
Interaction Profiles of Central Nervous System Active Drugs at Human Organic Cation Transporters 1–3 and Human Plasma Membrane Monoamine Transporter

 , , , and
, , , and
Abstract
:1. Introduction
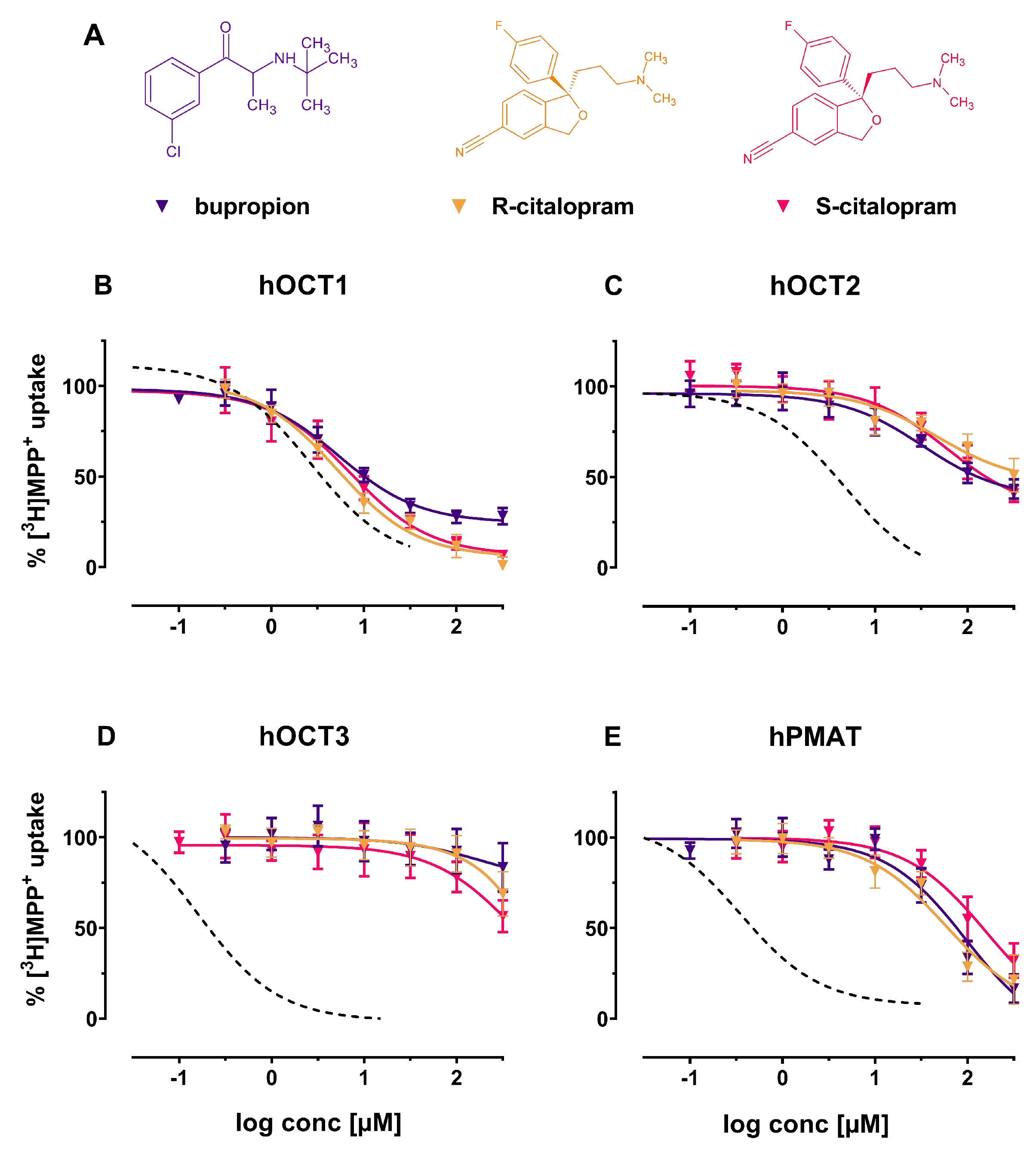
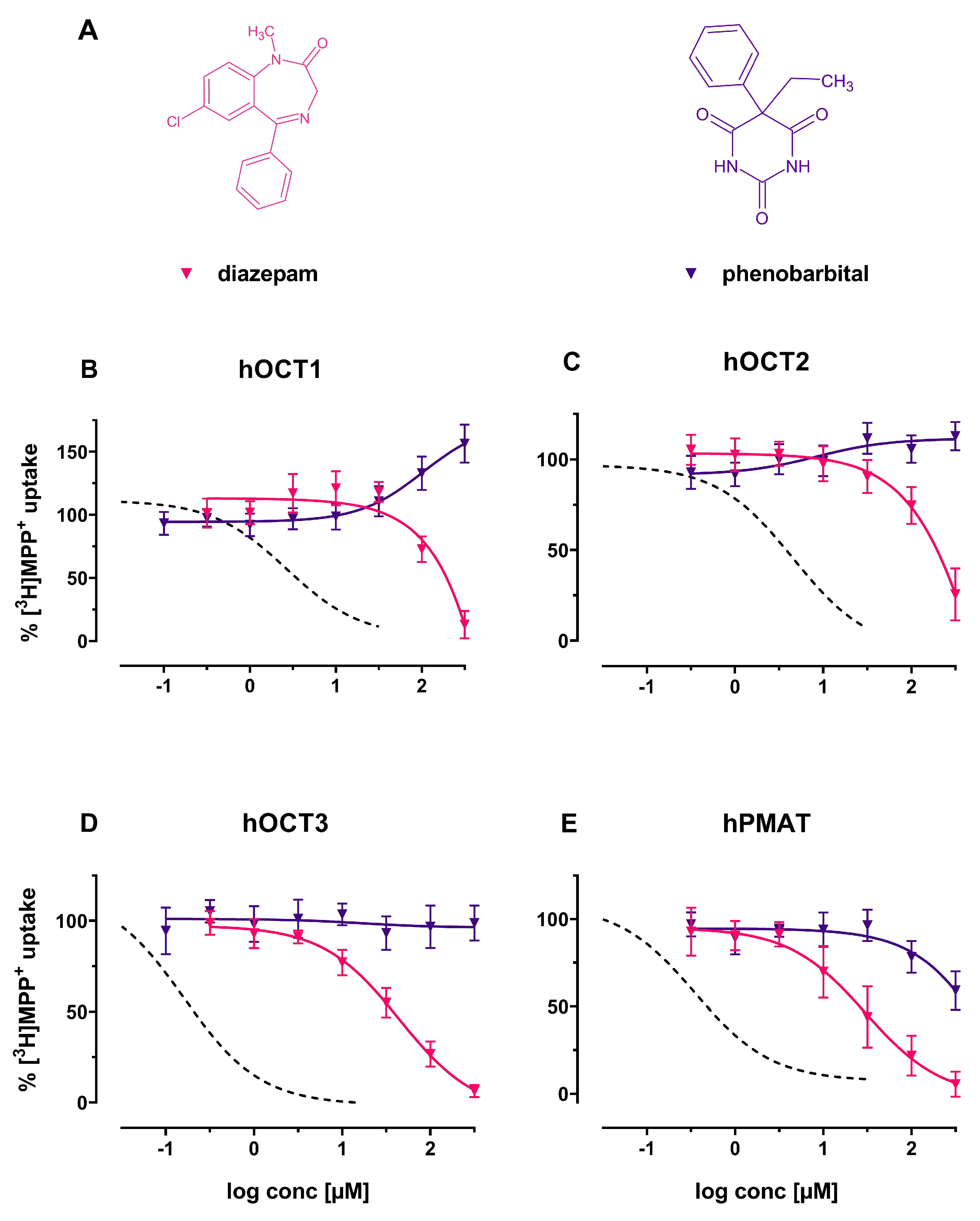
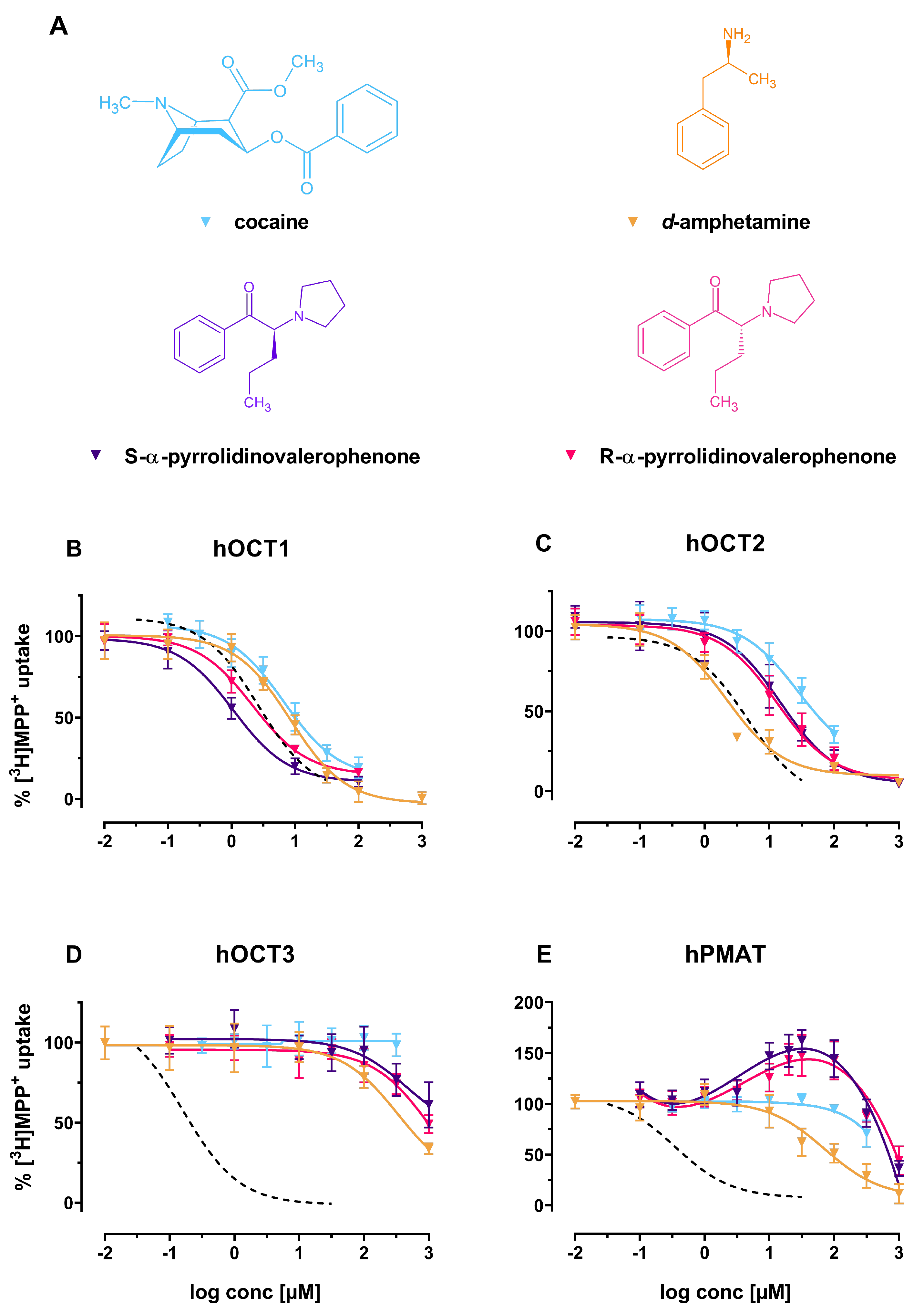
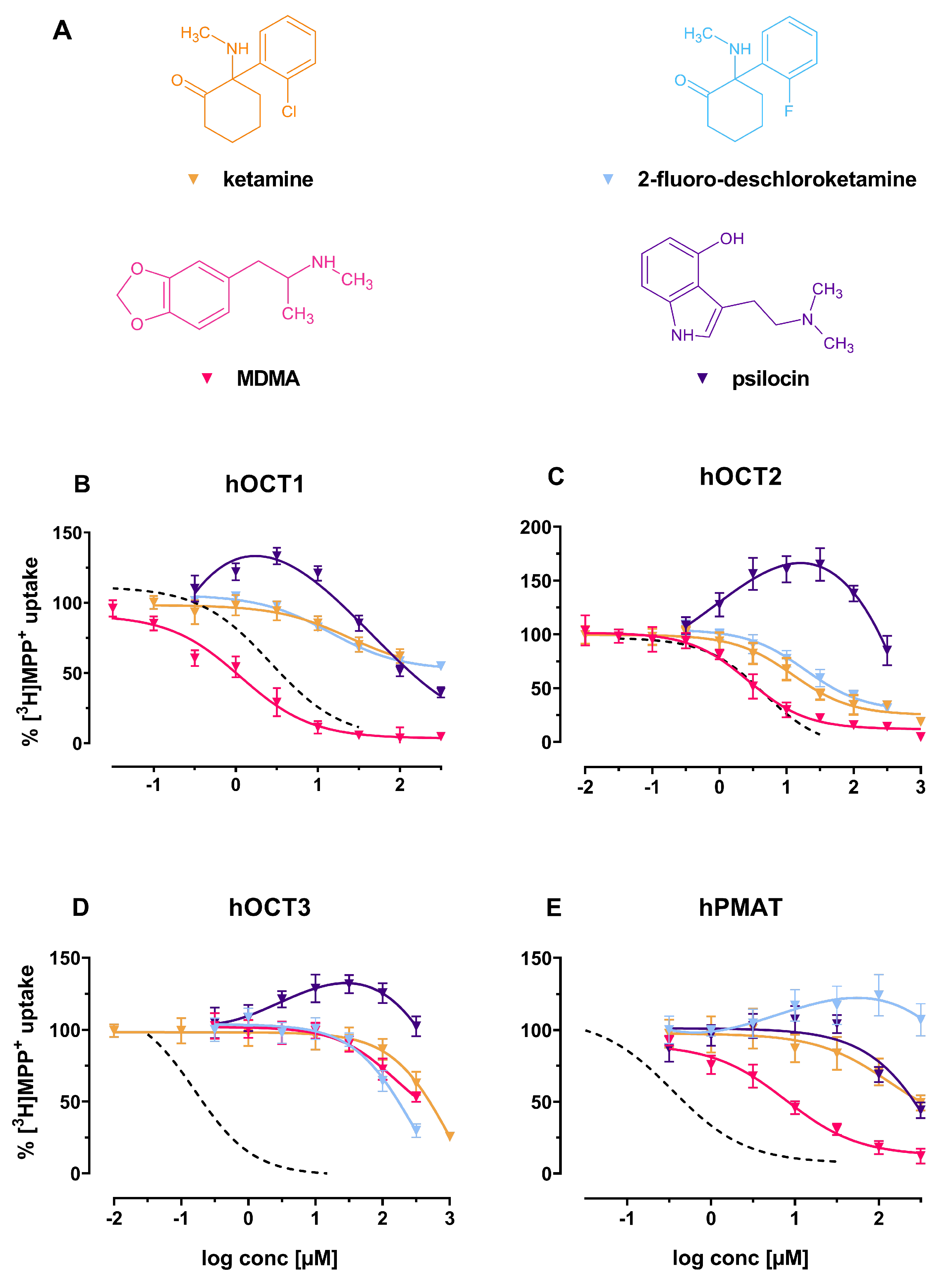
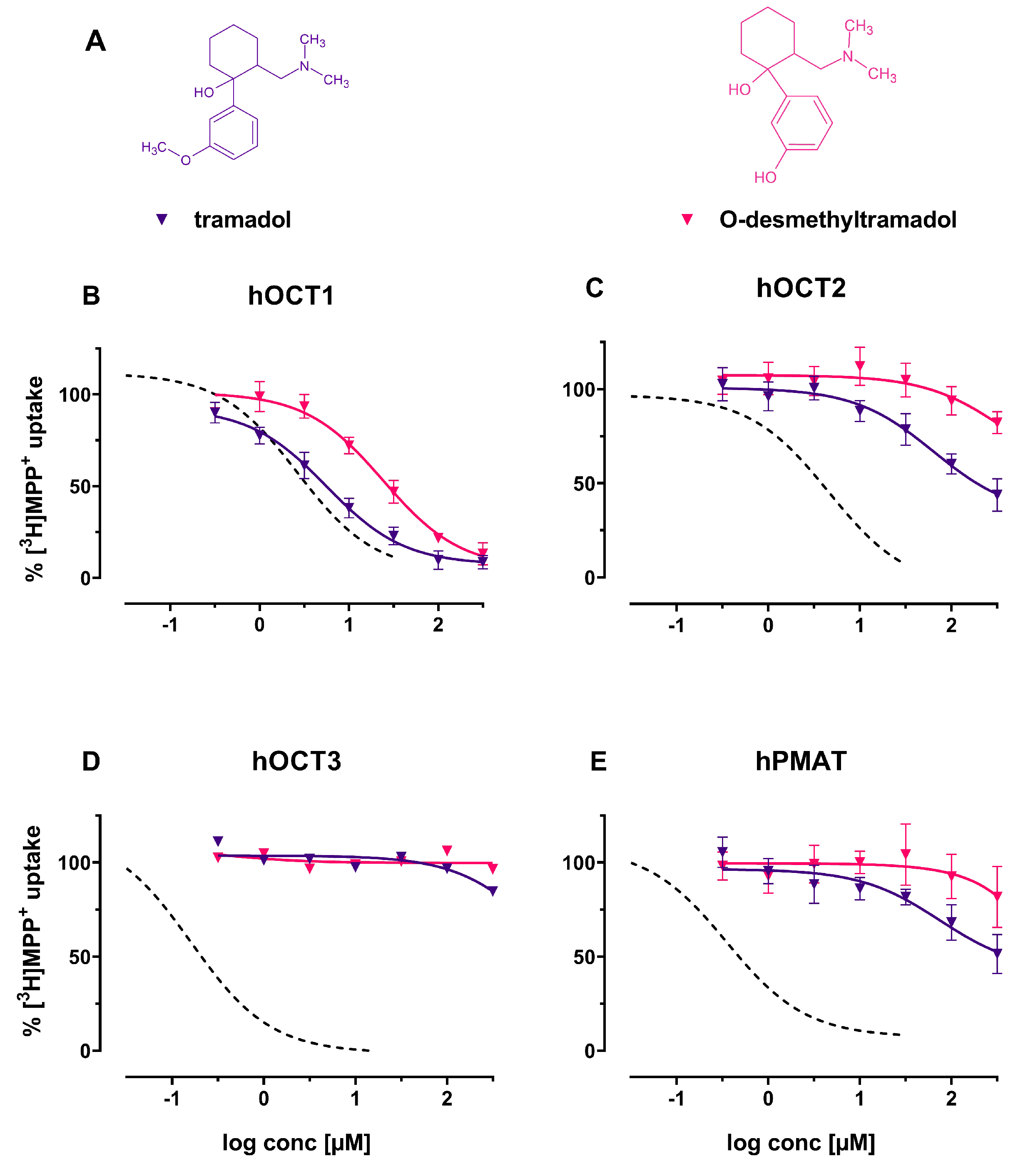
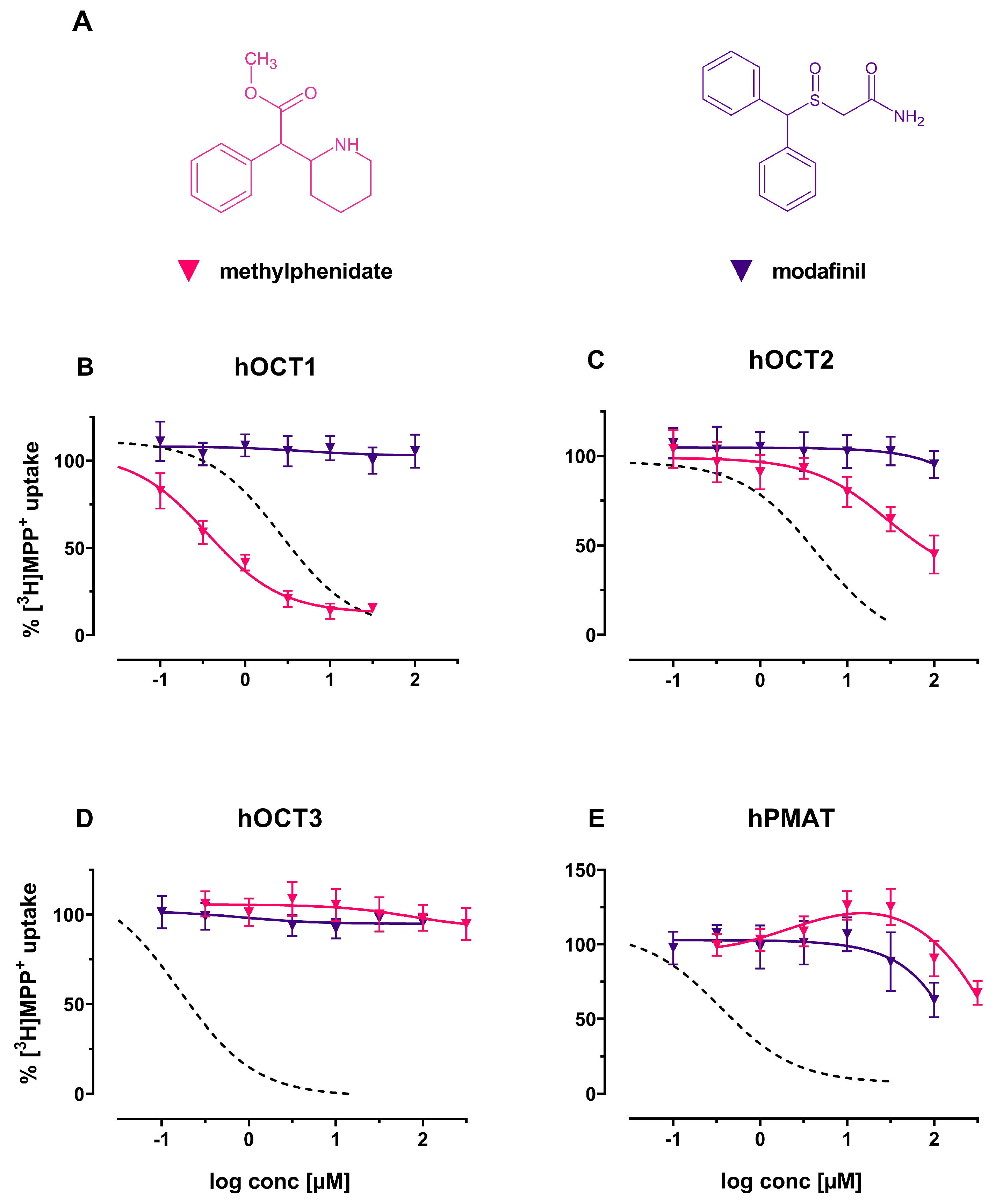
2. Results
3. Discussion
4. Materials and Methods
4.1. Chemicals and reagents
4.2. Cell Culture
4.3. Uptake Inhibition Assays
4.4. Data and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hayer-Zillgen, M.; Brüss, M.; Bönisch, H. Expression and pharmacological profile of the human organic cation transporters hOCT1, hOCT2 and hOCT3. Br. J. Pharmacol. 2002, 136, 829–836. [Google Scholar] [CrossRef] [Green Version]
- Maier, J.; Niello, M.; Rudin, D.; Daws, L.C.; Sitte, H.H. The Interaction of Organic Cation Transporters 1-3 and PMAT with Psychoactive Substances. Handb. Exp. Pharmacol. 2021, 3, 1–20. [Google Scholar] [CrossRef]
- Gasser, P.J. Organic Cation Transporters in Brain Catecholamine Homeostasis. In Handbook of Experimental Pharmacology; Springer: Cham, Switzerland, 2021; Volume 266, pp. 187–197. [Google Scholar] [CrossRef]
- Jonker, J.W.; Wagenaar, E.; van Eijl, S.; Schinkel, A.H. Deficiency in the Organic Cation Transporters 1 and 2 (Oct1/Oct2 [Slc22a1/Slc22a2]) in Mice Abolishes Renal Secretion of Organic Cations. Mol. Cell. Biol. 2003, 23, 7902–7908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonker, J.W.; Schinkel, A.H. Pharmacological and Physiological Functions of the Polyspecific Organic Cation Transporters: OCT1, 2, and 3 (SLC22A1-3). J. Pharmacol. Exp. Ther. 2004, 308, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Koepsell, H.; Lips, K.; Volk, C. Polyspecific organic cation transporters: Structure, function, physiological roles, and biopharmaceutical implications. Pharm. Res. 2007, 24, 1227–1251. [Google Scholar] [CrossRef]
- Gorboulev, V.; Ulzheimer, J.C.; Akhoundova, A.; Ulzheimer-Teuber, I.; Karbach, U.; Quester, S.; Baumann, C.; Lang, F.; Koepsell, H. Cloning and characterization of two human polyspecific organic cation transporters. DNA Cell Biol. 1997, 16, 871–881. [Google Scholar] [CrossRef] [PubMed]
- Vialou, V.; Balasse, L.; Callebert, J.; Launay, J.M.; Giros, B.; Gautron, S. Altered aminergic neurotransmission in the brain of organic cation transporter 3-deficient mice. J. Neurochem. 2008, 106, 1471–1482. [Google Scholar] [CrossRef]
- Couroussé, T.; Gautron, S. Role of organic cation transporters (OCTs) in the brain. Pharmacol. Ther. 2015, 146, 94–103. [Google Scholar] [CrossRef]
- Daws, L.C.; Koek, W.; Mitchell, N.C. Revisiting serotonin reuptake inhibitors and the therapeutic potential of “uptake-2” in psychiatric disorders. ACS Chem. Neurosci. 2013, 4, 16–21. [Google Scholar] [CrossRef] [Green Version]
- Wang, J. The plasma membrane monoamine transporter (PMAT): Structure, function, and role in organic cation disposition. Clin. Pharmacol. Ther. 2016, 100, 489–499. [Google Scholar] [CrossRef] [Green Version]
- Ciarimboli, G. Organic cation transporters. Xenobiotica 2008, 38, 936–971. [Google Scholar] [CrossRef]
- Fraser-Spears, R.; Krause-Heuer, A.M.; Basiouny, M.; Mayer, F.P.; Manishimwe, R.; Wyatt, N.A.; Dobrowolski, J.C.; Roberts, M.P.; Greguric, I.; Kumar, N.; et al. Comparative analysis of novel decynium-22 analogs to inhibit transport by the low-affinity, high-capacity monoamine transporters, organic cation transporters 2 and 3, and plasma membrane monoamine transporter. Eur. J. Pharmacol. 2019, 842, 351–364. [Google Scholar] [CrossRef]
- Sala-Rabanal, M.; Li, D.C.; Dake, G.R.; Kurata, H.T.; Inyushin, M.; Skatchkov, S.N.; Nichols, C.G. Polyamine transport by the polyspecific organic cation transporters OCT1, OCT2, and OCT3. Mol. Pharm. 2013, 10, 1450–1458. [Google Scholar] [CrossRef] [Green Version]
- Sitte, H.H.; Freissmuth, M. Amphetamines, new psychoactive drugs and the monoamine transporter cycle. Trends Pharmacol. Sci. 2015, 36, 41–50. [Google Scholar] [CrossRef] [Green Version]
- Haenisch, B.; Drescher, E.; Thiemer, L.; Xin, H.; Giros, B.; Gautron, S.; Bönisch, H. Interaction of antidepressant and antipsychotic drugs with the human organic cation transporters hOCT1, hOCT2 and hOCT3. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2012, 385, 1017–1023. [Google Scholar] [CrossRef]
- Jensen, O.; Rafehi, M.; Gebauer, L.; Brockmöller, J. Cellular Uptake of Psychostimulants–Are High- and Low-Affinity Organic Cation Transporters Drug Traffickers? Front. Pharmacol. 2021, 11, 1–14. [Google Scholar] [CrossRef]
- Massmann, V.; Edemir, B.; Schlatter, E.; Al-Monajjed, R.; Harrach, S.; Klassen, P.; Holle, S.K.; Sindic, A.; Dobrivojevic, M.; Pavenstädt, H.; et al. The organic cation transporter 3 (OCT3) as molecular target of psychotropic drugs: Transport characteristics and acute regulation of cloned murine OCT3. Pflugers Arch. Eur. J. Physiol. 2014, 466, 517–527. [Google Scholar] [CrossRef]
- Mayer, F.P.; Schmid, D.; Owens, W.A.; Gould, G.G.; Apuschkin, M.; Kudlacek, O.; Salzer, I.; Boehm, S.; Chiba, P.; Williams, P.H.; et al. An unsuspected role for organic cation transporter 3 in the actions of amphetamine. Neuropsychopharmacology 2018, 43, 2408–2417. [Google Scholar] [CrossRef]
- Tzvetkov, M.V.; Saadatmand, A.R.; Lötsch, J.; Tegeder, I.; Stingl, J.C.; Brockmöller, J. Genetically polymorphic OCT1: Another piece in the puzzle of the variable pharmacokinetics and pharmacodynamics of the opioidergic drug tramadol. Clin. Pharmacol. Ther. 2011, 90, 143–150. [Google Scholar] [CrossRef]
- Zhou, M.; Engel, K.; Wang, J. Evidence for significant contribution of a newly identified monoamine transporter (PMAT) to serotonin uptake in the human brain. Biochem. Pharmacol. 2007, 73, 147–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maier, J.; Rauter, L.; Rudin, D.; Niello, M.; Holy, M.; Schmid, D.; Wilson, J.; Blough, B.E.; Gannon, B.M.; Murnane, K.S.; et al. α-PPP and its derivatives are selective partial releasers at the human norepinephrine transporter: A pharmacological characterization of interactions between pyrrolidinopropiophenones and uptake1 and uptake2 monoamine transporters. Neuropharmacology 2021, 190, 108570. [Google Scholar] [CrossRef]
- Maier, J.; Mayer, F.P.; Brandt, S.D.; Sitte, H.H. DARK Classics in Chemical Neuroscience: Aminorex Analogues. ACS Chem. Neurosci. 2018, 9, 2484–2502. [Google Scholar] [CrossRef] [Green Version]
- Tzvetkov, M.V.; Dos Santos Pereira, J.N.; Meineke, I.; Saadatmand, A.R.; Stingl, J.C.; Brockmöller, J. Morphine is a substrate of the organic cation transporter OCT1 and polymorphisms in OCT1 gene affect morphine pharmacokinetics after codeine administration. Biochem. Pharmacol. 2013, 86, 666–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koepsell, H. Organic cation transporters in health and disease. Pharmacol. Rev. 2020, 72, 253–319. [Google Scholar] [CrossRef]
- Amphoux, A.; Vialou, V.; Drescher, E.; Brüss, M.; La Cour, C.M.; Rochat, C.; Millan, M.J.; Giros, B.; Bönisch, H.; Gautron, S. Differential pharmacological in vitro properties of organic cation transporters and regional distribution in rat brain. Neuropharmacology 2006, 50, 941–952. [Google Scholar] [CrossRef]
- Chen, Y.; Li, S.; Brown, C.; Cheatham, S.; Castro, R.A.; Leabman, M.K.; Urban, T.J.; Chen, L.; Yee, S.W.; Choi, J.H.; et al. Effect of genetic variation in the organic cation transporter 2 on the renal elimination of metformin. Pharmacogenetics Genom. 2009, 19, 497–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nigam, S.K. The SLC22 Transporter Family: A Paradigm for the Impact of Drug Transporters on Metabolic Pathways, Signaling, and Disease. Annu. Rev. Pharmacol. Toxicol. 2018, 58, 663–687. [Google Scholar] [CrossRef] [PubMed]
- Zamek-Gliszczynski, M.J.; Giacomini, K.M.; Zhang, L. Emerging Clinical Importance of Hepatic Organic Cation Transporter 1 (OCT1) in Drug Pharmacokinetics, Dynamics, Pharmacogenetic Variability, and Drug Interactions. Clin. Pharmacol. Ther. 2018, 103, 758–760. [Google Scholar] [CrossRef]
- Zhou, S.; Zeng, S.; Shu, Y. Drug-Drug Interactions at Organic Cation Transporter 1. Front. Pharmacol. 2021, 12, 1–17. [Google Scholar] [CrossRef]
- Goswami, S.; Gong, L.; Giacomini, K.; Altman, R.B.; Klein, T.E. PharmGKB summary: Very important pharmacogene information for SLC22A1. Pharmacogenetics Genom. 2014, 24, 324–328. [Google Scholar] [CrossRef] [Green Version]
- Damaj, M.I.; Carroll, F.I.; Eaton, J.B.; Navarro, H.A.; Blough, B.E.; Mirza, S.; Lukas, R.J.; Martin, B.R. Enantioselective effects of hydroxy metabolites of bupropion on behavior and on function of monoamine transporters and nicotinic receptors. Mol. Pharmacol. 2004, 66, 675–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-A, A.; Fiorenza, A.M.; Boschen, S.L.; Sugi, A.H.; Beckman, D.; Ferreira, S.T.; Lee, K.; Blaha, C.D.; Da Cunha, C. Diazepam Inhibits Electrically Evoked and Tonic Dopamine Release in the Nucleus Accumbens and Reverses the Effect of Amphetamine. ACS Chem. Neurosci. 2017, 8, 300–309. [Google Scholar] [CrossRef]
- Madras, B.K.; Xie, Z.; Lin, Z.; Jassen, A.; Panas, H.; Lynch, L.; Johnson, R.; Livni, E.; Spencer, T.J.; Bonab, A.A.; et al. Modafinil occupies dopamine and norepinephrine transporters in vivo and modulates the transporters and trace amine activity in vitro. J. Pharmacol. Exp. Ther. 2006, 319, 561–569. [Google Scholar] [CrossRef] [Green Version]
- Rickli, A.; Hoener, M.C.; Liechti, M.E. Monoamine transporter and receptor interaction profiles of novel psychoactive substances: Para-halogenated amphetamines and pyrovalerone cathinones. Eur. Neuropsychopharmacol. 2015, 25, 365–376. [Google Scholar] [CrossRef] [Green Version]
- Tatsumi, M.; Groshan, K.; Blakely, R.D.; Richelson, E. Pharmacological profile of antidepressants and related compounds at human monoamine transporters. Eur. J. Pharmacol. 1997, 340, 249–258. [Google Scholar] [CrossRef]
- Mato, E.P.M.; Guewo-Fokeng, M.; Faadiel Essop, M.; Owira, P.M.O. Genetic polymorphisms of organic cation transporters 1 (OCT1) and responses to metformin therapy in individuals with type 2 diabetes mellitus: A systematic review protocol. Syst. Rev. 2018, 7. [Google Scholar] [CrossRef]
- Arimany-Nardi, C.; Minuesa, G.; Keller, T.; Erkizia, I.; Koepsell, H.; Martinez-Picado, J.; Pastor-Anglada, M. Role of Human Organic Cation Transporter 1 (hOCT1) Polymorphisms in Lamivudine (3TC) Uptake and Drug-Drug Interactions. Front. Pharmacol. 2016, 7, 175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- FDA, U.S. Guidance for Industry: Clinical Drug Interaction Studies—Cytochrome P450 Enzyme- and Transporter-Mediated Drug Interactions. 2021; pp. 1–27. Available online: https://www.fda.gov/media/134581/download (accessed on 18 October 2020).
- EMA. Guideline on the Investigation of Drug Interactions Guideline on the Investigation of Drug Interactions. 2010. Available online: www.ema.europa.eu/contact (accessed on 29 November 2021).
- Ilic, M.; Holy, M.; Jaentsch, K.; Liechti, M.E.; Lubec, G.; Baumann, M.H.; Sitte, H.H.; Luethi, D. Cell-Based Radiotracer Binding and Uptake Inhibition Assays: A Comparison of In Vitro Methods to Assess the Potency of Drugs That Target Monoamine Transporters. Front. Pharmacol. 2020, 11, 1–11. [Google Scholar] [CrossRef]
- Gorboulev, V.; Shatskaya, N.; Volk, C.; Koepsell, H. Subtype-Specific Affinity for Corticosterone of Rat Organic Cation Transporters rOCT1 and rOCT2 Depends on Three Amino Acids within the Substrate Binding Region. Mol. Pharmacol. 2005, 67, 1612–1619. [Google Scholar] [CrossRef] [Green Version]
- Popp, C.; Gorboulev, V.; Müller, T.D.; Gorbunov, D.; Shatskaya, N.; Koepsell, H. Amino Acids Critical for Substrate Affinity of Rat Organic Cation Transporter 1 Line the Substrate Binding Region in a Model Derived from the Tertiary Structure of Lactose Permease. Mol. Pharmacol. 2005, 67, 1600–1611. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.C.; Goldenberg, A.; Chen, Y.; Lun, C.; Wu, W.; Bush, K.T.; Balac, N.; Rodriguez, P.; Abagyan, R.; Nigam, S.K. Molecular Properties of Drugs Interacting with SLC22 Transporters OAT1, OAT3, OCT1, and OCT2: A Machine-Learning Approachs. J. Pharmacol. Exp. Ther. 2016, 359, 215–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eshleman, A.J.; Wolfrum, K.M.; Reed, J.F.; Kim, S.O.; Swanson, T.; Johnson, R.A.; Janowsky, A. Structure-activity relationships of substituted cathinones, with transporter binding, uptake, and release. J. Pharmacol. Exp. Ther. 2017, 360, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Niello, M.; Cintulova, D.; Hellsberg, E.; Jäntsch, K.; Holy, M.; Ayatollahi, L.H.; Cozzi, N.V.; Freissmuth, M.; Sandtner, W.; Ecker, G.F.; et al. Para-Trifluoromethyl-methcathinone is an allosteric modulator of the serotonin transporter. Neuropharmacology 2019, 161. [Google Scholar] [CrossRef]
- Niello, M.; Cintulová, D.; Raithmayr, P.; Holy, M.; Jäntsch, K.; Colas, C.; Ecker, G.F.; Sitte, H.H.; Mihovilovic, M.D. Effects of Hydroxylated Mephedrone Metabolites on Monoamine Transporter Activity in vitro. Front. Pharmacol. 2021, 12, 545. [Google Scholar] [CrossRef]
- Niello, M.; Sideromenos, S.; Gradisch, R.; O’shea, R.; Schwazer, J.; Sandtner, W.; Maier, J.; Jäntsch, K.; Lupica, C.; Hoffman, A.; et al. Psychomotor stimulant effects of α-pyrrolidinovalerophenone (αPVP) enantiomers correlate with drug binding kinetics at the dopamine transporter. Res. Sq. 2021, 1–25. [Google Scholar] [CrossRef]
- Ahlin, G.; Karlsson, J.; Pedersen, J.M.; Gustavsson, L.; Larsson, R.; Matsson, P.; Norinder, U.; Bergström, C.A.; Artursson, P. Structural requirements for drug inhibition of the liver specific human organic cation transport protein 1. J. Med. Chem. 2008, 51, 5932–5942. [Google Scholar] [CrossRef]
- Niello, M.; Gradisch, R.; Loland, C.J.; Stockner, T.; Sitte, H.H. Allosteric Modulation of Neurotransmitter Transporters as a Therapeutic Strategy. Trends Pharmacol. Sci. 2020, 41, 446–463. [Google Scholar] [CrossRef]
- Keller, T.; Gorboulev, V.; Mueller, T.D.; Dötsch, V.; Bernhard, F.; Koepsell, H. Rat organic cation transporter 1 contains three binding sites for substrate 1-methyl-4-phenylpyridinium per monomer. Mol. Pharmacol. 2019, 95, 169–182. [Google Scholar] [CrossRef] [Green Version]
- Luethi, D.; Kolaczynska, K.E.; Docci, L.; Krähenbühl, S.; Hoener, M.C.; Liechti, M.E. Pharmacological profile of mephedrone analogs and related new psychoactive substances. Neuropharmacology 2018, 134, 4–12. [Google Scholar] [CrossRef] [Green Version]
- Kimko, H.C.; Cross, J.T.; Abernethy, D.R. Pharmacokinetics and clinical effectiveness of methylphenidate. Clin. Pharmacokinet. 1999, 37, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.; Mayer, F.P.; Luethi, D.; Holy, M.; Jäntsch, K.; Reither, H.; Hirtler, L.; Hoener, M.C.; Liechti, M.E.; Pifl, C.; et al. The psychostimulant (±)-cis-4,4’-dimethylaminorex (4,4’-DMAR) interacts with human plasmalemmal and vesicular monoamine transporters. Neuropharmacology 2018, 138, 282–291. [Google Scholar] [CrossRef] [PubMed]
- jetPRIME. Polyplus-Transfection® SA. 2021. Available online: https://www.polyplus-transfection.com/products/jetprime/ (accessed on 29 November 2021).
- ColorBrewer. ColorBrewer: Color Advice for Maps. 2015. Available online: http://colorbrewer2.org/ (accessed on 29 November 2021).
- Daws, L.C. Organic Cation Transporters in Psychiatric Disorders. Handb. Exp. Pharmacol. 2021. [Google Scholar] [CrossRef]









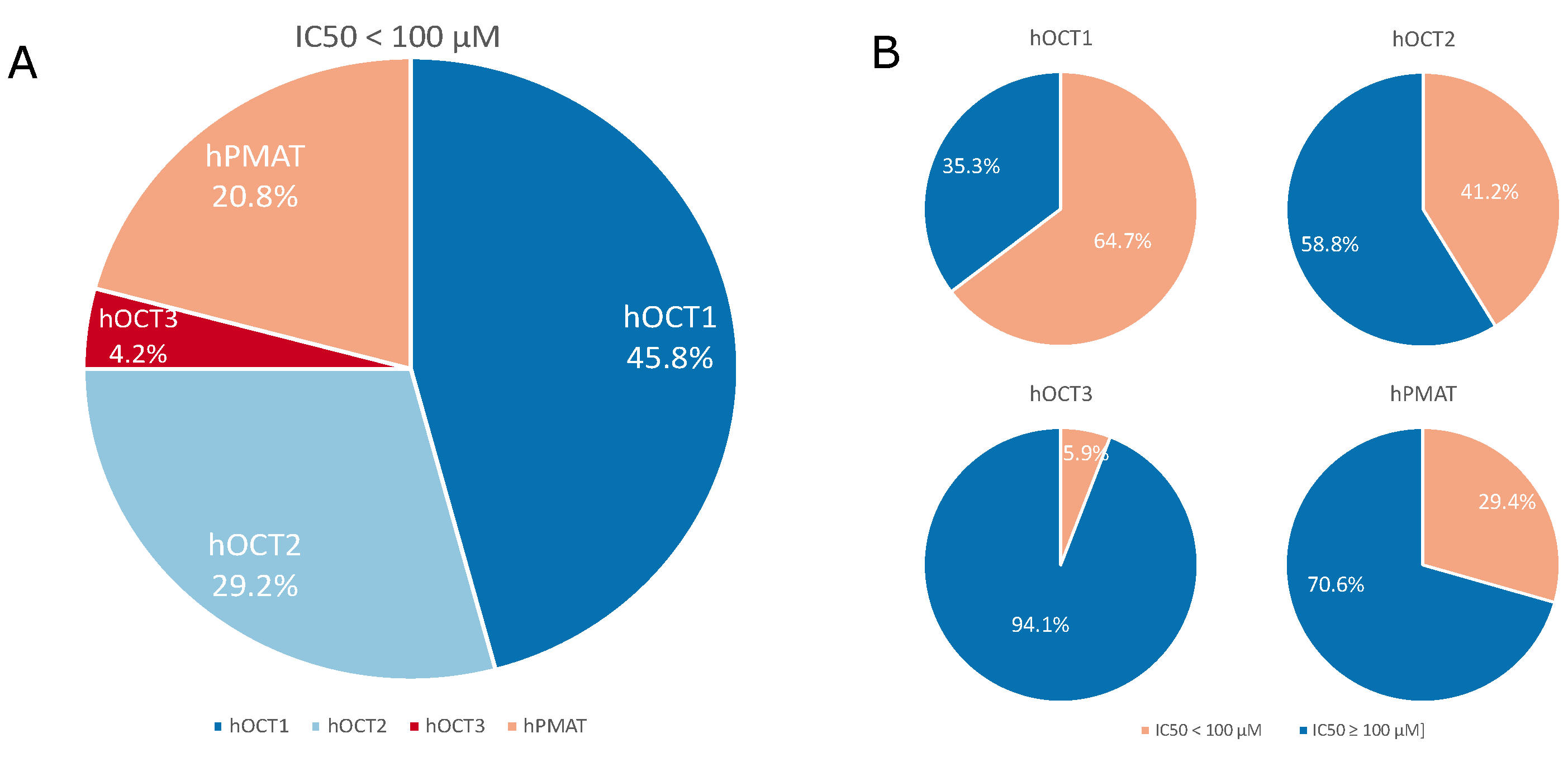{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substance | Transporters | |||
|---|---|---|---|---|
| hOCT1 | hOCT2 | hOCT3 | hPMAT | |
| Bupropion | 5.36 | 96.96 | ||
| S-Citalopram | 7.15 | |||
| R-Citalopram | 5.11 | 58.32 | ||
| Diazepam | 44.46 | 29.81 | ||
| Phenobarbital | ||||
| d-Amphetamine | 8.39 | 2.21 | 71.77 | |
| Cocaine | 6.66 | 27.80 | ||
| R--PVP | 2.15 | 13.09 | ||
| S--PVP | 1.07 | 15.02 | ||
| Ketamine | 12.46 | |||
| 2-Fluoro-deschloroketamin | 19.18 | |||
| MDMA | 1.14 | 2.71 | 7.77 | |
| Psilocin | ||||
| Tramadol | 5.60 | |||
| O-Desmethyltramadol | 24.16 | |||
| Metylphenidate | 0.36 | |||
| Modafinil | ||||
| Decynium-22 | 2.66 | 4.56 | 0.16 | 0.35 |

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Angenoorth, T.J.F.; Stankovic, S.; Niello, M.; Holy, M.; Brandt, S.D.; Sitte, H.H.; Maier, J. Interaction Profiles of Central Nervous System Active Drugs at Human Organic Cation Transporters 1–3 and Human Plasma Membrane Monoamine Transporter. Int. J. Mol. Sci. 2021, 22, 12995. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222312995
Angenoorth TJF, Stankovic S, Niello M, Holy M, Brandt SD, Sitte HH, Maier J. Interaction Profiles of Central Nervous System Active Drugs at Human Organic Cation Transporters 1–3 and Human Plasma Membrane Monoamine Transporter. International Journal of Molecular Sciences. 2021; 22(23):12995. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222312995
Chicago/Turabian StyleAngenoorth, Thomas J. F., Stevan Stankovic, Marco Niello, Marion Holy, Simon D. Brandt, Harald H. Sitte, and Julian Maier. 2021. "Interaction Profiles of Central Nervous System Active Drugs at Human Organic Cation Transporters 1–3 and Human Plasma Membrane Monoamine Transporter" International Journal of Molecular Sciences 22, no. 23: 12995. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222312995