
Toll-like Receptor Signaling Inhibitory Peptide Improves Inflammation in Animal Model and Human Systemic Lupus Erythematosus

 ,
,  , , , , and
, , , , and 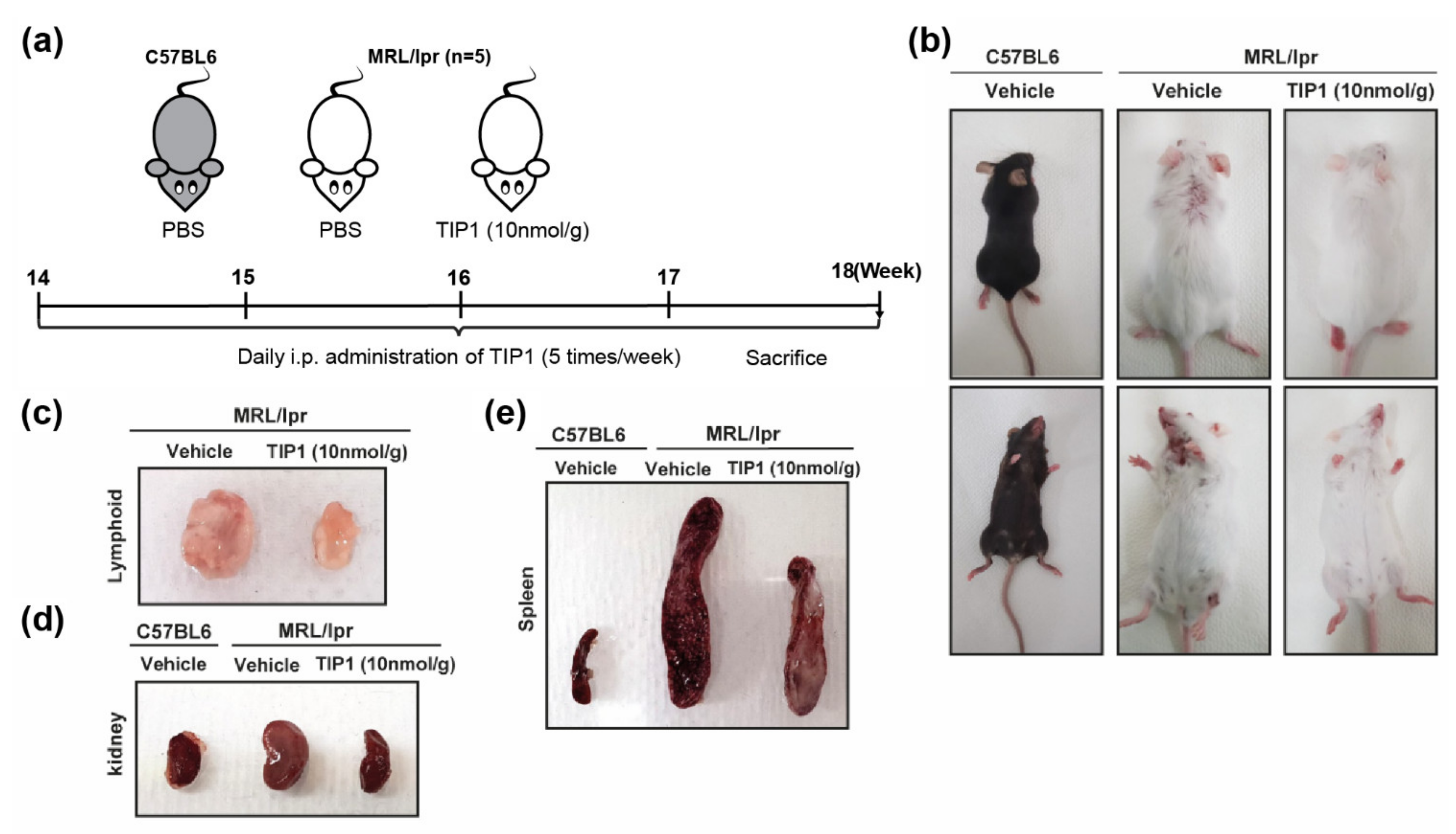{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. TIP1 Treatment Ameliorates Skin Damage in Lupus-Prone Mice (MRL/lpr)
2.2. TIP1 Treatment Reduces Nephromegaly, Splenomegaly, and Lymphadenopathy
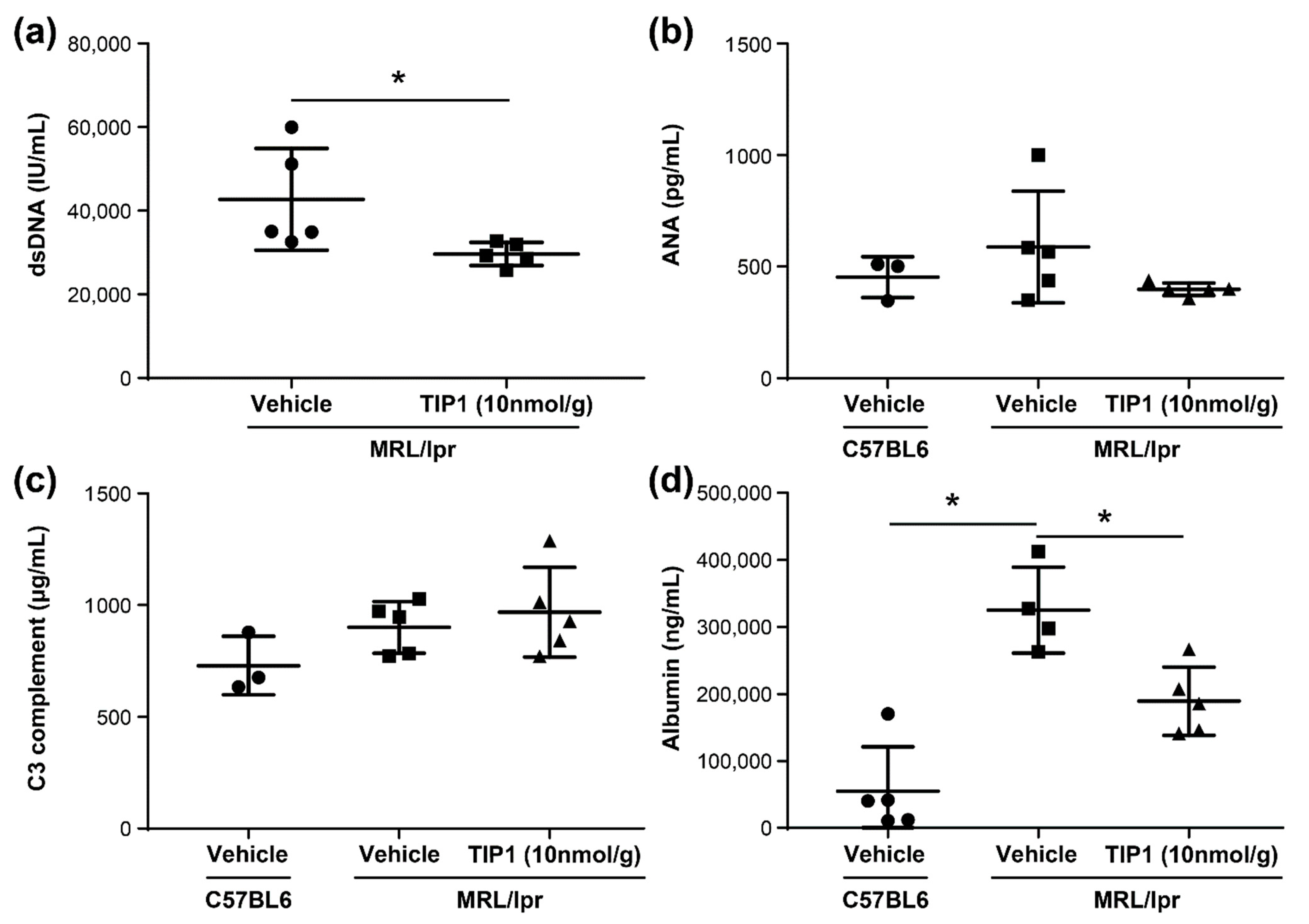
2.3. TIP1 Treatment Decreases Anti-dsDNA Antibody and Urine Albumin Levels
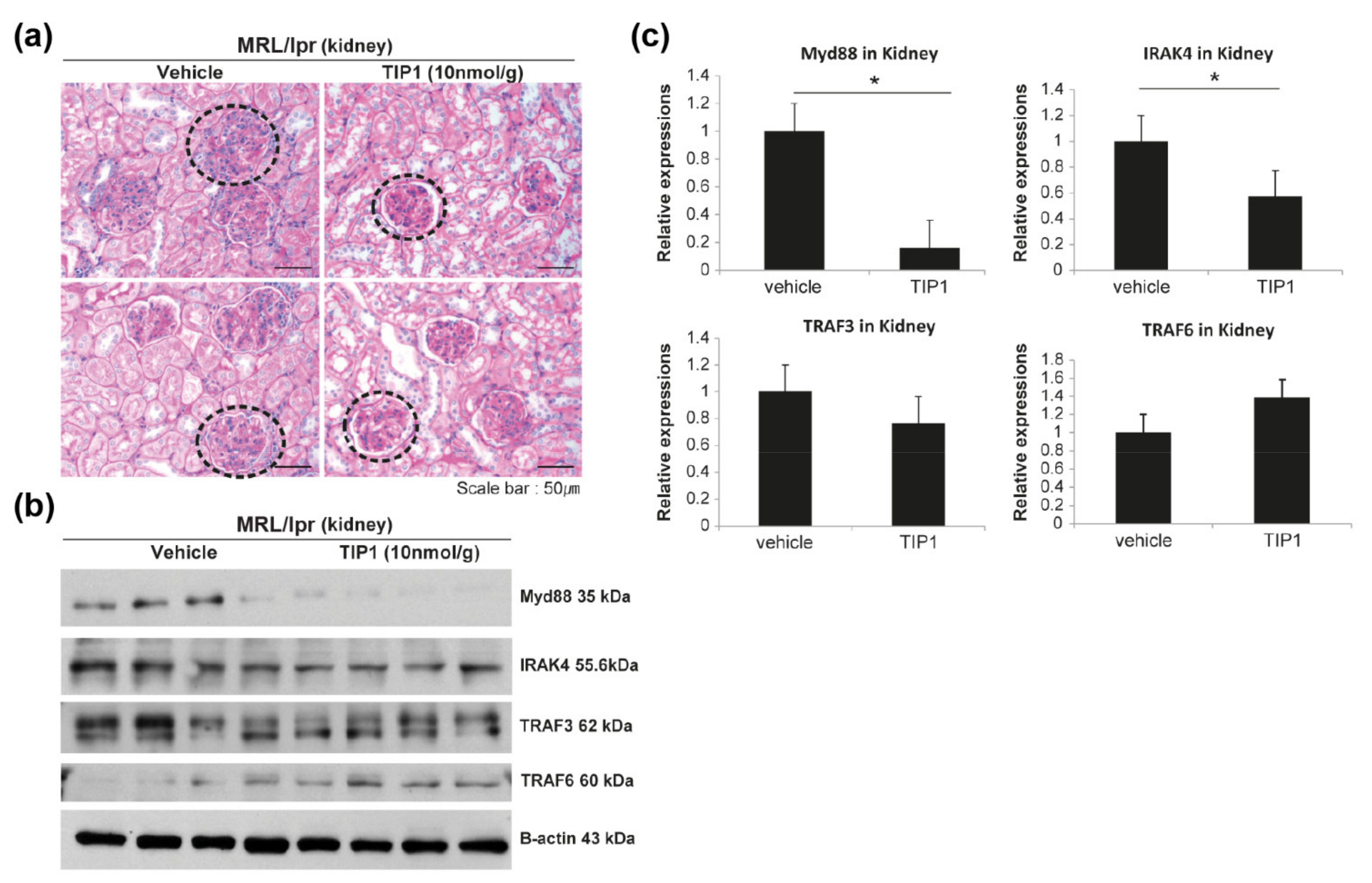
2.4. Therapeutic Effect of TIP1 in LN
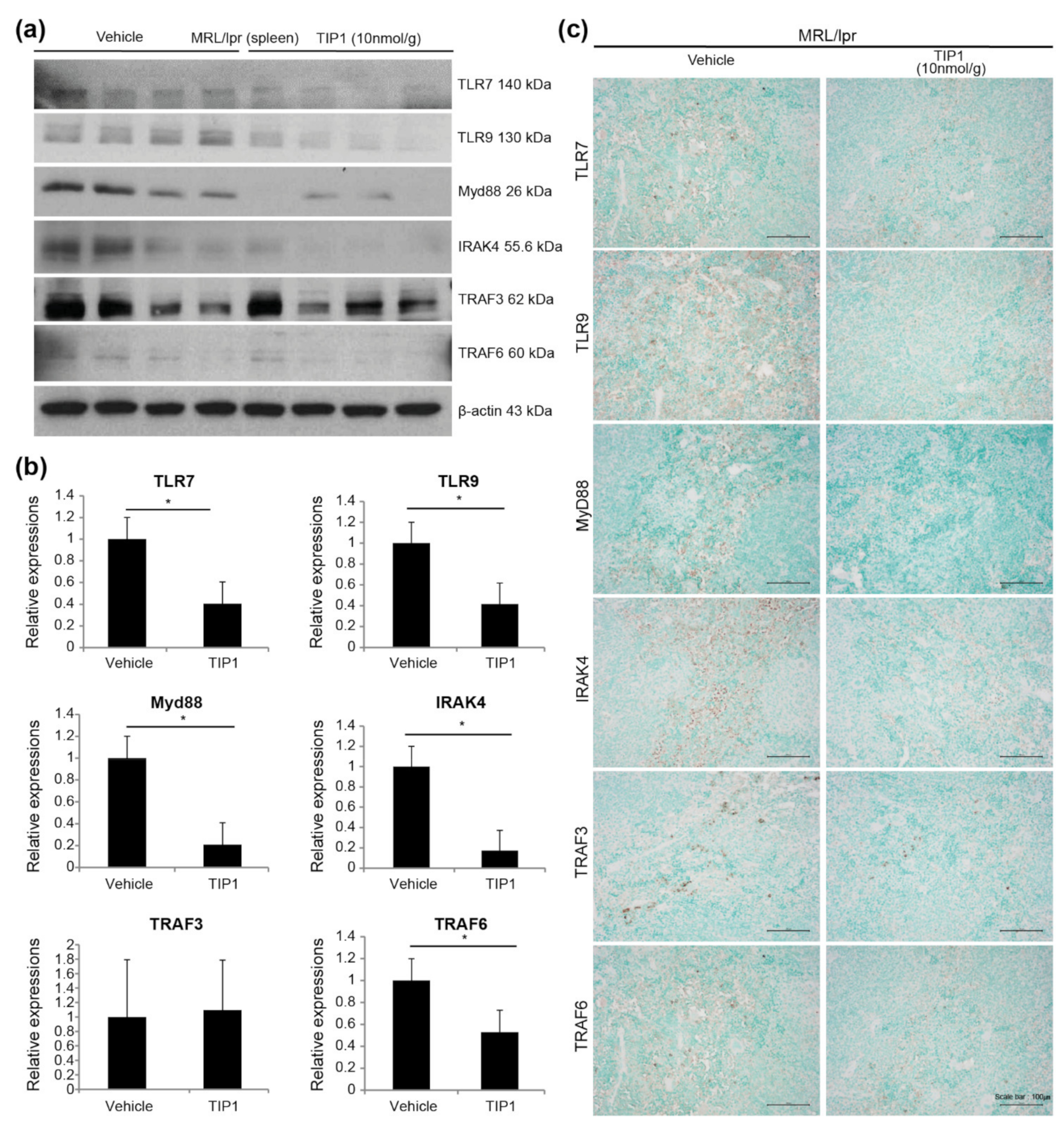
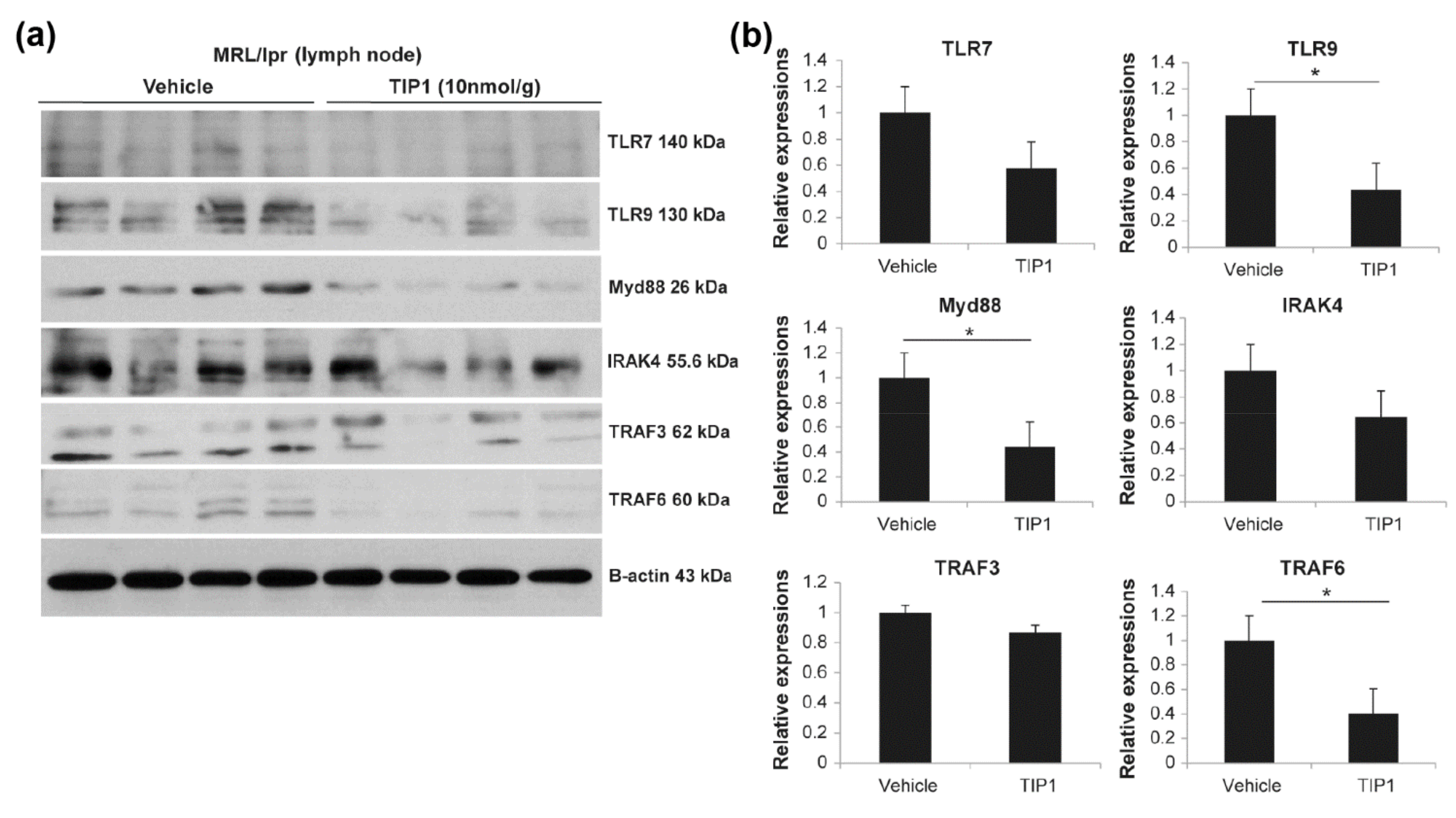
2.5. TIP1 Inhibits the TLR7/9 Signaling Pathway
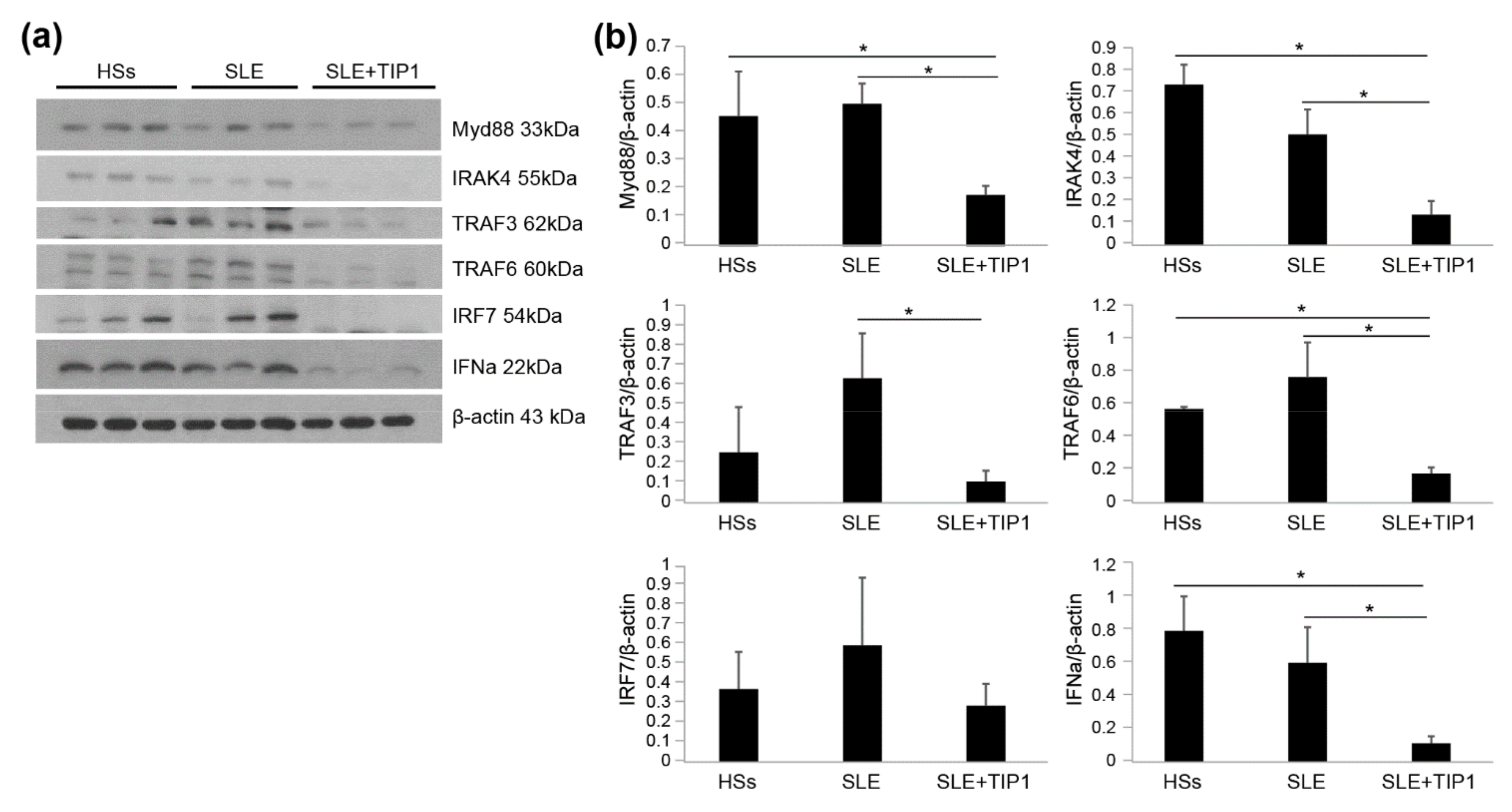
2.6. Inhibitory Effects of TIP1 on TLR Signaling Pathways Ex Vivo
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Measurement of Disease Markers of SLE
4.3. Histology and Immunohistochemistry of the Kidney Tissue
4.4. Western Blot Analysis
4.5. Ex Vivo Cell Culture
4.6. Data Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- D’Cruz, D.P.; Khamashta, M.A.; Hughes, G.R. Systemic lupus erythematosus. Lancet 2007, 369, 587–596. [Google Scholar] [CrossRef]
- Tsokos, G.C. Systemic lupus erythematosus. N. Engl. J. Med. 2011, 365, 2110–2121. [Google Scholar] [CrossRef] [Green Version]
- Rahman, A.; Isenberg, D.A. Systemic lupus erythematosus. N. Engl. J. Med. 2008, 358, 929–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceccarelli, F.; Perricone, C.; Borgiani, P.; Ciccacci, C.; Rufini, S.; Cipriano, E.; Alessandri, C.; Spinelli, F.R.; Sili Scavalli, A.; Novelli, G.; et al. Genetic Factors in Systemic Lupus Erythematosus: Contribution to Disease Phenotype. J. Immunol. Res. 2015, 2015, 745647. [Google Scholar] [CrossRef] [Green Version]
- Moser, K.L.; Kelly, J.A.; Lessard, C.J.; Harley, J.B. Recent insights into the genetic basis of systemic lupus erythematosus. Genes Immun. 2009, 10, 373–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrett-Sinha, L.A.; Kearly, A.; Satterthwaite, A.B. The Role of the Transcription Factor Ets1 in Lupus and Other Autoimmune Diseases. Crit. Rev. Immunol. 2016, 36, 485–510. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.J.; Lee, C.G.; Jung, J.Y.; Ghosh, A.; Hasan, S.N.; Hwang, S.M.; Kang, H.; Lee, C.; Kim, G.C.; Rudra, D.; et al. The Transcription Factor Ets1 Suppresses T Follicular Helper Type 2 Cell Differentiation to Halt the Onset of Systemic Lupus Erythematosus. Immunity 2018, 49, 1034–1048.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moulton, V.R.; Suarez-Fueyo, A.; Meidan, E.; Li, H.; Mizui, M.; Tsokos, G.C. Pathogenesis of Human Systemic Lupus Erythematosus: A Cellular Perspective. Trends Mol. Med. 2017, 23, 615–635. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janeway, C.A., Jr.; Medzhitov, R. Innate immune recognition. Annu. Rev. Immunol. 2002, 20, 197–216. [Google Scholar] [CrossRef] [Green Version]
- Medzhitov, R. Recognition of microorganisms and activation of the immune response. Nature 2007, 449, 819–826. [Google Scholar] [CrossRef] [PubMed]
- Ewald, S.E.; Barton, G.M. Nucleic acid sensing Toll-like receptors in autoimmunity. Curr. Opin. Immunol. 2011, 23, 3–9. [Google Scholar] [CrossRef] [Green Version]
- Berland, R.; Fernandez, L.; Kari, E.; Han, J.H.; Lomakin, I.; Akira, S.; Wortis, H.H.; Kearney, J.F.; Ucci, A.A.; Imanishi-Kari, T. Toll-like receptor 7-dependent loss of B cell tolerance in pathogenic autoantibody knockin mice. Immunity 2006, 25, 429–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawar, R.D.; Patole, P.S.; Zecher, D.; Segerer, S.; Kretzler, M.; Schlondorff, D.; Anders, H.J. Toll-like receptor-7 modulates immune complex glomerulonephritis. J. Am. Soc. Nephrol. 2006, 17, 141–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, C.; Chen, J.; Chu, F.; Zhu, J.; Jin, T. Inflammatory Role of TLR-MyD88 Signaling in Multiple Sclerosis. Front. Mol. Neurosci. 2019, 12, 314. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Toll-like receptor downstream signaling. Arthritis Res. Ther. 2005, 7, 12–19. [Google Scholar] [CrossRef] [Green Version]
- De Nardo, D.; Balka, K.R.; Cardona Gloria, Y.; Rao, V.R.; Latz, E.; Masters, S.L. Interleukin-1 receptor-associated kinase 4 (IRAK4) plays a dual role in myddosome formation and Toll-like receptor signaling. J. Biol. Chem. 2018, 293, 15195–15207. [Google Scholar] [CrossRef] [Green Version]
- Park, J.E.; Kim, Y.I.; Yi, A.K. Protein kinase D1 is essential for MyD88-dependent TLR signaling pathway. J. Immunol. 2009, 182, 6316–6327. [Google Scholar] [CrossRef]
- Uematsu, S.; Sato, S.; Yamamoto, M.; Hirotani, T.; Kato, H.; Takeshita, F.; Matsuda, M.; Coban, C.; Ishii, K.J.; Kawai, T.; et al. Interleukin-1 receptor-associated kinase-1 plays an essential role for Toll-like receptor (TLR)7- and TLR9-mediated interferon-{alpha} induction. J. Exp. Med. 2005, 201, 915–923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawai, T.; Sato, S.; Ishii, K.J.; Coban, C.; Hemmi, H.; Yamamoto, M.; Terai, K.; Matsuda, M.; Inoue, J.; Uematsu, S.; et al. Interferon-alpha induction through Toll-like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nat. Immunol. 2004, 5, 1061–1068. [Google Scholar] [CrossRef]
- Kwon, H.K.; Patra, M.C.; Shin, H.J.; Gui, X.; Achek, A.; Panneerselvam, S.; Kim, D.J.; Song, S.J.; Hong, R.; Kim, K.S.; et al. A cell-penetrating peptide blocks Toll-like receptor-mediated downstream signaling and ameliorates autoimmune and inflammatory diseases in mice. Exp. Mol. Med. 2019, 51, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohl, K.; Tenbrock, K. Inflammatory cytokines in systemic lupus erythematosus. J. Biomed. Biotechnol. 2011, 2011, 432595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Neill, L.A.; Golenbock, D.; Bowie, A.G. The history of Toll-like receptors—Redefining innate immunity. Nat. Rev. Immunol. 2013, 13, 453–460. [Google Scholar] [CrossRef]
- Lisnevskaia, L.; Murphy, G.; Isenberg, D. Systemic lupus erythematosus. Lancet 2014, 384, 1878–1888. [Google Scholar] [CrossRef]
- Gatto, M.; Zen, M.; Iaccarino, L.; Doria, A. New therapeutic strategies in systemic lupus erythematosus management. Nat. Rev. Rheumatol. 2019, 15, 30–48. [Google Scholar] [CrossRef] [PubMed]
- Lopate, G.; Pestronk, A. Chronic immune demyelinating neuropathies. Semin. Neurol. 1994, 14, 131–136. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Karypis, G.; Hippen, K.L.; Vegoe, A.L.; Ruiz, P.; Gilkeson, G.S.; Behrens, T.W. Genomic view of systemic autoimmunity in MRLlpr mice. Genes Immun. 2006, 7, 156–168. [Google Scholar] [CrossRef]
- Lemay, S.; Mao, C.; Singh, A.K. Cytokine gene expression in the MRL/lpr model of lupus nephritis. Kidney Int. 1996, 50, 85–93. [Google Scholar] [CrossRef] [Green Version]
- Arbuckle, M.R.; McClain, M.T.; Rubertone, M.V.; Scofield, R.H.; Dennis, G.J.; James, J.A.; Harley, J.B. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N. Engl. J. Med. 2003, 349, 1526–1533. [Google Scholar] [CrossRef] [Green Version]
- Wandstrat, A.E.; Carr-Johnson, F.; Branch, V.; Gray, H.; Fairhurst, A.M.; Reimold, A.; Karp, D.; Wakeland, E.K.; Olsen, N.J. Autoantibody profiling to identify individuals at risk for systemic lupus erythematosus. J. Autoimmun. 2006, 27, 153–160. [Google Scholar] [CrossRef]
- Gasparin, A.A.; Pamplona Bueno de Andrade, N.; Hax, V.; Tres, G.L.; Veronese, F.V.; Monticielo, O.A. Urinary biomarkers for lupus nephritis: The role of the vascular cell adhesion molecule-1. Lupus 2019, 28, 265–272. [Google Scholar] [CrossRef]
- Ahmed, N.; Shigidi, M.; Al Agib, A.N.; Abdelrahman, H.; Taha, E. Clinical features and antinuclear antibodies profile among adults with systemic lupus erythematosus and lupus nephritis: A cross-sectional study. Pan Afr. Med. J. 2017, 27, 114. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.M.; Li, C.F.; Ke, S.Y.; Piao, Y.R.; Han, T.X.; Kuang, W.Y.; Wang, J.; Deng, J.H.; Tan, X.H.; Li, C. Analysis of clinical manifestations and treatment in 26 children with fibrodysplasia ossificans progressiva in China. World J. Pediatr. 2020, 16, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Walport, M.J. Complement and systemic lupus erythematosus. Arthritis Res. 2002, 4, S279–S293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timlin, H.; Magder, L.; Petri, M. Clinical Outcomes Observed among Biopsy Proven Lupus Nephritis Patients Treated with Mycophenolate Mofetil as First-line Therapy. Cureus 2017, 9, e1907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clynes, R.; Dumitru, C.; Ravetch, J.V. Uncoupling of immune complex formation and kidney damage in autoimmune glomerulonephritis. Science 1998, 279, 1052–1054. [Google Scholar] [CrossRef] [PubMed]
- Conti, F.; Spinelli, F.R.; Truglia, S.; Miranda, F.; Alessandri, C.; Ceccarelli, F.; Bombardieri, M.; Giannakakis, K.; Valesini, G. Kidney Expression of Toll Like Receptors in Lupus Nephritis: Quantification and Clinicopathological Correlations. Mediators Inflamm. 2016, 2016, 7697592. [Google Scholar] [CrossRef]
- Yung, S.; Chan, T.M. Mechanisms of Kidney Injury in Lupus Nephritis—The Role of Anti-dsDNA Antibodies. Front. Immunol. 2015, 6, 475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yung, S.; Chan, T.M. Anti-DNA antibodies in the pathogenesis of lupus nephritis—The emerging mechanisms. Autoimmun. Rev. 2008, 7, 317–321. [Google Scholar] [CrossRef]
- Marshak-Rothstein, A. Toll-like receptors in systemic autoimmune disease. Nat. Rev. Immunol. 2006, 6, 823–835. [Google Scholar] [CrossRef] [PubMed]
- Baechler, E.C.; Batliwalla, F.M.; Karypis, G.; Gaffney, P.M.; Ortmann, W.A.; Espe, K.J.; Shark, K.B.; Grande, W.J.; Hughes, K.M.; Kapur, V.; et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc. Natl. Acad. Sci. USA 2003, 100, 2610–2615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Celhar, T.; Lu, H.K.; Benso, L.; Rakhilina, L.; Lee, H.Y.; Tripathi, S.; Zharkova, O.; Ong, W.Y.; Yasuga, H.; Au, B.; et al. TLR7 Protein Expression in Mild and Severe Lupus-Prone Models Is Regulated in a Leukocyte, Genetic, and IRAK4 Dependent Manner. Front. Immunol. 2019, 10, 1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baek, W.Y.; Woo, J.M.; Kim, H.A.; Jung, J.Y.; Suh, C.H. Polymorphisms of MFGE8 are associated with susceptibility and clinical manifestations through gene expression modulation in Koreans with systemic lupus erythematosus. Sci. Rep. 2019, 9, 18565. [Google Scholar] [CrossRef] [PubMed]
- Hennessy, E.J.; Parker, A.E.; O’Neill, L.A. Targeting Toll-like receptors: Emerging therapeutics? Nat. Rev. Drug Discov. 2010, 9, 293–307. [Google Scholar] [CrossRef] [PubMed]
- Mills, K.H. TLR-dependent T cell activation in autoimmunity. Nat. Rev. Immunol. 2011, 11, 807–822. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, M.E. DAMPs, PAMPs and alarmins: All we need to know about danger. J. Leukoc. Biol. 2007, 81, 1–5. [Google Scholar] [CrossRef]
- Uematsu, S.; Akira, S. Toll-like receptors and Type I interferons. J. Biol. Chem. 2007, 282, 15319–15323. [Google Scholar] [CrossRef] [Green Version]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [Green Version]
- Ye, H.; Arron, J.R.; Lamothe, B.; Cirilli, M.; Kobayashi, T.; Shevde, N.K.; Segal, D.; Dzivenu, O.K.; Vologodskaia, M.; Yim, M.; et al. Distinct molecular mechanism for initiating TRAF6 signalling. Nature 2002, 418, 443–447. [Google Scholar] [CrossRef]
- Tan, E.M.; Cohen, A.S.; Fries, J.F.; Masi, A.T.; McShane, D.J.; Rothfield, N.F.; Schaller, J.G.; Talal, N.; Winchester, R.J. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1982, 25, 1271–1277. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baek, W.-Y.; Choi, Y.-S.; Lee, S.-W.; Son, I.-O.; Jeon, K.-W.; Choi, S.-D.; Suh, C.-H. Toll-like Receptor Signaling Inhibitory Peptide Improves Inflammation in Animal Model and Human Systemic Lupus Erythematosus. Int. J. Mol. Sci. 2021, 22, 12764. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222312764
Baek W-Y, Choi Y-S, Lee S-W, Son I-O, Jeon K-W, Choi S-D, Suh C-H. Toll-like Receptor Signaling Inhibitory Peptide Improves Inflammation in Animal Model and Human Systemic Lupus Erythematosus. International Journal of Molecular Sciences. 2021; 22(23):12764. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222312764
Chicago/Turabian StyleBaek, Wook-Young, Yang-Seon Choi, Sang-Won Lee, In-Ok Son, Ki-Woong Jeon, Sang-Dun Choi, and Chang-Hee Suh. 2021. "Toll-like Receptor Signaling Inhibitory Peptide Improves Inflammation in Animal Model and Human Systemic Lupus Erythematosus" International Journal of Molecular Sciences 22, no. 23: 12764. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222312764