auger, F.O.A.; GuéRin, S.L.; Germain, L. Impact of Cell Source on Human Cornea Reconstructed by Tissue Engineering. Investig. Opthalmology Vis. Sci. 2009, 50, 2645. [Google Scholar] [CrossRef] [Green Version]
- Germain, L.; Auger, F.A.; Grandbois, E.; Guignard, R.; Giasson, M.; Boisjoly, H.; Guerin, S.L. Reconstructed human cornea produced in vitro by tissue engineering. Pathobiology 1999, 67, 140–147. [Google Scholar] [CrossRef]
- Lake, J.; Zaniolo, K.; Gaudreault, M.; Carrier, P.; Deschambault, A.; Bazin, R.; Germain, L.; Salesse, C.; Guerin, S.L. Expression of the α5 integrin gene in corneal epithelial cells cultured on tissue-engineered human extracellular matrices. Biomaterials 2013, 34, 6367–6376. [Google Scholar] [CrossRef]
- Lake, J.; Zaniolo, K.; Gingras, M.E.; Couture, C.; Salesse, C.; Guerin, S.L. Functional Impact of Collagens on the Activity Directed by the Promoter of the α5 Integrin Subunit Gene in Corneal Epithelial Cells. Investig. Ophthalmol. Vis. Sci. 2015, 56, 6217–6232. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.Y.; Kao, W.W. Corneal Epithelial Wound Healing. Prog. Mol. Biol. Transl. Sci. 2015, 134, 61–71. [Google Scholar] [CrossRef]
- Humphreys, D.T.; Carver, J.A.; Easterbrook-Smith, S.B.; Wilson, M.R. Clusterin has chaperone-like activity similar to that of small heat shock proteins. J. Biol. Chem. 1999, 274, 6875–6881. [Google Scholar] [CrossRef] [Green Version]
- Trougakos, I.P.; Lourda, M.; Antonelou, M.H.; Kletsas, D.; Gorgoulis, V.G.; Papassideri, I.S.; Zou, Y.; Margaritis, L.H.; Boothman, D.A.; Gonos, E.S. Intracellular clusterin inhibits mitochondrial apoptosis by suppressing p53-activating stress signals and stabilizing the cytosolic Ku70-Bax protein complex. Clin. Cancer Res. 2009, 15, 48–59. [Google Scholar] [CrossRef] [Green Version]
- Kim, N.; Yoo, J.C.; Han, J.Y.; Hwang, E.M.; Kim, Y.S.; Jeong, E.Y.; Sun, C.H.; Yi, G.S.; Roh, G.S.; Kim, H.J.; et al. Human nuclear clusterin mediates apoptosis by interacting with Bcl-XL through C-terminal coiled coil domain. J. Cell Physiol. 2012, 227, 1157–1167. [Google Scholar] [CrossRef]
- Silkensen, J.R.; Skubitz, K.M.; Skubitz, A.P.; Chmielewski, D.H.; Manivel, J.C.; Dvergsten, J.A.; Rosenberg, M.E. Clusterin promotes the aggregation and adhesion of renal porcine epithelial cells. J. Clin. Investig. 1995, 96, 2646–2653. [Google Scholar] [CrossRef] [Green Version]
- Millis, A.J.; Luciani, M.; McCue, H.M.; Rosenberg, M.E.; Moulson, C.L. Clusterin regulates vascular smooth muscle cell nodule formation and migration. J. Cell Physiol. 2001, 186, 210–219. [Google Scholar] [CrossRef]
- Shin, Y.J.; Kang, S.W.; Jeong, S.Y.; Shim, Y.J.; Kim, Y.H.; Kim, B.M.; Kee, S.H.; Park, J.J.; Park, I.S.; Min, B.H. Clusterin enhances proliferation of primary astrocytes through extracellular signal-regulated kinase activation. Neuroreport 2006, 17, 1871–1875. [Google Scholar] [CrossRef]
- Bettuzzi, S.; Scorcioni, F.; Astancolle, S.; Davalli, P.; Scaltriti, M.; Corti, A. Clusterin (SGP-2) transient overexpression decreases proliferation rate of SV40-immortalized human prostate epithelial cells by slowing down cell cycle progression. Oncogene 2002, 21, 4328–4334. [Google Scholar] [CrossRef] [Green Version]
- Trougakos, I.P.; Gonos, E.S. Clusterin/apolipoprotein J in human aging and cancer. Int. J. Biochem. Cell Biol. 2002, 34, 1430–1448. [Google Scholar] [CrossRef]
- Shannan, B.; Seifert, M.; Leskov, K.; Willis, J.; Boothman, D.; Tilgen, W.; Reichrath, J. Challenge and promise: Roles for clusterin in pathogenesis, progression and therapy of cancer. Cell Death Differ. 2006, 13, 12–19. [Google Scholar] [CrossRef]
- Park, S.; Mathis, K.W.; Lee, I.K. The physiological roles of apolipoprotein J/clusterin in metabolic and cardiovascular diseases. Rev. Endocr. Metab. Disord. 2014, 15, 45–53. [Google Scholar] [CrossRef]
- Jurkunas, U.V.; Bitar, M.S.; Rawe, I.; Harris, D.L.; Colby, K.; Joyce, N.C. Increased clusterin expression in Fuchs’ endothelial dystrophy. Investig. Ophthalmol. Vis. Sci. 2008, 49, 2946–2955. [Google Scholar] [CrossRef] [Green Version]
- Nishida, K.; Quantock, A.J.; Dota, A.; Choi-Miura, N.H.; Kinoshita, S. Apolipoproteins J and E co-localise with amyloid in gelatinous drop-like and lattice type I corneal dystrophies. Br. J. Ophthalmol. 1999, 83, 1178–1182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koltai, T. Clusterin: A key player in cancer chemoresistance and its inhibition. OncoTargets Ther. 2014, 7, 447–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.H.; Kim, J.H.; Jun, H.O.; Yu, Y.S.; Min, B.H.; Park, K.H.; Kim, K.W. Protective effect of clusterin from oxidative stress-induced apoptosis in human retinal pigment epithelial cells. Investig. Ophthalmol. Vis. Sci. 2010, 51, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.; Kutty, R.K.; Darrow, R.M.; Shivaram, S.; Kutty, G.; Fletcher, R.T.; Wiggert, B.; Chader, G.; Organisciak, D.T. Changes in clusterin expression associated with light-induced retinal damage in rats. Biochem. Cell Biol. 1994, 72, 499–503. [Google Scholar] [CrossRef]
- Vargas, A.; Kim, H.S.; Baral, E.; Yu, W.Q.; Craft, C.M.; Lee, E.J. Protective effect of clusterin on rod photoreceptor in rat model of retinitis pigmentosa. PLoS ONE 2017, 12, e0182389. [Google Scholar] [CrossRef] [Green Version]
- Ricci, F.; Pucci, S.; Sesti, F.; Missiroli, F.; Cerulli, L.; Spagnoli, L.G. Modulation of Ku70/80, clusterin/ApoJ isoforms and Bax expression in indocyanine-green-mediated photo-oxidative cell damage. Ophthalmic Res. 2007, 39, 164–173. [Google Scholar] [CrossRef]
- Bonacini, M.; Coletta, M.; Ramazzina, I.; Naponelli, V.; Modernelli, A.; Davalli, P.; Bettuzzi, S.; Rizzi, F. Distinct promoters, subjected to epigenetic regulation, drive the expression of two clusterin mRNAs in prostate cancer cells. Biochim. Biophys. Acta 2015, 1849, 44–54. [Google Scholar] [CrossRef]
- Cervellera, M.; Raschella, G.; Santilli, G.; Tanno, B.; Ventura, A.; Mancini, C.; Sevignani, C.; Calabretta, B.; Sala, A. Direct transactivation of the anti-apoptotic gene apolipoprotein J (clusterin) by B-MYB. J. Biol. Chem. 2000, 275, 21055–21060. [Google Scholar] [CrossRef] [Green Version]
- Jin, G.; Howe, P.H. Regulation of Clusterin Gene Expression by Transforming Growth Factor β. J. Biol. Chem. 1997, 272, 26620–26626. [Google Scholar] [CrossRef] [Green Version]
- Jin, G.; Howe, P.H. Transforming growth factor beta regulates clusterin gene expression via modulation of transcription factor c-Fos. Eur. J. Biochem. 1999, 263, 534–542. [Google Scholar] [CrossRef]
- Shiota, M.; Zardan, A.; Takeuchi, A.; Kumano, M.; Beraldi, E.; Naito, S.; Zoubeidi, A.; Gleave, M.E. Clusterin mediates TGF-beta-induced epithelial-mesenchymal transition and metastasis via Twist1 in prostate cancer cells. Cancer Res. 2012, 72, 5261–5272. [Google Scholar] [CrossRef] [Green Version]
- Shiota, M.; Zoubeidi, A.; Kumano, M.; Beraldi, E.; Naito, S.; Nelson, C.C.; Sorensen, P.H.; Gleave, M.E. Clusterin is a critical downstream mediator of stress-induced YB-1 transactivation in prostate cancer. Mol. Cancer Res. 2011, 9, 1755–1766. [Google Scholar] [CrossRef] [Green Version]
- Gutacker, C.; Klock, G.; Diel, P.; Koch-Brandt, C. Nerve growth factor and epidermal growth factor stimulate clusterin gene expression in PC12 cells. Biochem. J. 1999, 339 Pt 3, 759–766. [Google Scholar] [CrossRef]
- Park, J.; Park, S.Y.; Shin, E.; Lee, S.H.; Kim, Y.S.; Lee, D.H.; Roh, G.S.; Kim, H.J.; Kang, S.S.; Cho, G.J.; et al. Hypoxia inducible factor-1α directly regulates nuclear clusterin transcription by interacting with hypoxia response elements in the clusterin promoter. Mol. Cells 2014, 37, 178–186. [Google Scholar] [CrossRef] [Green Version]
- Herault, Y.; Chatelain, G.; Brun, G.; Michel, D. V-src-induced-transcription of the avian clusterin gene. Nucleic Acids Res. 1992, 20, 6377–6383. [Google Scholar] [CrossRef] [Green Version]
- Michel, D.; Chatelain, G.; North, S.; Brun, G. Stress-induced transcription of the clusterin/apoJ gene. Biochem. J. 1997, 328 Pt 1, 45–50. [Google Scholar] [CrossRef] [Green Version]
- Wong, P.; Taillefer, D.; Lakins, J.; Pineault, J.; Chader, G.; Tenniswood, M. Molecular characterization of human TRPM-2/clusterin, a gene associated with sperm maturation, apoptosis and neurodegeneration. Eur. J. Biochem. 1994, 221, 917–925. [Google Scholar] [CrossRef]
- Andersen, C.L.; Schepeler, T.; Thorsen, K.; Birkenkamp-Demtroder, K.; Mansilla, F.; Aaltonen, L.A.; Laurberg, S.; Orntoft, T.F. Clusterin expression in normal mucosa and colorectal cancer. Mol. Cell Proteom. 2007, 6, 1039–1048. [Google Scholar] [CrossRef] [Green Version]
- Prochnow, H.; Gollan, R.; Rohne, P.; Hassemer, M.; Koch-Brandt, C.; Baiersdorfer, M. Non-secreted clusterin isoforms are translated in rare amounts from distinct human mRNA variants and do not affect Bax-mediated apoptosis or the NF-kappaB signaling pathway. PLoS ONE 2013, 8, e75303. [Google Scholar] [CrossRef]
- Leskov, K.S.; Klokov, D.Y.; Li, J.; Kinsella, T.J.; Boothman, D.A. Synthesis and functional analyses of nuclear clusterin, a cell death protein. J. Biol. Chem. 2003, 278, 11590–11600. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.; Deng, J.H.; Wang, Z.; Cao, K.Y.; Zhou, L.; Wan, H. Knockdown of clusterin inhibits the growth and migration of renal carcinoma cells and leads to differential gene expression. Mol. Med. Rep. 2013, 8, 35–40. [Google Scholar] [CrossRef] [Green Version]
- Gingras, M.-E.; Larouche, K.; Larouche, N.; Leclerc, S.; Salesse, C.; Guérin, S.L. Regulation of the integrin subunit α5 gene promoter by the transcription factors Sp1/Sp3 is influenced by the cell density in rabbit corneal epithelial cells. Investig. Ophthalmol. Vis. Sci. 2003, 44, 3742–3755. [Google Scholar] [CrossRef] [Green Version]
- Gingras, M.E.; Masson-Gadais, B.; Zaniolo, K.; Leclerc, S.; Drouin, R.; Germain, L.; Guerin, S.L. Differential binding of the transcription factors Sp1, AP-1, and NFI to the promoter of the human α5 integrin gene dictates its transcriptional activity. Investig. Ophthalmol. Vis. Sci. 2009, 50, 57–67. [Google Scholar] [CrossRef]
- Larouche, K.; Leclerc, S.; Salesse, C.; Guérin, S.L. Expression of the α5 Integrin Subunit Gene Promoter Is Positively Regulated by the Extracellular Matrix Component Fibronectin through the Transcription Factor Sp1 in Corneal Epithelial Cells in Vitro. J. Biol. Chem. 2000, 275, 39182–39192. [Google Scholar] [CrossRef] [Green Version]
- Gaudreault, M.; Carrier, P.; Larouche, K.; Leclerc, S.; Giasson, M.; Germain, L.; Guerin, S.L. Influence of sp1/sp3 expression on corneal epithelial cells proliferation and differentiation properties in reconstructed tissues. Investig. Ophthalmol. Vis. Sci. 2003, 44, 1447–1457. [Google Scholar] [CrossRef] [Green Version]
- Gaudreault, M.; Gingras, M.-E.; Lessard, M.; Leclerc, S.; Guérin, S.L. Electrophoretic Mobility Shift Assays for the Analysis of DNA-Protein Interactions. In DNA-Protein Interactions; Humana Press: Totowa, NJ, USA, 2009; pp. 15–35. [Google Scholar] [CrossRef]
- Gaudreault, M.; Vigneault, F.O.; Leclerc, S.; Guérin, S.L. Laminin Reduces Expression of the Human α6 Integrin Subunit Gene by Altering the Level of the Transcription Factors Sp1 and Sp3. Investig. Opthalmol. Vis. Sci. 2007, 48, 3490. [Google Scholar] [CrossRef] [Green Version]
- Gain, P.; Jullienne, R.; He, Z.; Aldossary, M.; Acquart, S.; Cognasse, F.; Thuret, G. Global Survey of Corneal Transplantation and Eye Banking. JAMA Ophthalmol. 2016, 134, 167–173. [Google Scholar] [CrossRef] [Green Version]
- Jung, G.S.; Kim, M.K.; Jung, Y.A.; Kim, H.S.; Park, I.S.; Min, B.H.; Lee, K.U.; Kim, J.G.; Park, K.G.; Lee, I.K. Clusterin attenuates the development of renal fibrosis. J. Am. Soc. Nephrol. 2012, 23, 73–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, S.; Ledee, D.R.; Gordon, G.M.; Itakura, T.; Patel, N.; Martin, A.; Fini, M.E. Interaction of clusterin and matrix metalloproteinase-9 and its implication for epithelial homeostasis and inflammation. Am. J. Pathol. 2012, 180, 2028–2039. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Wang, Y.; Zhang, K.; Jiao, X.; Yan, B.; Liang, J. Antisense oligonucleotide against clusterin regulates human hepatocellular carcinoma invasion through transcriptional regulation of matrix metalloproteinase-2 and E-cadherin. Int. J. Mol. Sci. 2012, 13, 10594–10607. [Google Scholar] [CrossRef] [Green Version]
- Nishida, T.; Tanaka, T. Extracellular matrix and growth factors in corneal wound healing. Curr. Opin. Ophthalmol. 1996, 7, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Ljubimov, A.V.; Saghizadeh, M. Progress in corneal wound healing. Prog. Retin. Eye Res. 2015, 49, 17–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Hazmi, N.; Thomas, G.J.; Speight, P.M.; Whawell, S.A. The 120 kDa cell-binding fragment of fibronectin up-regulates migration of αvβ6-expressing cells by increasing matrix metalloproteinase-2 and -9 secretion. Eur. J. Oral Sci. 2007, 115, 454–458. [Google Scholar] [CrossRef] [PubMed]
- Thomas-Salgar, S.; Millis, A.J. Clusterin expression in differentiating smooth muscle cells. J. Biol. Chem. 1994, 269, 17879–17885. [Google Scholar] [CrossRef]
- Landreville, S.; Vigneault, F.; Bergeron, M.A.; Leclerc, S.; Gaudreault, M.; Morcos, M.; Mouriaux, F.; Salesse, C.; Guerin, S.L. Suppression of α5 gene expression is closely related to the tumorigenic properties of uveal melanoma cell lines. Pigment Cell Melanoma Res. 2011, 24, 643–655. [Google Scholar] [CrossRef]
- Halazonetis, T.D.; Georgopoulos, K.; Greenberg, M.E.; Leder, P. c-Jun dimerizes with itself and with c-Fos, forming complexes of different DNA binding affinities. Cell 1988, 55, 917–924. [Google Scholar] [CrossRef]
- O’Shea, E.K.; Rutkowski, R.; Kim, P.S. Mechanism of specificity in the Fos-Jun oncoprotein heterodimer. Cell 1992, 68, 699–708. [Google Scholar] [CrossRef]
- Bakiri, L.; Matsuo, K.; Wisniewska, M.; Wagner, E.F.; Yaniv, M. Promoter specificity and biological activity of tethered AP-1 dimers. Mol. Cell. Biol. 2002, 22, 4952–4964. [Google Scholar] [CrossRef] [Green Version]
- Grondin, B.; Lefrancois, M.; Tremblay, M.; Saint-Denis, M.; Haman, A.; Waga, K.; Bedard, A.; Tenen, D.G.; Hoang, T. c-Jun homodimers can function as a context-specific coactivator. Mol. Cell. Biol. 2007, 27, 2919–2933. [Google Scholar] [CrossRef] [Green Version]
- Chiu, R.; Angel, P.; Karin, M. Jun-B differs in its biological properties from, and is a negative regulator of, c-Jun. Cell 1989, 59, 979–986. [Google Scholar] [CrossRef]
- Vartanian, R.; Masri, J.; Martin, J.; Cloninger, C.; Holmes, B.; Artinian, N.; Funk, A.; Ruegg, T.; Gera, J. AP-1 regulates cyclin D1 and c-MYC transcription in an AKT-dependent manner in response to mTOR inhibition: Role of AIP4/Itch-mediated JUNB degradation. Mol. Cancer Res. 2011, 9, 115–130. [Google Scholar] [CrossRef] [Green Version]
- Rylski, M.; Amborska, R.; Zybura, K.; Michaluk, P.; Bielinska, B.; Konopacki, F.A.; Wilczynski, G.M.; Kaczmarek, L. JunB is a repressor of MMP-9 transcription in depolarized rat brain neurons. Mol. Cell. Neurosci. 2009, 40, 98–110. [Google Scholar] [CrossRef]
- Fuchs, S.Y.; Dolan, L.; Davis, R.J.; Ronai, Z. Phosphorylation-dependent targeting of c-Jun ubiquitination by Jun N-kinase. Oncogene 1996, 13, 1531–1535. [Google Scholar]
- Bossis, G.; Ferrara, P.; Acquaviva, C.; Jariel-Encontre, I.; Piechaczyk, M. c-Fos proto-oncoprotein is degraded by the proteasome independently of its own ubiquitinylation in vivo. Mol. Cell. Biol. 2003, 23, 7425–7436. [Google Scholar] [CrossRef] [Green Version]
- Bossis, G.; Malnou, C.E.; Farras, R.; Andermarcher, E.; Hipskind, R.; Rodriguez, M.; Schmidt, D.; Muller, S.; Jariel-Encontre, I.; Piechaczyk, M. Down-regulation of c-Fos/c-Jun AP-1 dimer activity by sumoylation. Mol. Cell. Biol. 2005, 25, 6964–6979. [Google Scholar] [CrossRef] [Green Version]
- Treier, M.; Staszewski, L.M.; Bohmann, D. Ubiquitin-dependent c-Jun degradation in vivo is mediated by the delta domain. Cell 1994, 78, 787–798. [Google Scholar] [CrossRef]
- Cuevas, B.D.; Uhlik, M.T.; Garrington, T.P.; Johnson, G.L. MEKK1 regulates the AP-1 dimer repertoire via control of JunB transcription and Fra-2 protein stability. Oncogene 2005, 24, 801–809. [Google Scholar] [CrossRef] [Green Version]
- Salvat, C.; Jariel-Encontre, I.; Acquaviva, C.; Omura, S.; Piechaczyk, M. Differential directing of c-Fos and c-Jun proteins to the proteasome in serum-stimulated mouse embryo fibroblasts. Oncogene 1998, 17, 327–337. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.J.; Lin, X.J.; Yang, X.J.; Zhou, L.; He, S.; Zhuang, S.M.; Yang, J. A novel AP-1/miR-101 regulatory feedback loop and its implication in the migration and invasion of hepatoma cells. Nucleic Acids Res. 2014, 42, 12041–12051. [Google Scholar] [CrossRef] [Green Version]
- Franklin, C.C.; McCulloch, A.V.; Kraft, A.S. In vitro association between the Jun protein family and the general transcription factors, TBP and TFIIB. Biochem. J. 1995, 305 Pt 3, 967–974. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.S.; See, R.H.; Deng, T.; Shi, Y. Adenovirus E1A downregulates cJun- and JunB-mediated transcription by targeting their coactivator p300. Mol. Cell. Biol. 1996, 16, 4312–4326. [Google Scholar] [CrossRef] [Green Version]
- Canettieri, G.; Coni, S.; Della Guardia, M.; Nocerino, V.; Antonucci, L.; Di Magno, L.; Screaton, R.; Screpanti, I.; Giannini, G.; Gulino, A. The coactivator CRTC1 promotes cell proliferation and transformation via AP-1. Proc. Natl. Acad. Sci. USA 2009, 106, 1445–1450. [Google Scholar] [CrossRef] [Green Version]
- Arias, J.; Alberts, A.S.; Brindle, P.; Claret, F.X.; Smeal, T.; Karin, M.; Feramisco, J.; Montminy, M. Activation of cAMP and mitogen responsive genes relies on a common nuclear factor. Nature 1994, 370, 226–229. [Google Scholar] [CrossRef]
- Claret, F.X.; Hibi, M.; Dhut, S.; Toda, T.; Karin, M. A new group of conserved coactivators that increase the specificity of AP-1 transcription factors. Nature 1996, 383, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Moreau, A.; Yotov, W.V.; Glorieux, F.H.; St-Arnaud, R. Bone-specific expression of the alpha chain of the nascent polypeptide-associated complex, a coactivator potentiating c-Jun-mediated transcription. Mol. Cell. Biol. 1998, 18, 1312–1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, C.C.; Chakraborty, A.; Diefenbacher, M.E.; Skehel, M.; Behrens, A. Arginine methylation of the c-Jun coactivator RACO-1 is required for c-Jun/AP-1 activation. EMBO J. 2013, 32, 1556–1567. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Pratt, H.; Gao, M.; Wei, F.; Weng, Z.; Struhl, K. YAP and TAZ are transcriptional co-activators of AP-1 proteins and STAT3 during breast cellular transformation. eLife 2021, 10. [Google Scholar] [CrossRef]
- Thompson, B.J. YAP/TAZ: Drivers of Tumor Growth, Metastasis, and Resistance to Therapy. Bioessays 2020, 42, e1900162. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, K.; Halle, J.P.; Stelzer, G.; Meisterernst, M.; Kawakami, K. Involvement of negative cofactor NC2 in active repression by zinc finger-homeodomain transcription factor AREB6. Mol. Cell. Biol. 1998, 18, 10–18. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.; Spann, N.J.; Hui, T.Y.; Davis, R.A. ARP-1/COUP-TF II determines hepatoma phenotype by acting as both a transcriptional repressor of microsomal triglyceride transfer protein and an inducer of CYP7A1. J. Biol. Chem. 2003, 278, 30478–30486. [Google Scholar] [CrossRef] [Green Version]
- Liang, B.; Wang, J. EVI1 in Leukemia and Solid Tumors. Cancers 2020, 12, 2667. [Google Scholar] [CrossRef]
- Suzuki, K.; Matsumoto, M.; Katoh, Y.; Liu, L.; Ochiai, K.; Aizawa, Y.; Nagatomi, R.; Okuno, H.; Itoi, E.; Igarashi, K. Bach1 promotes muscle regeneration through repressing Smad-mediated inhibition of myoblast differentiation. PLoS ONE 2020, 15, e0236781. [Google Scholar] [CrossRef]
- Wilson, M.; Tsakraklides, V.; Tran, M.; Xiao, Y.Y.; Zhang, Y.; Perkins, A.S. EVI1 Interferes with Myeloid Maturation via Transcriptional Repression of Cebpa, via Binding to Two Far Downstream Regulatory Elements. J. Biol. Chem. 2016, 291, 13591–13607. [Google Scholar] [CrossRef] [Green Version]
- Yusoff, F.M.; Maruhashi, T.; Kawano, K.I.; Nakashima, A.; Chayama, K.; Tashiro, S.; Igarashi, K.; Higashi, Y. Bach1 plays an important role in angiogenesis through regulation of oxidative stress. Microvasc. Res. 2021, 134, 104126. [Google Scholar] [CrossRef]
- Bisson, F.; Rochefort, É.; Lavoie, A.; Larouche, D.; Zaniolo, K.; Simard-Bisson, C.; Damour, O.; Auger, F.; Guérin, S.; Germain, L. Irradiated Human Dermal Fibroblasts Are as Efficient as Mouse Fibroblasts as a Feeder Layer to Improve Human Epidermal Cell Culture Lifespan. Int. J. Mol. Sci. 2013, 14, 4684–4704. [Google Scholar] [CrossRef]
- Laplante, A.F.; Germain, L.; Auger, F.A.; Moulin, V. Mechanisms of wound reepithelialization: Hints from a tissue-engineered reconstructed skin to long-standing questions. FASEB J. 2001, 15, 2377–2389. [Google Scholar] [CrossRef] [Green Version]
- Pothier, F.; Ouellet, M.; Julien, J.P.; Guérin, S.L. An improved CAT assay for promoter analysis in either transgenic mice or tissue culture cells. DNA Cell Biol. 1992, 11. [Google Scholar] [CrossRef]
- Zaniolo, K.; Desnoyers, S.; Leclerc, S.; Guérin, S.L. Regulation of poly(ADP-ribose) polymerase-1 (PARP-1) gene expression through the post-translational modification of Sp1: A nuclear target protein of PARP-1. BMC Mol. Biol. 2007, 8, 96. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gross, C.; Le-Bel, G.; Desjardins, P.; Benhassine, M.; Germain, L.; Guérin, S.L. Contribution of the Transcription Factors Sp1/Sp3 and AP-1 to Clusterin Gene Expression during Corneal Wound Healing of Tissue-Engineered Human Corneas. Int. J. Mol. Sci. 2021, 22, 12426. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222212426
Gross C, Le-Bel G, Desjardins P, Benhassine M, Germain L, Guérin SL. Contribution of the Transcription Factors Sp1/Sp3 and AP-1 to Clusterin Gene Expression during Corneal Wound Healing of Tissue-Engineered Human Corneas. International Journal of Molecular Sciences. 2021; 22(22):12426. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222212426
Chicago/Turabian StyleGross, Christelle, Gaëtan Le-Bel, Pascale Desjardins, Manel Benhassine, Lucie Germain, and Sylvain L. Guérin. 2021. "Contribution of the Transcription Factors Sp1/Sp3 and AP-1 to Clusterin Gene Expression during Corneal Wound Healing of Tissue-Engineered Human Corneas" International Journal of Molecular Sciences 22, no. 22: 12426. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222212426