
Cytokine Signalling at the Microglial Penta-Partite Synapse


1
Brain and Mind Centre, Faculty of Medicine and Health, The University of Sydney, Sydney, NSW 2006, Australia
2
Faculty of Medicine, Health & Human Sciences, Macquarie Medical School, Macquarie University, Sydney, NSW 2109, Australia
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2021, 22(24), 13186; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222413186
Submission received: 7 November 2021
/
Revised: 1 December 2021
/
Accepted: 2 December 2021
/
Published: 7 December 2021
(This article belongs to the Section Molecular Neurobiology)
Abstract
:Microglial cell processes form part of a subset of synaptic contacts that have been dubbed microglial tetra-partite or quad-partite synapses. Since tetrapartite may also refer to the presence of extracellular matrix components, we propose the more precise term microglial penta-partite synapse for synapses that show a microglial cell process in close physical proximity to neuronal and astrocytic synaptic constituents. Microglial cells are now recognised as key players in central nervous system (CNS) synaptic changes. When synaptic plasticity involving microglial penta-partite synapses occurs, microglia may utilise their cytokine arsenal to facilitate the generation of new synapses, eliminate those that are not needed anymore, or modify the molecular and structural properties of the remaining synaptic contacts. In addition, microglia–synapse contacts may develop de novo under pathological conditions. Microglial penta-partite synapses have received comparatively little attention as unique sites in the CNS where microglial cells, cytokines and other factors they release have a direct influence on the connections between neurons and their function. It concerns our understanding of the penta-partite synapse where the confusion created by the term “neuroinflammation” is most counterproductive. The mere presence of activated microglia or the release of their cytokines may occur independent of inflammation, and penta-partite synapses are not usually active in a neuroimmunological sense. Clarification of these details is the main purpose of this review, specifically highlighting the relationship between microglia, synapses, and the cytokines that can be released by microglial cells in health and disease.
1. Glia Interact with Synapses in Health and Disease
The ability to adapt and protect neuronal circuitry throughout an animal’s lifespan is a remarkable feature of the CNS. Synaptic plasticity refers to the process by which the nervous system generates new synapses, eliminates unneeded ones and modifies the electrophysiological, molecular and structural properties of existing synapses in response to stimuli.
Synaptic plasticity generally depends on reciprocal interactions between neurons and glial cells and the extracellular matrix (ECM) in what has been described as the ‘tetrapartite synapse’ [1,2]. The concept of the synapse has evolved over the last two decades from involving pre- and post- synaptic neuronal elements to including a third element—astrocytes—resulting in a ‘tripartite’ synapse [3]. This further evolved to include a fourth component—that is, the ECM [2], which is difficult to discern in CNS parenchyma—to form the ‘tetrapartite synapse’. In this ensemble, astrocytic processes engulf pre- and post-synaptic elements and express EAAT1-EAAT2 glutamate transporters that facilitate rapid re-uptake of excess glutamate limiting excitatory synaptic stimulation. Astrocytes also release gliotransmitters such as ATP and GABA [4]. The ECM provides scaffolding, which is important for maintaining synaptic integrity. Specifically, in the context of the tetrapartite synapse, the ECM serves to retain neuronal connectivity [5], which impacts the diffusion and localisation of key molecules such as neurotransmitters, ions and membrane receptors, and it also contains matrix molecules such as chondroitin sulfate proteoglycans (CSPGs), which inhibit neurite and axon growth [6]. In addition to the growing literature on the role of the ECM, astrocytes and NG2 cells [7] on synaptic plasticity, microglial cells are now also considered essential players. De Leo, Tawfik and LaCroix-Fralish [1] deserve credit for drawing attention to the presence of the tips of microglial cell processes at a subset of CNS synapses and coining the term tetrapartite synapse originally.
Microglia and brain macrophages, which can be derived from microglia as well as other sources, are traditionally understood to play a role in innate immune responses. This has resulted in associating microglial activity with immunological reactions by default and microglial relations with synapses to be referred to as ‘neuroimmunological’ [8]. This is incorrect. Microglia in healthy brain have been shown to interact with synapses in the absence of any inflammation as well as in pathological conditions [9]. Some of the earliest evidence for microglia-derived cytokines regulating synaptic function pin-pointed TNF-α as a regulator of the amount of AMPA receptors and synaptic strength in the hippocampus [10,11,12]. Transcriptomic analysis revealed that microglia rather than astrocytes produce TNF-α transcripts [13]. Fractalkine, interleukins, interferons and growth factors such as brain-derived neurotrophic factor (BDNF) derived from microglia have also been shown to have a role in synaptic plasticity [14,15,16,17]. Abnormally functioning microglia displaying dysregulated cytokine levels have been associated with abnormal synaptic structure and function in neurodegenerative disease models of Alzheimer’s Disease (AD) and autism (ASD) [18,19,20]. More recently, abnormal cellular bioenergetic states such as caloric restriction and ketogenic diet as well as the glycolytic inhibitor 2-deoxyglucose (2DG) have been demonstrated to influence microglial transcription of cytokine genes [21,22]. York et al. [23] reported metabolic reprogramming of microglia stimulated by lipopolysaccharide (LPS), which resulted in glycolysis necessary for IL-1β production. This process could be reversed by the addition of 2DG, which resulted in the inhibition of HIF-1α accumulation and IL-1β production, and rescued long-term potentiation (LTP) of synaptic plasticity. Overall, microglia and their cytokines are now recognised to play an indispensable role in plasticity of the tetra-partite synapse.
Since tetrapartite may also refer to the presence of extracellular matrix components, we propose the more precise term microglial penta-partite synapse for synaptic contacts that show a microglial cell process in close physical proximity to neuronal and astrocytic synaptic elements. Thus, microglial penta-partite synapses comprise a smaller subset of synapses. Precise topoanatomical data on which neuronal circuits if any are preferentially involved are currently lacking. We also do not know whether microglial cell processes have a clear affinity for specific types of synapses and whether they contact the same synapses again and again, i.e., how transient any topoanatomical penta-partite synapse maps are.
Furthermore, we discuss the controversial term ‘neuroinflammation’. AD, Parkinson’s Disease (PD) and schizophrenia (SCZ) are often conceptually linked to ‘neuroinflammation’. Yet, typical inflammation pathology is missing in these diseases, and molecular genetic evidence in fact shows that they are categorically non-inflammatory conditions [24]. We propose instead that dysfunction of the microglial penta-partite synapse is of key relevance to CNS disorders that feature microglial activation.
2. Microglial Activation Is Not (Neuro) Inflammation
2.1. What Is Inflammation?
Inflammation is a multicellular, multistage and dynamic response to tissue damage, characterised by the infiltration of cells of the adaptive immune system [25,26]. The CNS is no exception to this relatively common pathology, which can affect all tissues of the body. The specific tissue characteristics of inflammation are very well established and the exclusion or confirmation of their presence forms part of the routine diagnostic microscopic algorithm in histopathology. Classical examples of inflammation in the CNS include bacterial or viral infections, autoimmune conditions such as multiple sclerosis but also ischaemia/stroke.
A number of authors refer to inflammation as “neuroinflammation” when it occurs in the nervous system. In other words, “neuro” refers to topology only in this case and is strictly speaking tautological. Other authors, however, apply the term “neuroinflammation” to entirely different pathological processes that are specific to the nervous system, lack participation of peripheral immune cells and display microglial activation. Milatovic et al. [27] defined “neuroinflammation” as the activation of microglia and astrocytes accompanied by the upregulation of several pro-inflammatory cytokines, such as interleukin (IL)-1β, IL-6, and tumour necrosis factor (TNF-α), and chemokines such as CCL2, CCL5, CXCL1, secondary messengers (NO and prostaglandins) and reactive oxygen species [28,29]. However, this is not sufficient for a diagnosis of inflammation. Examples of cases in the literature where the term “neuroinflammation” has been extended in such a way include Alzheimer’s Disease (AD), Parkinson’s Disease (PD), schizophrenia (SZ), as well as etiologically highly diverse conditions such as obesity, autism, major depression, bipolar disorder, anxiety, air pollution and epilepsy amongst others. Neither infiltrating innate nor adaptive immune cells are typically found in Alzheimer’s Disease (AD), Parkinson’s Disease (PD) and schizophrenia (SZ) to any significant extent. Recently, Gate et al. [30] suggested a role for the adaptive immune system in blood and cerebrospinal fluid in AD based on a small number of clonal, antigen-experienced specialised CD8+ T effector memory CD45RA+ (TEMRA) cells patrolling intrathecal cerebrospinal fluid (CSF), as well as CD8+ T cells in the hippocampus near beta-amyloid plaques. In general, neither AD, PD nor SZ demonstrate perivascular mononuclear cell infiltrates either, but they have been assigned to the category of ‘neuroinflammation’ by some authors on the basis of observed microglial activation, particularly with reference to translocator protein positron emission tomography (TSPO PET) imaging results [22]. There is no generally accepted neuropathological tissue correlate of ‘neuroinflammation’ [24], and many if not most publications that use the term do not define it.
Applying the same scientific term, inflammation, to categorically different disease entities represents a logical impossibility. The term “neuroinflammation” is therefore not viable. Unfortunately, as long as the confusion persists, scientific progress will be inhibited. Most relevant in our view, the ambiguity of “neuroinflammation” inhibits research interest in what happens at the microglial penta-partite synapse.
2.2. Microglia Are Never-Resting Sensors of Pathology
Microglia are considered activated when there is, at a minimum, increased expression of markers that are absent from microglia in healthy CNS tissue. This was first described for certain lectins in 1987 [31] and complement receptors in 1988 [32] but may also include the production of inflammatory mediators such as cytokines [33]. As discussed by Cherry et al. [34], concepts concerning microglial phenotype polarisation have been carried over from literature on peripheral macrophages throughout the last century. One particular such concept that is of concern is that microglia in the healthy CNS exist in a resting state [35]. This concept has been translated into the idea that cytokine expression by microglia is reflective of activation. This is problematic primarily because (1) microglia, as we know today, should never be considered “resting”—they are constantly “surveying” the microenvironment—and (2) cytokine expression occurs in non-activated microglia as well.
Some studies have explored the role of microglial activation in ALS [36,37], AD [38,39] and MS [40,41,42]. While these studies report significantly increased microglial activation, as inferred from increased TSPO binding, some studies have demonstrated no correlation between microglial activation and increased TSPO binding [40,41,42]. Owen et al. [43] reported that TSPO expression did not increase with classical pro-inflammatory activation in primary human microglia, unlike in rodent microglia. Hafizi et al. [44] reported no significant differences between patients and healthy volunteers with regard to microglial activation using [18F] FEPPA in either the dorsolateral prefrontal cortex or hippocampus. Nutma et al. [45] found that although TSPO expression may provide a general marker for glial activation in multiple sclerosis, such interpretations depended on pathological context. As with AD [46], increased TSPO PET signal may reflect differences in microglia and astrocyte abundance [45]. Increased TSPO expression was not associated with solely pro- or anti- inflammatory microglia phenotypes in MS, but rather with an intermediate activation state demonstrated through the expression of both CD40 and CD206 [45]. Sneeboer et al. [47] found no positive correlation between TSPO expression and microglial activation markers in either patients or controls for schizophrenia. Microglia constantly require ATP as an energy source for their baseline dynamics and maintenance of cell morphology [48]. A positive correlation between TSPO and increased ATP production suggest that TSPO expression reflects an increased demand for energy [49]. This had been previously demonstrated by Banati et al. [50], who reported TSPO−/− microglia producing significantly less ATP. Taken together, the findings suggest that interpreting TSPO imaging as a reflection of microglial activation needs to be treated with caution, particularly given that increased ATP production in microglia does not necessarily suggest inflammation-induced activation.
Molecules associated with immune functions in peripheral organs such as certain cytokines can have additional CNS-specific roles independent of their conventional roles in generating an inflammatory and/or immune response. A number of such molecules, IL-1β, IL-γ, CCL2, major histocompatibility complex (MHC) proteins, DAP-12, and the CR3 complement receptors have been implicated in synaptic plasticity [51]. The role of some major cytokines in microglial-mediated synaptic plasticity has been reviewed by Werneburg et al. [52] and thus will not be discussed further here.
2.3. Cytokines Are Present in Unstimulated Microglia
Cytokine levels in microglia are typically increased in response to pathological stimuli [53]. For instance, both CNS neurons and glial cells express low levels of interleukin-6 (IL-6) and IL-6R in physiological conditions [54]. In contrast, under pathological conditions, such as following peripheral nerve injury, cytokines and their receptors, such as IL-6/IL-6R, have been shown to be increased with microglial activation [55]. IL-6 utilises glycoprotein 130 (gp130) signalling via binding with IL-6R, which activates intracellular Janus kinase (JAK) tyrosine-kinase, which then activates the signal transducer and activator of transcription (STAT) [56]. Figure 1 illustrates this process in the context of miR-21-5p mediated neuroprotection following traumatic brain injury [56,57].
Microglia release cytokines that can have neuroprotective roles without stimulation [58]. Studies have identified cytokine expression in unstimulated microglia [59,60,61]. In the absence of recognised stimulatory signals, microglia are the primary source and secretors of cytokines in the CNS. Lee, Nagai and Kim [59] reported mRNA transcripts for interleukin-1β (IL-1β) -6, -8, -10, -12, -15, tumor necrosis factor-α (TNF-α), macrophage inflammatory protein-1α (MIP-1α), MIP-1β, and monocyte chemoattractant protein-1 (MCP-1) in unstimulated human microglia.
Elkabes, DiCicco-Bloom and Black [61] demonstrated neurotrophins NGF, BDNF, NT-3 and NT-4 in microglia regulating microglial proliferation. Other studies have demonstrated the expression of several cytokines in different CNS cell types in both humans and animal models under physiological conditions, including retina Muller glial cells [62], cerebellar neurons and glia [63], as well as microglia obtained from human brain tissues [64,65]. Under non-stimulated conditions, human microglia express various interleukin family cytokines, TNF-α, macrophage inflammatory protein (MIP)-1α, MIP-1β and MCP-1, as well as receptors for some of these cytokines, as shown in Table 1 below [66]. CXC chemokines such as fractalkine (CX3CL1/CX3CR1), SDF-1α, and CXCL12/CXCR4/CXCR7 have been reported in the healthy CNS of mice and human microglia [67,68] and neurons [69,70,71,72,73]. Similarly, β-chemokines (or CC chemokines) such as MCP-1 (CCL2), MIP-1α (CCL3), MIP-1β (CCL4), RANTES (CCL5) and C10/MRP-1 (CCL6) have also been identified in both healthy neurons [69,70,71,72,73] and microglia [74].
Several studies have reported MIP-1α (CCL3), MIP-1β (CCL4) and RANTES (CCL5) mRNA expression across several brain regions in healthy animals, such as the cerebral cortex [75], thalamus, striatum, hippocampus, midbrain [76,77], ventromedial hypothalamus [78] and cerebellum [79]. Ciechanowska et al. [76], however, observed no statistically significant changes in CCL3 protein expression levels when compared to the thalamus of traumatic brain injury (TBI) mice. Several studies have also reported microglia as the primary source of CCL3 and CCL6, as well as neurons [80]. CCR3 has also been demonstrated in both neurons and microglia of several CNS regions, such as the hippocampus, cerebral cortex, brain stem, cerebellum and spinal cord [81,82,83,84]. Lanfranco et al. [77] identified CCL5 mRNA expression by glia cells, but not by neurons, in the corpus callosum. CCL5mRNA was detected more predominantly in neurons than in glial cells of the cerebral cortex, hippocampus and midbrain [77]. In summary, these studies suggest that several cytokines associated with inflammation in peripheral organs such as fractalkine (CX3CL1/CX3CR1), SDF-1α, CXCL12/CXCR4/CXCR7, MCP-1 (CCL2), MIP-1α (CCL3), MIP-1β (CCL4), RANTES (CCL5) and C10/MRP-1 (CCL6) are expressed in the normal CNS as well as during the inflammatory response.
In recent times, microglia have been shown to reprogram their cellular metabolism to accommodate functions needing cytoskeletal changes, functional remodelling and cytokine synthesis [85,86,87]. Murine brain microglia express all genes required for glycolytic and oxidative energy metabolism, including the expression of transporters for all energy substrates [13,88,89]. In the absence of glucose such as during anti-inflammatory states, microglia utilise oxidative phosphorylation by metabolising alternate carbon sources such as glutamine [90], and fatty acids from lipid droplets (LDs) in glucose-deprived microglial cells [91,92]. During pro-inflammatory conditions, microglia use aerobic glycolysis for energy production. Glucose fluctuations activate signalling pathways such as MAPKs and PI3K/Akt, which form the stress that alters NF-κB activity in microglia [93]. Yamada et al. [94] demonstrated using real-time RT-PCR that miR-16-5p, miR-196b-5p, and miR-218-5p expression levels were significantly higher in caloric-restricted mice compared with controls. Specifically, miR-16-5p was found to suppress mRNA expression of IL-1β, IL-6, and TNF-α, further suggesting that caloric restriction suppresses cytokines through transcriptional control [94]. Bernhart et al. [95] proposed that upregulation of glycolytic enzymes in response to lysophosphatidic acid (LPA) treatment of microglia could serve two purposes: (i) generating a survival signal by generating sufficient amounts of ATP and (ii) a source of cellular energy to fuel plasma membrane located, ATP-dependent ion pumps. Wang et al. [96] found that GLUT1 was significantly increased to promote anaerobic glycolysis following LPS + IFNγ stimulation, and that blocking GLUT1 reduced anaerobic glycolysis, reprogrammed microglial metabolism to aerobic glycolysis and suppressed phagocytosis in microglia. These findings suggest that microglial mitochondrial reprogramming control cytokine-mediated microglial functions.
Taken together, the increased appreciation of the role of microglial cytokines in physiological conditions independently necessitates the need to reconsider usage of the term “neuroinflammation”. The “neuroinflammatory” paradigm wrongly disregards the active role microglia play in physiological functions.
3. The Microglial Penta-Partite Synapse
TSPO PET imaging and histopathological studies of diseased brains have revealed microglial activation across a wide range of brain pathologies. What we do not know with certainty yet, although this seems likely, is whether the microglia activated in these conditions all have cell processes that are in direct contact with synapses, at least temporarily. Microglial cells are known to “check” on synaptic integrity in regular time intervals [48,97], reaching some synapses. Similarly, astrocyte processes are not present at all synapses (cf. below). Figure 2 provides a schematic drawing of a microglial penta-partite synapse.
3.1. Neuron-Glia Bidirectional Communication Is Mediated by Purinergic Signalling
Astrocyte–neuron bidirectional communication mediated by purinergic signalling has been well documented by in vivo as well as ex vivo studies [98]. There is substantial evidence suggesting that increased neuronal activity, as evidenced by neurotransmitter release, results in increased GPCR-mediated intracellular astrocytic Ca2+ signalling and astrocyte glutamate release [99,100,101].
Purinergic receptors and purines have been implicated in neuron–glia interactions during various physiological and pathological conditions. Neuronal release of ATP has been shown to directly act on astrocytic purinergic receptors as either ATP or as a breakdown product, such as ADP or adenosine [102]. Juaristi et al. [103] found that astrocytes upregulate glycolysis, lactate and pyruvate production in mitochondria in response to intracellular Ca2+ increases following ATP and glutamate stimulation of P2YR. Shigetomi et al. [104] reported that increased P2Y1R expression triggered increased intracellular Ca2+ in astrocytes. Recently, Monai et al. [105] reported astrocytic GPCR activation as the prevalent mechanism of transcranial direct current stimulation (tDCS)-induced Ca2+ surges. Muller and Taylor [106] found that ATP-evoked Ca2+ signals in cultured human fetal astrocytes are mediated by P2Y1 and P2Y2 receptors.
Microglial communication with both neurons and astrocytes is well documented. For instance, microglia are known to alter excitatory neurotransmission through an ATP-dependent astrocyte communication mechanism [107]. Microglia are also known to be stimulated by neuronal activity-dependent mechanisms to eliminate excessive synapses during neurodevelopment [108]. Purinergic receptors found on microglia have been demonstrated to mediate microglial communication with astrocytes and neurons. Janks et al. [109] found that cultured human microglia express P2X7Rs that stimulate ATP-gated cationic currents. Beneventano et al. [110] reported rapid microvesicle shedding from BV2 microglia following P2X7R activation. Liu et al. [111] showed that P2X7R inhibition reduced IL-1β expression, P38 phosphorylation, and glial activation in the cerebral cortex and improved neurobehavioural outcomes following traumatic brain injury. Several purinergic receptor subtypes have also been identified in microglia, specifically: P2Y1, P2Y2, P2Y2/4, P2Y6, and P2Y12, with P2Y6 and P2Y12 receptors appearing to be functionally important [112]. Several P2YRs have also been identified in astrocytes, such as the above and other subtypes: P2Y11, P2Y13, and P2Y14 receptors [112]. Pascual, Achour, Rostaing, Triller and Bessis [107] found that in vitro microglia enhance excitatory postsynaptic neurotransmission by releasing ATP which stimulates astrocyte P2Y1R. Jiang et al. [113] reported both ATP and ADP mediated mechanical stimulation-induced intercellular Ca2+ wave communication via P2Y12/13 receptors in BV-2 microglia. Quintas et al. [114] found that the microglial P2YR inhibition of astrocyte proliferation occurs via the release of IL-1β, IL-1α and TNF-α. Furthermore, the absence of P2Y12 has been shown to lead to impaired microglia process motility following injury [48]. Microglial expression of P2Y6 receptors has also been shown to trigger microglial phagocytosis following hippocampal excitotoxicity [115]. Collectively, these findings suggest that purinergic receptors may play an important role at the microglia penta-partite synapse. Table 2 provides a summary of microglial mediators and their receptors that have been shown to be involved in synaptic plasticity.
3.2. Astrocytes and Microglia Are Not Present at All Synapses
Although astrocytes can contribute to the processing, transfer and storage of information, they are not present at all synapses. Most studies report a ‘non-uniform distribution’ of perisynaptic astrocyte processes along synapses with a subset of synapses (around 40%) undergoing astrocyte-induced potentiation following post-synaptic depolarisation in the hippocampus [116]. In vivo two-photon imaging has shown that microglia processes make frequent, direct and transient contact with synapses in the uninjured CNS [97]. Akiyoshi et al. [117] further characterised the microglia–synapse contact as resulting in increased neuronal activity in vivo with awake mice. Tremblay, Lowery and Majewska [9] further described this relationship and demonstrated protrusions of microglial processes apposing dendritic spines, presynaptic terminals, the synaptic cleft and perisynaptic astrocyte processes.
Augusto-Oliveira et al. [118] suggest that astrocyte process contact with neuronal synapses ranges from 50 to 60% of all synaptic structures, depending on CNS region. Hippocampal synapses have demonstrated astroglial coverage ranging from 60 to 90%; specifically, Witcher et al. [119] found astrocyte processes in 40–60% of thin, mushroom spine-axon synapses in the hippocampus, while 60–99% of larger mushroom spines and spines with perforated synapses were found to be in contact with astrocyte processes. Ventura and Harris [120] demonstrated that 57 ± 11% of the hippocampal excitatory synapses in mature rats had astrocytic processes apposed to them. Astrocytic processes in this synapse subset surrounded less than half of the entire synaptic interface. In contrast, the number of synaptic spines contacted by microglia is significantly lower. Weinhard et al. [121] found that only 3% of spines were in contact with microglia, taken from a total of 8900 spines from secondary dendrites of GFP+ neurons in the CA1 stratum radiatum of fixed hippocampal tissue at P15. About 1.9% of spines contacted by microglia presented with minor microglia-spine co-localisation [121].
In the cerebellum, Xu-Friedman et al. [122] found that glial processes cover approximately 90% of climbing fibre synapses and approximately 65% of parallel fibre synapses on Purkinje neurons. Spacek [123] reported that 74% of cerebellar Purkinje cell synapses were ensheathed by astrocytes as opposed to 29% of dendritic spines in the mouse visual cortex. Broadhead et al. [124] identified glial presence in around 56% of synapses in the mouse spinal cord. Bernardinelli et al. [125] reported astrocytic process contact as high as 90% in the somatosensory cortex layer IV, particularly with the post-synaptic element [126]. Collectively, these observations suggest that synapses featuring astrocytes represent a subset. The subset of penta-partite microglial synapses is less frequent. In addition, the latter may be significantly modulated by microglial activation.
Since glial cell processes are not present at all synapses, the question arises under what circumstances can we expect their presence? Astrocytes have been shown to discriminate between the activity of different synapses belonging to different axons, suggesting synapse-specificity among astrocytes. Earlier work by Perea and Araque [127] found that rises in Ca2+ in astrocytes can be evoked by acetylcholine and glutamate released from Schaffer collaterals, but not glutamate released by alveus stimulation. Quantitative 3D analyses by Ventura and Harris [120] identified astrocytes at 88% of perforated synapses, 61% of single synapse boutons (SSBs), 52% of macular synapses, and 40% of multisynaptic boutons (MSBs). Xu-Friedman, Harris and Regehr [122] discovered differences in astrocyte ensheathment between parallel fibre synapses and climbing fibre synapses of Purkinje cells, suggesting a preference of astrocytes for climbing fibre synapses as opposed to parallel fibre synapses. Witcher, Kirov and Harris [119] found that the post-synaptic density (PSD) and mean synapse size were greater when astroglia was present at the post-synaptic dendritic spine, suggesting that larger synapses were more likely to have astrocytes than smaller synapses. With regard to microglia, Tremblay, Lowery and Majewska [9] found that microglial processes preferentially localised to small and structurally dynamic dendritic spines rather than larger dendritic spines in the primary visual cortex (V1) of juvenile mice. Another suggestion is that glia are present at active synapses where glutamate is present. This ties in with the former suggestion that astrocytes may have a preference for synapse size because local efficacy of glutamate uptake is lower at large spines [128]. Henneberger et al. [129] found that LTP triggered the withdrawal of astroglial processes from potentiated synapses, thereby boosting glutamate spillover and NMDA-receptor-mediated inter-synaptic cross-talk. Microglial contact with synaptic elements have also been shown to be modulated by sensory deprivation [97]. Umpierre et al. [130] recently found that hyperactive shifts in neuronal activity triggered increased microglial process calcium signalling and process extension in awake mice. It is likely that a higher number of penta-partite synapses exists in the living brain compared to tissue slices (cf. Conclusions and [119]).
3.3. Ultrastructural Analysis of Microglia Penta-Partite Synapses
Microglia cell somata can be found in juxtaposition to neuronal cell bodies, particularly in the cerebral cortex (“satellite microglia”), and their processes may also appose to dendrites, dendritic spines, pre- and post-synaptic elements and even patrol the synaptic cleft, especially under pathological conditions [131,132]. In one of the earliest ultrastructural studies on axotomised facial nuclei, Blinzinger and Kreutzberg [132] identified microglial somata and stem processes surrounding large surface areas of motorneuron cell bodies. Tremblay et al. [133] identified microglial cell bodies typically surrounded by pockets of extracellular space, with large and small processes in direct juxtaposition to synaptic elements such as dendritic spines, axon terminals and perisynaptic astrocytic processes in saline-injected mice. Savage et al. [134] similarly observed pockets of extracellular space around microglial cell bodies in both saline- and LPS- injected mice. In another study, Savage et al. [131] found that wild-type microglia from the dorsomedial striatum interacted with excitatory synapses irrespective of age. More recently, the microglia relationships with synapses have been further characterised by trogocytosis or ‘nibbling’ of presynaptic boutons and axons rather than by (large-scale) phagocytosis [121]. It is proposed that the microglial trogocytosis of presynaptic boutons and axons are aimed at reducing or remodelling axonal processes [121].
3.4. Factors Influcening the State of the Penta-Partite Synapse
Studies on experience-dependent synaptic plasticity have demonstrated that the highly dynamic remodelling of synapses occurs across the entire lifespan of an organism and not just during the neurodevelopmental stages [135,136]. The in vivo two-photon imaging of fluorescent-labelled neurons and microglia by Wake et al. [97] demonstrated neuronal activity-dependent synaptic contact by “patrolling” microglial processes at a frequency of about once per hour in mice. Altering light exposure has also been shown to induce changes in contact between microglia processes and synaptic profiles in the visual cortex of juvenile mice [9]. Studies examining the role of microglia in monocular dominance (MD) have demonstrated a shrinkage and loss of thalamocortical synapses in the mouse primary visual cortex (V1) [137,138,139]. Sipe et al. [138] demonstrated in vivo that microglia depend on the G-inhibitory-protein purinergic receptor, P2Y12R, when responding to changes in cortical activity by increasing process arborisation, reducing process motility and increasing interactions with synaptic elements. Stowell et al. [140] had shown that pharmacologic stimulation of β2-adrenergic receptors in microglia disrupted experience-dependent plasticity.
Recent studies have also explored the role of key microglial microRNAs (miRNAs) that regulate the expression of several proteins in synaptic stability and microglial function. Prada et al. [141] observed microglia-to-neuron miR-146a-5p transfer and Syt1 and Nlg1 downregulation that resulted in a significant decrease in dendritic spine density in rat hippocampal neurons in vivo and in vitro. Interestingly, Prada et al. [141] found that the administration of IL-1β, TNF-α, and IFN-γ promoted extracellular vesicle (EV) release from microglia, which delivered miR-146a-5p. Sun et al. [142] reported the downregulatory role of miR-324-5p on astrocyte CCL5 expression, which resulted in the synapse loss of mice neurons. Tsai et al. [143] reported that drosophilia larvae lacking miR-274 released by glia did not develop complete synaptic boutons due to Sprouty (Sty) expression. In addition to the miRNAs mentioned, several studies have also contributed to an understanding of the role of miR-135 and miR-137 in regulatory control of key active zone proteins [144,145,146]. In a similar way to which microglial extracellular vesicles deliver miRNA to neurons, neurons have also been identified to communicate with glia in the same way [147,148]. Peng, Harvey, Richards and Nixon [147] recently isolated neuron-derived EVs (NDEVs) on microglia from rat cortical neurons and added them to cultured rat microglia, which resulted in improved microglia viability and reduced gene expression of TNF-α, IL-6, MCP-1 and iNOS. Yang et al. [148] further demonstrated that neuronal EV transfer of miR-98 to microglia after ischemic stroke prevented certain neurons from microglial phagocytosis. Future work in this domain should confirm the role of other glia-derived miRNAs essential for synaptic plasticity through ultrastructural data.
In vivo studies have demonstrated a role for microglia in shaping structural synaptic plasticity by phagocytosing the perisynaptic ECM [149,150]. Biswas et al. [151] found that interaction with astrocyte-derived ECM via β1-containing integrin influenced microglial activation. In mice retina, microglia are shown to be hyperactivated while microglial TGF-β1 expression is down-regulated in the absence of γ3-containing laminins [151]. Stoyanov et al. [152] found that chondroitinase ABC (ChABC) treatment promoted microglial recruitment to a lesion site three days following the induction of damage in 5xFAD mice but reduced migration in control animals. Histological analyses have revealed that activated microglia engulf damaged perineuronal nets (PNNs) in the 5xFAD brain, with PNN material identified in both mouse and human microglia [153]. Further, Strackeljan et al. [154] observed that the depletion of microglia increased perisynaptic ECM proteoglycan brevican expression, accompanied by elevated expression of presynaptic marker vGluT1 and increased dendritic spine density. Collectively, these studies suggest an interplay between synaptic elements including microglia process tips, the ECM, and key molecules in glia-neuron communication, in regulating structural synaptic plasticity.
3.5. Phagocytic Elimination of Synapses by Microglia Impact Balance between Excitatory and Inhibitory Synapses
In vivo studies over the last decade support the concept that astrocytes contribute to higher brain functions such as learning and memory [155], as well as pathophysiologies such as mood and emotional disorders [156]. Initially, astrocytes were considered to play only less complex roles, such as providing trophic and metabolic support, supporting neuronal survival, differentiation, neuronal guidance, neurite outgrowth and synaptogenesis, while actual brain function would arise exclusively from neuronal activity. Growing evidence has emerged over the last decade to suggest that brain function may arise from neuron–glia interactions.
Microglia create transient contacts with astrocytes, oligodendrocytes as well as neuronal synapses via their motile processes in physiological conditions. Wake, Moorhouse, Jinno, Kohsaka and Nabekura [97] found that the contact time between microglial processes and synapses increases during prolonged ischemia, thereby increasing the likelihood of synaptic elimination. The observation of increased microglial contact time with synapses have been reported in long-term potentiation (LTP). Pfeiffer et al. [157] observed that in mice hippocampal LTP, microglia increased the number of their processes and prolonged their physical contacts with dendritic spines. In LTP, high calcium levels or higher neuronal activity has been demonstrated to result in an increased expression of synaptic AMPA receptors [11,158]. Recently, Saw et al. [159] reported that PI3K in microglia is regulated by epigenetic factors which alter expression of BDNF and thereby downregulate LTP. Sa de Almeida et al. [160] found that Sirt2-deficiency in microglia resulted in LTP impairment in mice hippocampal slices. Hoshino et al. [161] demonstrated that impaired LTP induced by minocycline was alleviated following IL-1 receptor antagonist injection in mice hippocampal sepsis-induced encephalopathy. These findings suggest that prolonged contact time between microglia and synapses may be indicative of long-term synaptic activity.
In persistent pain, glia have been shown to change phenotypes and release chemical mediators resulting in signalling cascades that contribute toward neuronal hypersensitivity. TNF, IL-1β, IFNγ, CCL2 and reactive oxygen species (ROS) have been shown to directly modulate excitatory synaptic transmission by enhancing glutamate release in neuropathic models. Cytokines have been previously shown to be required for normal surface expression of AMPA receptors at synapses. Increased TNF-α concentrations cause a rapid exocytosis of AMPA receptors in hippocampal pyramidal neurons, resulting in increased excitatory synaptic strength [10,11,158,162]. Yu et al. [163] recently identified a microglial P2Y12 receptor-dependent GTP-RhoA/ROCK2 signalling pathway that upregulates excitatory synaptic transmission in the spinal cord dorsal horn, resulting in nociceptive allodynia. Reischer et al. [164] demonstrated that prolonged IFN-γ treatment increases synaptic strength at C-fibre synapses in spinal lamina I rat neurons mediated by microglial activation. Thus, microglial release of chemical mediators may shape synaptic strength. We also note that microglia-derived cytokines can induce neuronal death by phagocytosis [165,166,167], thus influencing the balance between excitatory and inhibitory synaptic transmission.
An imbalance in the ratio of excitatory to inhibitory (E/I) synaptic activity has emerged as a common pathophysiological mechanism in several neuropathological conditions such as Alzheimer’s Disease (AD), autism spectrum disorder (ASD), epilepsy and schizophrenia (SZ). In fact, loss of synaptic connections are considered one of the earliest pathologic hallmarks of AD [168]. Fatemi et al. [169] demonstrated reduced expression of GAD65, GAD67 and receptor subunits GABARA1, GABARA2, GABARA3 and GABARB3 in the parietal cortex and cerebellum of human autism cases compared with controls. Ultrastructural studies of schizophrenia cases have demonstrated an increased density of excitatory synapses in several areas of the basal ganglia and substantia nigra as well as a decreased density of inhibitory synapses [170,171]. Mitew et al. [172] identified a reduction in excitatory vesicular glutamate transporter 1 (VGlut1) boutons in end-stage AD cases and in preclinical AD. In contrast, inhibitory GABAergic synapses were found to be preserved in both human AD and mouse transgenic samples [172]. Recent evidence is suggesting, however, that this may not be the case for all; Kurucu et al. [173] used high-resolution array tomography imaging to demonstrate plaque-associated loss of inhibitory synapses and accumulation of Aβ plaques in a small subset of inhibitory presynaptic terminals. The difference in these findings can be attributed to the methodology used—that is, proteomic studies of synaptic fractions from human AD cases have not been able to yield any changes in inhibitory synaptic proteins. Petrache et al. [174] reported decreased canonical Wnt signalling activity affecting the lateral entorhinal cortex (LEC) with synaptic hyperexcitation, disrupted excitatory–inhibitory inputs onto principal cells, a synaptic imbalance and reduction in the number of parvalbumin-containing (PV) interneurons, and a reduction in the somatic inhibitory axon terminals in the LEC. Petrache et al. [174] also found that targeting GABAA receptors on PV cells restored the identified excitatory–inhibitory imbalance, suggesting a reversible role of excitatory to inhibitory synaptic ratios in determining the progression of AD pathophysiology. Davenport et al. [175] identified a role for CYFIP1 in regulating synapse number and the excitatory–inhibitory synapse ratio in ASD and schizophrenia. More specifically, Davenport, Szulc, Drew, Taylor, Morgan, Higgs, López-Doménech and Kittler [175] found that CYFIP1/CYFIP2 upregulation increased excitatory synapse number while decreasing the size and amplitude of inhibitory synapses and their miniature inhibitory post-synaptic currents (mIPSCs). Furthermore, in vivo knockout in neocortical principal cells increased the expression of post-synaptic GABAA receptor β2/3-subunits and neuroligin 3, thus enhancing synaptic inhibition. Howell and Smith [176] have provided a detailed review of several synaptic structural proteins, such as SHANK, Gephyrin, Homer, PSD-95, and SAPAP, that regulate the excitatory–inhibitory synaptic balance across various brain regions in ASD.
Taken together, abnormal glial function is increasingly recognised as an early pathological feature commonly observed in neurodegenerative and psychiatric diseases. Both astrocytes and microglia have been demonstrated to influence synapse formation and elimination under various physiological conditions [177,178,179,180]. For example, Choudhury, Miyanishi, Takeda, Islam, Matsuoka, Kubo, Matsumoto, Kunieda, Nomoto and Yano [180] found that microglia eliminate assumed weak synapses opsonized with eat-me signals C3 or MFG-E8 during every sleep phase via phagocytosis. In contrast to physiological conditions, glial dysfunction—as observed in pathological contexts—is proposed to result in the disruption of the excitatory/inhibitory synaptic balance [181,182,183]. In colony-stimulating factor 1 receptor (Csf1r) knockout mice, microglia-mediated climbing fiber (CF) elimination was found to be inhibited, thereby attenuating inhibitory GABAergic synaptic transmission [184]. Cantaut-Belarif et al. [185] have shown that microglia differentially control GABAergic and glycinergic inhibition in the spinal cord. Ren et al. [186] found that the microglia-specific Trem2R47H Alzheimer-linked variant increased glutamatergic neuronal transmission and suppressed LTP by enhancing brain TNF-α concentrations in rats. Thus, the role of microglia in influencing the excitatory to inhibitory synaptic ratio appears to present an example where the penta-partite microglial synapse is most relevant in brain pathologies where microglial activation has been described on imaging and/or histologically.
4. Conclusions
Over the course of the last decade, microglia have undoubtedly become a force at the CNS synapse that needs to be reckoned with. The concept of a microglial contribution to normal synaptic function should provide a stimulus for additional studies into the detailed tissue distribution of microglial penta-partite synapses, their role in CNS physiology as well as their changes under pathological conditions. Microglial penta-partite synapses represent only a subset of synapses but likely one that has significance for the symptoms reported under conditions of “neuroinflammation”. The latter term is misleading and should be abandoned, but the microglial activation that characterises “neuroinflammation” deserves further scrutiny. Penta-partite aptly describes the type of synapse to look out for, but we agree with the authors of a recent paper on the “active milieu” [187], a new integrating morpho-functional concept of the synapse, that a holistic view is preferrable to an enumeration of synaptic components.
Author Contributions
Conceptualization, J.A.A. and M.B.G.; software, J.A.A.; validation, M.B.G. and M.M.; data curation, J.A.A. and M.B.G.; writing—original draft preparation, J.A.A. and M.B.G.; writing—review and editing, J.A.A., A.V.-I., M.M. and M.B.G.; visualization, J.A.A. and M.B.G.; supervision, M.B.G. and M.M.; funding acquisition, M.B.G. and M.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Australian Research Council (ARC) grants, DP150104472 and DP210103469.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- De Leo, J.A.; Tawfik, V.L.; LaCroix-Fralish, M.L. The tetrapartite synapse: Path to CNS sensitization and chronic pain. Pain 2006, 122, 17–21. [Google Scholar] [CrossRef]
- Dityatev, A.; Rusakov, D.A. Molecular signals of plasticity at the tetrapartite synapse. Curr. Opin. Neurobiol. 2011, 21, 353–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araque, A.; Parpura, V.; Sanzgiri, R.P.; Haydon, P.G. Tripartite synapses: Glia, the unacknowledged partner. Trends Neurosci. 1999, 22, 208–215. [Google Scholar] [CrossRef]
- Chelini, G.; Pantazopoulos, H.; Durning, P.; Berretta, S. The tetrapartite synapse: A key concept in the pathophysiology of schizophrenia. Eur. Psychiatry 2018, 50, 60–69. [Google Scholar] [CrossRef]
- Bikbaev, A.; Frischknecht, R.; Heine, M. Brain extracellular matrix retains connectivity in neuronal networks. Sci. Rep. 2015, 5, 1–12. [Google Scholar]
- Zhai, J.; Kim, H.; Han, S.B.; Manire, M.; Yoo, R.; Pang, S.; Smith, G.M.; Son, Y.-J. Co-targeting myelin inhibitors and CSPGs markedly enhances regeneration of GDNF-stimulated, but not conditioning-lesioned, sensory axons into the spinal cord. Elife 2021, 10, e63050. [Google Scholar] [CrossRef]
- Kabdesh, I.M.; Arkhipova, S.S.; Mukhamedshina, Y.O.; James, V.; Rizvanov, A.A.; Chelyshev, Y.A. The function of NG2/CSPG4-expressing cells in the rat spinal cord injury: An immunoelectron microscopy study. Neuroscience 2021, 467, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Lenz, M.; Vlachos, A. The neuroimmunological synapse: From synaptic homeostasis to brain disease. Neuroforum 2019, 25, 163–171. [Google Scholar] [CrossRef]
- Tremblay, M.-È.; Lowery, R.L.; Majewska, A.K. Microglial interactions with synapses are modulated by visual experience. PLoS Biol. 2010, 8, e1000527. [Google Scholar] [CrossRef] [Green Version]
- Stellwagen, D. The contribution of TNFalpha to synaptic plasticity and nervous system function. Adv. Exp. Med. Biol. 2011, 691, 541–557. [Google Scholar] [CrossRef]
- Beattie, E.C.; Stellwagen, D.; Morishita, W.; Bresnahan, J.C.; Ha, B.K.; Von Zastrow, M.; Beattie, M.S.; Malenka, R.C. Control of synaptic strength by glial TNFalpha. Science 2002, 295, 2282–2285. [Google Scholar] [CrossRef] [PubMed]
- Raffaele, S.; Lombardi, M.; Verderio, C.; Fumagalli, M. TNF Production and Release from Microglia via Extracellular Vesicles: Impact on Brain Functions. Cells 2020, 9, 2145. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-Sequencing Transcriptome and Splicing Database of Glia, Neurons, and Vascular Cells of the Cerebral Cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.; García-García, E.; Straccia, M.; Comella-Bolla, A.; Miguez, A.; Masana, M.; Alberch, J.; Canals, J.M.; Rodríguez, M.J. Reduced Fractalkine Levels Lead to Striatal Synaptic Plasticity Deficits in Huntington’s Disease. Front. Cell Neurosci. 2020, 14, 163. [Google Scholar] [CrossRef] [PubMed]
- Freria, C.M.; Hall, J.C.; Wei, P.; Guan, Z.; McTigue, D.M.; Popovich, P.G. Deletion of the fractalkine receptor, CX3CR1, improves endogenous repair, axon sprouting, and synaptogenesis after spinal cord injury in mice. J. Neurosci. 2017, 37, 3568–3587. [Google Scholar] [CrossRef] [Green Version]
- Kowiański, P.; Lietzau, G.; Czuba, E.; Waśkow, M.; Steliga, A.; Moryś, J. BDNF: A key factor with multipotent impact on brain signaling and synaptic plasticity. Cell Mol. Neurobiol. 2018, 38, 579–593. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, F.R.; Musella, A.; De Vito, F.; Fresegna, D.; Bullitta, S.; Vanni, V.; Guadalupi, L.; Stampanoni Bassi, M.; Buttari, F.; Mandolesi, G. Tumor necrosis factor and interleukin-1β modulate synaptic plasticity during neuroinflammation. Neural Plast. 2018, 2018, 8430123-12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Varvel, N.H.; Konerth, M.E.; Xu, G.; Cardona, A.E.; Ransohoff, R.M.; Lamb, B.T. CX3CR1 deficiency alters microglial activation and reduces beta-amyloid deposition in two Alzheimer’s disease mouse models. Am. J. Pathol. 2010, 177, 2549–2562. [Google Scholar] [CrossRef]
- Li, Y.; Tan, M.-S.; Jiang, T.; Tan, L. Microglia in Alzheimer’s disease. BioMed Res. Int. 2014, 2014, 437483–437487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tetreault, N.A.; Hakeem, A.Y.; Jiang, S.; Williams, B.A.; Allman, E.; Wold, B.J.; Allman, J.M. Microglia in the cerebral cortex in autism. J. Autism Dev. Disord. 2012, 42, 2569–2584. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Castillo, E.; Frias, E.S.; Swanson, R.A. Bioenergetic regulation of microglia. Glia 2018, 66, 1200–1212. [Google Scholar] [CrossRef] [PubMed]
- Longo, V.D.; Mattson, M.P. Fasting: Molecular mechanisms and clinical applications. Cell Metab. 2014, 19, 181–192. [Google Scholar] [CrossRef] [Green Version]
- York, E.M.; Zhang, J.; Choi, H.B.; MacVicar, B.A. Neuroinflammatory inhibition of synaptic long-term potentiation requires immunometabolic reprogramming of microglia. Glia 2021, 69, 567–578. [Google Scholar] [CrossRef] [PubMed]
- Filiou, M.D.; Arefin, A.S.; Moscato, P.; Graeber, M.B. ‘Neuroinflammation’ differs categorically from inflammation: Transcriptomes of Alzheimer’s disease, Parkinson’s disease, schizophrenia and inflammatory diseases compared. Neurogenetics 2014, 15, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Sochocka, M.; Diniz, B.S.; Leszek, J. Inflammatory response in the CNS: Friend or foe? Mol. Neurobiol. 2017, 54, 8071–8089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Headland, S.E.; Norling, L.V. The resolution of inflammation: Principles and challenges. In Seminars in Immunology. Elsevier: Amsterdam, The Netherlands, 2015; Volume 27, pp. 149–160. [Google Scholar]
- Milatovic, D.; Zaja-Milatovic, S.; Breyer, R.M.; Aschner, M.; Montine, T.J. Neuroinflammation and oxidative injury in developmental neurotoxicity. In Reproductive and Developmental Toxicology; Elsevier: Amsterdam, The Netherlands, 2017; pp. 1051–1061. [Google Scholar]
- Norden, D.M.; Trojanowski, P.J.; Villanueva, E.; Navarro, E.; Godbout, J.P. Sequential activation of microglia and astrocyte cytokine expression precedes increased iba-1 or GFAP immunoreactivity following systemic immune challenge. Glia 2016, 64, 300–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Dasgupta, C.; Huang, L.; Meng, X.; Zhang, L. MiRNA-210 induces microglial activation and regulates microglia-mediated neuroinflammation in neonatal hypoxic-ischemic encephalopathy. Cell Mol. Immunol. 2020, 17, 976–991. [Google Scholar] [CrossRef]
- Gate, D.; Saligrama, N.; Leventhal, O.; Yang, A.C.; Unger, M.S.; Middeldorp, J.; Chen, K.; Lehallier, B.; Channappa, D.; De Los Santos, M.B.; et al. Clonally expanded CD8 T cells patrol the cerebrospinal fluid in Alzheimer’s disease. Nature 2020, 577, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J.; Kreutzberg, G.W. Lectin binding by resting and reactive microglia. J. Neurocytol. 1987, 16, 249–260. [Google Scholar] [CrossRef] [PubMed]
- Law, S.K. C3 receptors on macrophages. J. Cell Sci. Suppl. 1988, 9, 67–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, M.L.; Viaene, A.N. What are activated and reactive glia and what is their role in neurodegeneration? Neurobiol. Dis. 2021, 148, 105172. [Google Scholar] [CrossRef]
- Cherry, J.D.; Olschowka, J.A.; O’Banion, M.K. Are “resting” microglia more “m2”? Front. Immunol. 2014, 5, 594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cone, W. Acute pathologic changes in neuroglia and in microglia. Arch. Neurol. Psychiatry 1928, 20, 34–72. [Google Scholar] [CrossRef]
- Tondo, G.; Iaccarino, L.; Cerami, C.; Vanoli, G.E.; Presotto, L.; Masiello, V.; Coliva, A.; Salvi, F.; Bartolomei, I.; Mosca, L. 11C-PK11195 PET–based molecular study of microglia activation in SOD1 amyotrophic lateral sclerosis. Ann. Clin. Transl. Neurol. 2020, 7, 1513–1523. [Google Scholar] [CrossRef] [PubMed]
- Radford, R.A.; Morsch, M.; Rayner, S.L.; Cole, N.J.; Pountney, D.L.; Chung, R.S. The established and emerging roles of astrocytes and microglia in amyotrophic lateral sclerosis and frontotemporal dementia. Front. Cell Neurosci. 2015, 9, 414. [Google Scholar] [CrossRef] [Green Version]
- Gui, Y.; Marks, J.D.; Das, S.; Hyman, B.T.; Serrano-Pozo, A. Characterization of the 18 kDa translocator protein (TSPO) expression in post-mortem normal and Alzheimer’s disease brains. Brain Pathol. 2020, 30, 151–164. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.; Pan, D.; Wang, Y.; Bao, W.; Zuo, C.; Guan, Y.; Hua, F.; Yang, M.; Zhao, J. PET Imaging for Dynamically Monitoring Neuroinflammation in APP/PS1 Mouse Model Using [18F] DPA714. Front. Neurosci. 2020, 14, 810. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, D.S.; Mainero, C.; Ichijo, E.; Ward, N.; Granziera, C.; Zürcher, N.R.; Akeju, O.; Bonnier, G.; Price, J.; Hooker, J.M. Imaging of neuroinflammation in migraine with aura: A [11C] PBR28 PET/MRI study. Neurology 2019, 92, e2038–e2050. [Google Scholar] [CrossRef] [PubMed]
- Barletta, V.T.; Herranz, E.; Treaba, C.A.; Ouellette, R.; Mehndiratta, A.; Loggia, M.L.; Klawiter, E.C.; Ionete, C.; Jacob, S.A.; Mainero, C. Evidence of diffuse cerebellar neuroinflammation in multiple sclerosis by 11C-PBR28 MR-PET. Mult. Scler. J. 2020, 26, 668–678. [Google Scholar] [CrossRef]
- Herranz, E.; Louapre, C.; Treaba, C.A.; Govindarajan, S.T.; Ouellette, R.; Mangeat, G.; Loggia, M.L.; Cohen-Adad, J.; Klawiter, E.C.; Sloane, J.A. Profiles of cortical inflammation in multiple sclerosis by 11C-PBR28 MR-PET and 7 Tesla imaging. Mult. Scler. J. 2020, 26, 1497–1509. [Google Scholar] [CrossRef]
- Owen, D.R.; Narayan, N.; Wells, L.; Healy, L.; Smyth, E.; Rabiner, E.A.; Galloway, D.; Williams, J.B.; Lehr, J.; Mandhair, H. Pro-inflammatory activation of primary microglia and macrophages increases 18 kDa translocator protein expression in rodents but not humans. J. Cereb. Blood Flow Metab. 2017, 37, 2679–2690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hafizi, S.; Tseng, H.-H.; Rao, N.; Selvanathan, T.; Kenk, M.; Bazinet, R.P.; Suridjan, I.; Wilson, A.A.; Meyer, J.H.; Remington, G. Imaging microglial activation in untreated first-episode psychosis: A PET study with [18F] FEPPA. Am. J. Psychiatry 2017, 174, 118–124. [Google Scholar] [CrossRef] [Green Version]
- Nutma, E.; Stephenson, J.A.; Gorter, R.P.; De Bruin, J.; Boucherie, D.M.; Donat, C.K.; Breur, M.; Van Der Valk, P.; Matthews, P.M.; Owen, D.R. A quantitative neuropathological assessment of translocator protein expression in multiple sclerosis. Brain 2019, 142, 3440–3455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okello, A.; Edison, P.; Archer, H.A.; Turkheimer, F.E.; Kennedy, J.; Bullock, R.; Walker, Z.; Kennedy, A.; Fox, N.; Rossor, M.; et al. Microglial activation and amyloid deposition in mild cognitive impairment: A PET study. Neurology 2009, 72, 56–62. [Google Scholar] [CrossRef] [Green Version]
- Sneeboer, M.A.; van der Doef, T.; Litjens, M.; Psy, N.; Melief, J.; Hol, E.M.; Kahn, R.S.; de Witte, L.D. Microglial activation in schizophrenia: Is translocator 18 kDa protein (TSPO) the right marker? Schizophr. Res. 2020, 215, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Davalos, D.; Grutzendler, J.; Yang, G.; Kim, J.V.; Zuo, Y.; Jung, S.; Littman, D.R.; Dustin, M.L.; Gan, W.B. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 2005, 8, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.-J.; Middleton, R.J.; Kam, W.W.-Y.; Chin, D.Y.; Hatty, C.R.; Chan, R.H.; Banati, R.B. Functional gains in energy and cell metabolism after TSPO gene insertion. Cell Cycle 2017, 16, 436–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banati, R.B.; Middleton, R.J.; Chan, R.; Hatty, C.R.; Kam, W.W.-Y.; Quin, C.; Graeber, M.B.; Parmar, A.; Zahra, D.; Callaghan, P. Positron emission tomography and functional characterization of a complete PBR/TSPO knockout. Nat. Commun. 2014, 5, 1–12. [Google Scholar] [CrossRef]
- Shatz, C.J. MHC class I: An unexpected role in neuronal plasticity. Neuron 2009, 64, 40–45. [Google Scholar] [CrossRef] [Green Version]
- Werneburg, S.; Feinberg, P.A.; Johnson, K.M.; Schafer, D.P. A microglia-cytokine axis to modulate synaptic connectivity and function. Curr. Opin. Neurobiol. 2017, 47, 138–145. [Google Scholar] [CrossRef]
- Hanisch, U.K. Microglia as a source and target of cytokines. Glia 2002, 40, 140–155. [Google Scholar] [CrossRef]
- West, P.K.; Viengkhou, B.; Campbell, I.L.; Hofer, M.J. Microglia responses to interleukin-6 and type I interferons in neuroinflammatory disease. Glia 2019, 67, 1821–1841. [Google Scholar] [CrossRef]
- Bobbo, V.C.; Jara, C.P.; Mendes, N.F.; Morari, J.; Velloso, L.A.; Araujo, E.P. Interleukin-6 expression by hypothalamic microglia in multiple inflammatory contexts: A systematic review. BioMed Res. Int. 2019, 2019, 1365210-11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Huang, S.; Zhu, J.; Hu, T.; Han, Z.; Zhang, S.; Zhao, J.; Chen, F.; Lei, P. Exosomes from MiR-21-5p-Increased Neurons Play a Role in Neuroprotection by Suppressing Rab11a-Mediated Neuronal Autophagy In Vitro After Traumatic Brain Injury. Med. Sci. Monit. 2019, 25, 1871–1885. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Chen, F.; Ge, X.; Tan, J.; Lei, P.; Zhang, J. miR-21 alleviated apoptosis of cortical neurons through promoting PTEN-Akt signaling pathway in vitro after experimental traumatic brain injury. Brain Res. 2014, 1582, 12–20. [Google Scholar] [CrossRef]
- Parkhurst, C.N.; Yang, G.; Ninan, I.; Savas, J.N.; Yates III, J.R.; Lafaille, J.J.; Hempstead, B.L.; Littman, D.R.; Gan, W.-B. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 2013, 155, 1596–1609. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.B.; Nagai, A.; Kim, S.U. Cytokines, chemokines, and cytokine receptors in human microglia. J. Neurosci. Res. 2002, 69, 94–103. [Google Scholar] [CrossRef]
- Lam, D.; Lively, S.; Schlichter, L.C. Responses of rat and mouse primary microglia to pro- and anti-inflammatory stimuli: Molecular profiles, K(+) channels and migration. J. Neuroinflamm. 2017, 14, 166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elkabes, S.; DiCicco-Bloom, E.M.; Black, I.B. Brain microglia/macrophages express neurotrophins that selectively regulate microglial proliferation and function. J. Neurosci. 1996, 16, 2508–2521. [Google Scholar] [CrossRef] [Green Version]
- Eastlake, K.; Banerjee, P.; Angbohang, A.; Charteris, D.; Khaw, P.; Limb, G. Müller glia as an important source of cytokines and inflammatory factors present in the gliotic retina during proliferative vitreoretinopathy. Glia 2016, 64, 495–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clausen, B.H.; Wirenfeldt, M.; Høgedal, S.S.; Frich, L.H.; Nielsen, H.H.; Schrøder, H.D.; Østergaard, K.; Finsen, B.; Kristensen, B.W.; Lambertsen, K.L. Characterization of the TNF and IL-1 systems in human brain and blood after ischemic stroke. Acta Neuropathol. Commun. 2020, 8, 81. [Google Scholar] [CrossRef] [PubMed]
- Hammond, T.R.; Dufort, C.; Dissing-Olesen, L.; Giera, S.; Young, A.; Wysoker, A.; Walker, A.J.; Gergits, F.; Segel, M.; Nemesh, J. Single-cell RNA sequencing of microglia throughout the mouse lifespan and in the injured brain reveals complex cell-state changes. Immunity 2019, 50, 253–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dulken, B.W.; Buckley, M.T.; Navarro Negredo, P.; Saligrama, N.; Cayrol, R.; Leeman, D.S.; George, B.M.; Boutet, S.C.; Hebestreit, K.; Pluvinage, J.V.; et al. Single-cell analysis reveals T cell infiltration in old neurogenic niches. Nature 2019, 571, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.U.; de Vellis, J. Microglia in health and disease. J. Neurosci. Res. 2005, 81, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Gyoneva, S.; Hosur, R.; Gosselin, D.; Zhang, B.; Ouyang, Z.; Cotleur, A.C.; Peterson, M.; Allaire, N.; Challa, R.; Cullen, P.; et al. Cx3cr1-deficient microglia exhibit a premature aging transcriptome. Life Sci. Alliance 2019, 2(6), 201900453. [Google Scholar] [CrossRef] [Green Version]
- Olah, M.; Patrick, E.; Villani, A.C.; Xu, J.; White, C.C.; Ryan, K.J.; Piehowski, P.; Kapasi, A.; Nejad, P.; Cimpean, M.; et al. A transcriptomic atlas of aged human microglia. Nat. Commun. 2018, 9, 539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, A.K.; Yip, P.K.; Malcangio, M. The liberation of fractalkine in the dorsal horn requires microglial cathepsin S. J. Neurosci. 2009, 29, 6945–6954. [Google Scholar] [CrossRef] [Green Version]
- Stumm, R.K.; Rummel, J.; Junker, V.; Culmsee, C.; Pfeiffer, M.; Krieglstein, J.; Höllt, V.; Schulz, S. A dual role for the SDF-1/CXCR4 chemokine receptor system in adult brain: Isoform-selective regulation of SDF-1 expression modulates CXCR4-dependent neuronal plasticity and cerebral leukocyte recruitment after focal ischemia. J. Neurosci. 2002, 22, 5865–5878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, K.; Yao, X.; Wu, W.; Yu, Z.; Li, Z.; Ma, Z.; Liu, D. HIF-1α/SDF-1/CXCR4 axis reduces neuronal apoptosis via enhancing the bone marrow-derived mesenchymal stromal cell migration in rats with traumatic brain injury. Exp. Mol. Pathol. 2020, 114, 104416. [Google Scholar] [CrossRef]
- Chia, K.; Mazzolini, J.; Mione, M.; Sieger, D. Tumor initiating cells induce Cxcr4-mediated infiltration of pro-tumoral macrophages into the brain. Elife 2018, 7, e31918. [Google Scholar] [CrossRef] [PubMed]
- Banisadr, G.; Podojil, J.R.; Miller, S.D.; Miller, R.J. Pattern of CXCR7 gene expression in mouse brain under normal and inflammatory conditions. J. Neuroimmune Pharmacol. 2016, 11, 26–35. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Wang, H.; Xu, M.; Frank, J.A.; Luo, J. Role of MCP-1 and CCR2 in ethanol-induced neuroinflammation and neurodegeneration in the developing brain. J. Neuroinflamm. 2018, 15, 197. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, S.; Huang, Y.; Sun, L. miR-21 protects neonatal rats from hypoxic-ischemic brain damage by targeting CCL3. Apoptosis 2020, 25, 275–289. [Google Scholar] [CrossRef]
- Ciechanowska, A.; Popiolek-Barczyk, K.; Pawlik, K.; Ciapała, K.; Oggioni, M.; Mercurio, D.; De Simoni, M.-G.; Mika, J. Changes in macrophage inflammatory protein-1 (MIP-1) family members expression induced by traumatic brain injury in mice. Immunobiology 2020, 225, 151911. [Google Scholar] [CrossRef] [PubMed]
- Lanfranco, M.F.; Mocchetti, I.; Burns, M.P.; Villapol, S. Glial-and neuronal-specific expression of CCL5 mRNA in the rat brain. Front. Neuroanat. 2018, 11, 137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, S.-Y.; Ajoy, R.; Changou, C.A.; Hsieh, Y.-T.; Wang, Y.-K.; Hoffer, B. CCL5/RANTES contributes to hypothalamic insulin signaling for systemic insulin responsiveness through CCR5. Sci. Rep. 2016, 6, 37659. [Google Scholar] [CrossRef] [Green Version]
- Campbell, L.A.; Avdoshina, V.; Rozzi, S.; Mocchetti, I. CCL5 and cytokine expression in the rat brain: Differential modulation by chronic morphine and morphine withdrawal. Brain Behav. Immun. 2013, 34, 130–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arisi, G.M.; Foresti, M.L.; Katki, K.; Shapiro, L.A. Increased CCL2, CCL3, CCL5, and IL-1β cytokine concentration in piriform cortex, hippocampus, and neocortex after pilocarpine-induced seizures. J. Neuroinflamm. 2015, 12, 129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.H.; Long, L.; Tang, Y.C.; Zhang, J.T.; Hut, H.T.; Tang, F.R. CCR3, CCR2A and macrophage inflammatory protein (MIP)-1a, monocyte chemotactic protein-1 (MCP-1) in the mouse hippocampus during and after pilocarpine-induced status epilepticus (PISE). Neuropathol. Appl. Neurobiol. 2009, 35, 496–514. [Google Scholar] [CrossRef]
- Zhu, C.; Xu, B.; Sun, X.; Zhu, Q.; Sui, Y. Targeting CCR3 to Reduce Amyloid-β Production, Tau Hyperphosphorylation, and Synaptic Loss in a Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2017, 54, 7964–7978. [Google Scholar] [CrossRef]
- van der Meer, P.; Ulrich, A.M.; Gonźalez-Scarano, F.; Lavi, E. Immunohistochemical analysis of CCR2, CCR3, CCR5, and CXCR4 in the human brain: Potential mechanisms for HIV dementia. Exp. Mol. Pathol. 2000, 69, 192–201. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, H.; Sherbini, O.; Ling-Lin Pai, E.; Kang, S.U.; Kwon, J.S.; Yang, J.; He, W.; Wang, H.; Eacker, S.M.; et al. High-Content Genome-Wide RNAi Screen Reveals CCR3 as a Key Mediator of Neuronal Cell Death. eNeuro 2016, 3(5), ENEURO.0185-16.2016. [Google Scholar] [CrossRef] [Green Version]
- Lynch, M.A. Can the emerging field of immunometabolism provide insights into neuroinflammation? Prog. Neurobiol. 2020, 184, 101719. [Google Scholar] [CrossRef]
- Jarc, E.; Petan, T. A twist of FATe: Lipid droplets and inflammatory lipid mediators. Biochimie 2020, 169, 69–87. [Google Scholar] [CrossRef]
- Boucher, D.M.; Vijithakumar, V.; Ouimet, M. Lipid Droplets as Regulators of Metabolism and Immunity. Immunometabolism 2021, 3, e210021. [Google Scholar]
- Kalsbeek, M.J.; Mulder, L.; Yi, C.X. Microglia energy metabolism in metabolic disorder. Mol. Cell Endocrinol. 2016, 438, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Wang, Z.; Liang, X.; Wang, Y.; Gao, L.; Ma, C. Monocarboxylate transporter 1 promotes classical microglial activation and pro-inflammatory effect via 6-phosphofructo-2-kinase/fructose-2, 6-biphosphatase 3. J. Neuroinflamm. 2019, 16, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Bernier, L.-P.; York, E.M.; Kamyabi, A.; Choi, H.B.; Weilinger, N.L.; MacVicar, B.A. Microglial metabolic flexibility supports immune surveillance of the brain parenchyma. Nat. Commun. 2020, 11, 1–17. [Google Scholar] [CrossRef]
- Khatchadourian, A.; Bourque, S.D.; Richard, V.R.; Titorenko, V.I.; Maysinger, D. Dynamics and regulation of lipid droplet formation in lipopolysaccharide (LPS)-stimulated microglia. Biochim. Biophys. Acta BBA-Mol. Cell Biol. Lipids 2012, 1821, 607–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Churchward, M.A.; Tchir, D.R.; Todd, K.G. Microglial function during glucose deprivation: Inflammatory and neuropsychiatric implications. Mol. Neurobiol. 2018, 55, 1477–1487. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, C.F.; Liu, C.K.; Lee, C.T.; Yu, L.E.; Wang, J.Y. Acute glucose fluctuation impacts microglial activity, leading to inflammatory activation or self-degradation. Sci. Rep. 2019, 9, 840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, K.; Takizawa, S.; Ohgaku, Y.; Asami, T.; Furuya, K.; Yamamoto, K.; Takahashi, F.; Hamajima, C.; Inaba, C.; Endo, K. MicroRNA 16-5p is upregulated in calorie-restricted mice and modulates inflammatory cytokines of macrophages. Gene 2020, 725, 144191. [Google Scholar] [CrossRef] [PubMed]
- Bernhart, E.; Kollroser, M.; Rechberger, G.; Reicher, H.; Heinemann, A.; Schratl, P.; Hallström, S.; Wintersperger, A.; Nusshold, C.; DeVaney, T. Lysophosphatidic acid receptor activation affects the C13NJ microglia cell line proteome leading to alterations in glycolysis, motility, and cytoskeletal architecture. Proteomics 2010, 10, 141–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Pavlou, S.; Du, X.; Bhuckory, M.; Xu, H.; Chen, M. Glucose transporter 1 critically controls microglial activation through facilitating glycolysis. Mol. Neurodegener. 2019, 14, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wake, H.; Moorhouse, A.J.; Jinno, S.; Kohsaka, S.; Nabekura, J. Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J. Neurosci. 2009, 29, 3974–3980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kofuji, P.; Araque, A. G-protein-coupled receptors in astrocyte–neuron communication. Neuroscience 2021, 456, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Navarrete, M.; Perea, G.; de Sevilla, D.F.; Gómez-Gonzalo, M.; Núñez, A.; Martín, E.D.; Araque, A. Astrocytes mediate in vivo cholinergic-induced synaptic plasticity. PLoS Biol. 2012, 10, e1001259. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, R.; Huang, B.S.; Venugopal, S.; Johnston, A.D.; Chai, H.; Zeng, H.; Golshani, P.; Khakh, B.S. Ca 2+ signaling in astrocytes from Ip3r2−/− mice in brain slices and during startle responses in vivo. Nat. Neurosci. 2015, 18, 708–717. [Google Scholar] [CrossRef] [Green Version]
- Ding, F.; O’Donnell, J.; Thrane, A.S.; Zeppenfeld, D.; Kang, H.; Xie, L.; Wang, F.; Nedergaard, M. α1-Adrenergic receptors mediate coordinated Ca2+ signaling of cortical astrocytes in awake, behaving mice. Cell Calcium 2013, 54, 387–394. [Google Scholar] [CrossRef] [Green Version]
- Fields, R.D.; Burnstock, G. Purinergic signalling in neuron–glia interactions. Nat. Rev. Neurosci. 2006, 7, 423–436. [Google Scholar] [CrossRef]
- Juaristi, I.; Llorente-Folch, I.; Satrústegui, J.; Del Arco, A. Extracellular ATP and glutamate drive pyruvate production and energy demand to regulate mitochondrial respiration in astrocytes. Glia 2019, 67, 759–774. [Google Scholar] [CrossRef] [PubMed]
- Shigetomi, E.; Hirayama, Y.J.; Ikenaka, K.; Tanaka, K.F.; Koizumi, S. Role of purinergic receptor P2Y1 in spatiotemporal Ca2+ dynamics in astrocytes. J. Neurosci. 2018, 38, 1383–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monai, H.; Ohkura, M.; Tanaka, M.; Oe, Y.; Konno, A.; Hirai, H.; Mikoshiba, K.; Itohara, S.; Nakai, J.; Iwai, Y. Calcium imaging reveals glial involvement in transcranial direct current stimulation-induced plasticity in mouse brain. Nat. Commun. 2016, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Muller, M.S.; Taylor, C.W. ATP evokes Ca2+ signals in cultured foetal human cortical astrocytes entirely through G protein-coupled P2Y receptors. J. Neurochem. 2017, 142, 876–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascual, O.; Achour, S.B.; Rostaing, P.; Triller, A.; Bessis, A. Microglia activation triggers astrocyte-mediated modulation of excitatory neurotransmission. Proc. Natl. Acad. Sci. USA 2012, 109, E197–E205. [Google Scholar] [CrossRef] [Green Version]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janks, L.; Sharma, C.V.; Egan, T.M. A central role for P2X7 receptors in human microglia. J. Neuroinflamm. 2018, 15, 1–18. [Google Scholar] [CrossRef]
- Beneventano, M.; Spampinato, S.F.; Merlo, S.; Chisari, M.; Platania, P.; Ragusa, M.; Purrello, M.; Nicoletti, F.; Sortino, M.A. Shedding of microvesicles from microglia contributes to the effects induced by metabotropic glutamate receptor 5 activation on neuronal death. Front. Pharmacol. 2017, 8, 812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Zhao, Z.; Ji, R.; Zhu, J.; Sui, Q.-Q.; Knight, G.E.; Burnstock, G.; He, C.; Yuan, H.; Xiang, Z. Inhibition of P2X7 receptors improves outcomes after traumatic brain injury in rats. Purinergic Signal. 2017, 13, 529–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Köles, L.; Leichsenring, A.; Rubini, P.; Illes, P. P2 receptor signaling in neurons and glial cells of the central nervous system. Adv. Pharmacol. 2011, 61, 441–493. [Google Scholar]
- Jiang, P.; Xing, F.; Guo, B.; Yang, J.; Li, Z.; Wei, W.; Hu, F.; Lee, I.; Zhang, X.; Pan, L. Nucleotide transmitters ATP and ADP mediate intercellular calcium wave communication via P2Y12/13 receptors among BV-2 microglia. PLoS ONE 2017, 12, e0183114. [Google Scholar] [CrossRef] [Green Version]
- Quintas, C.; Vale, N.; Gonçalves, J.; Queiroz, G. Microglia P2Y13 receptors prevent astrocyte proliferation mediated by P2Y1 receptors. Front. Pharmacol. 2018, 9, 418. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, S.; Shigemoto-Mogami, Y.; Nasu-Tada, K.; Shinozaki, Y.; Ohsawa, K.; Tsuda, M.; Joshi, B.V.; Jacobson, K.A.; Kohsaka, S.; Inoue, K. UDP acting at P2Y 6 receptors is a mediator of microglial phagocytosis. Nature 2007, 446, 1091–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perea, G.; Araque, A. Astrocytes potentiate transmitter release at single hippocampal synapses. Science 2007, 317, 1083–1086. [Google Scholar] [CrossRef]
- Akiyoshi, R.; Wake, H.; Kato, D.; Horiuchi, H.; Ono, R.; Ikegami, A.; Haruwaka, K.; Omori, T.; Tachibana, Y.; Moorhouse, A.J.; et al. Microglia Enhance Synapse Activity to Promote Local Network Synchronization. eNeuro 2018, 5(5), ENEURO.0088-182018. [Google Scholar] [CrossRef] [PubMed]
- Augusto-Oliveira, M.; Arrifano, G.P.; Takeda, P.Y.; Lopes-Araújo, A.; Santos-Sacramento, L.; Anthony, D.C.; Verkhratsky, A.; Crespo-Lopez, M.E. Astroglia-specific contributions to the regulation of synapses, cognition and behaviour. Neurosci. Biobehav. Rev. 2020, 118, 331–357. [Google Scholar] [CrossRef]
- Witcher, M.R.; Kirov, S.A.; Harris, K.M. Plasticity of perisynaptic astroglia during synaptogenesis in the mature rat hippocampus. Glia 2007, 55, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Ventura, R.; Harris, K.M. Three-dimensional relationships between hippocampal synapses and astrocytes. J. Neurosci. 1999, 19, 6897–6906. [Google Scholar] [CrossRef]
- Weinhard, L.; Di Bartolomei, G.; Bolasco, G.; Machado, P.; Schieber, N.L.; Neniskyte, U.; Exiga, M.; Vadisiute, A.; Raggioli, A.; Schertel, A. Microglia remodel synapses by presynaptic trogocytosis and spine head filopodia induction. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Xu-Friedman, M.A.; Harris, K.M.; Regehr, W.G. Three-dimensional comparison of ultrastructural characteristics at depressing and facilitating synapses onto cerebellar Purkinje cells. J. Neurosci. 2001, 21, 6666–6672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spacek, J. Three-dimensional analysis of dendritic spines. III. Glial sheath. Anat. Embryol. 1985, 171, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Broadhead, M.J.; Bonthron, C.; Arcinas, L.; Bez, S.; Zhu, F.; Goff, F.; Nylk, J.; Dholakia, K.; Gunn-Moore, F.; Grant, S.G.N.; et al. Nanostructural Diversity of Synapses in the Mammalian Spinal Cord. Sci. Rep. 2020, 10, 8189. [Google Scholar] [CrossRef]
- Bernardinelli, Y.; Randall, J.; Janett, E.; Nikonenko, I.; König, S.; Jones, E.V.; Flores, C.E.; Murai, K.K.; Bochet, C.G.; Holtmaat, A.; et al. Activity-dependent structural plasticity of perisynaptic astrocytic domains promotes excitatory synapse stability. Curr. Biol. 2014, 24, 1679–1688. [Google Scholar] [CrossRef] [Green Version]
- Papouin, T.; Dunphy, J.; Tolman, M.; Foley, J.C.; Haydon, P.G. Astrocytic control of synaptic function. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372, 20160154. [Google Scholar] [CrossRef] [Green Version]
- Perea, G.; Araque, A. Properties of synaptically evoked astrocyte calcium signal reveal synaptic information processing by astrocytes. J. Neurosci. 2005, 25, 2192–2203. [Google Scholar] [CrossRef]
- Herde, M.K.; Bohmbach, K.; Domingos, C.; Vana, N.; Komorowska-Müller, J.A.; Passlick, S.; Schwarz, I.; Jackson, C.J.; Dietrich, D.; Schwarz, M.K. Local efficacy of glutamate uptake decreases with synapse size. Cell Rep. 2020, 32, 108182. [Google Scholar] [CrossRef] [PubMed]
- Henneberger, C.; Bard, L.; Panatier, A.; Reynolds, J.P.; Kopach, O.; Medvedev, N.I.; Minge, D.; Herde, M.K.; Anders, S.; Kraev, I. LTP induction boosts glutamate spillover by driving withdrawal of perisynaptic astroglia. Neuron 2020, 108, 919–936. [Google Scholar] [CrossRef] [PubMed]
- Umpierre, A.D.; Bystrom, L.L.; Ying, Y.; Liu, Y.U.; Worrell, G.; Wu, L.-J. Microglial calcium signaling is attuned to neuronal activity in awake mice. Elife 2020, 9, e56502. [Google Scholar] [CrossRef] [PubMed]
- Savage, J.C.; St-Pierre, M.-K.; Carrier, M.; El Hajj, H.; Novak, S.W.; Sanchez, M.G.; Cicchetti, F.; Tremblay, M.-È. Microglial physiological properties and interactions with synapses are altered at presymptomatic stages in a mouse model of Huntington’s disease pathology. J. Neuroinflamm. 2020, 17, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Blinzinger, K.; Kreutzberg, G. Displacement of synaptic terminals from regenerating motoneurons by microglial cells. Z. Fur Zellforsch. Mikrosk. Anat. 1968, 85, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, M.-È.; Marker, D.F.; Puccini, J.M.; Muly, E.C.; Lu, S.-M.; Gelbard, H.A. Ultrastructure of microglia-synapse interactions in the HIV-1 Tat-injected murine central nervous system. Commun. Integr. Biol. 2013, 6, e27670. [Google Scholar] [CrossRef] [PubMed]
- Savage, J.C.; St-Pierre, M.-K.; Hui, C.W.; Tremblay, M.-E. Microglial Ultrastructure in the Hippocampus of a Lipopolysaccharide-Induced Sickness Mouse Model. Front. Neurosci. 2019, 13, 1340. [Google Scholar] [CrossRef] [Green Version]
- Stevens, B.; Allen, N.J.; Vazquez, L.E.; Howell, G.R.; Christopherson, K.S.; Nouri, N.; Micheva, K.D.; Mehalow, A.K.; Huberman, A.D.; Stafford, B.; et al. The classical complement cascade mediates CNS synapse elimination. Cell 2007, 131, 1164–1178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef] [Green Version]
- Schecter, R.W.; Maher, E.E.; Welsh, C.A.; Stevens, B.; Erisir, A.; Bear, M.F. Experience-dependent synaptic plasticity in V1 occurs without microglial CX3CR1. J. Neurosci. 2017, 37, 10541–10553. [Google Scholar] [CrossRef] [PubMed]
- Sipe, G.; Lowery, R.; Tremblay, M.-È.; Kelly, E.; Lamantia, C.; Majewska, A. Microglial P2Y12 is necessary for synaptic plasticity in mouse visual cortex. Nat. Commun. 2016, 7, 1–15. [Google Scholar] [CrossRef]
- Lowery, R.L.; Tremblay, M.E.; Hopkins, B.E.; Majewska, A.K. The microglial fractalkine receptor is not required for activity-dependent plasticity in the mouse visual system. Glia 2017, 65, 1744–1761. [Google Scholar] [CrossRef]
- Stowell, R.D.; Sipe, G.O.; Dawes, R.P.; Batchelor, H.N.; Lordy, K.A.; Whitelaw, B.S.; Stoessel, M.B.; Bidlack, J.M.; Brown, E.; Sur, M. Noradrenergic signaling in the wakeful state inhibits microglial surveillance and synaptic plasticity in the mouse visual cortex. Nat. Neurosci. 2019, 22, 1782–1792. [Google Scholar] [CrossRef] [PubMed]
- Prada, I.; Gabrielli, M.; Turola, E.; Iorio, A.; D’Arrigo, G.; Parolisi, R.; De Luca, M.; Pacifici, M.; Bastoni, M.; Lombardi, M.; et al. Glia-to-neuron transfer of miRNAs via extracellular vesicles: A new mechanism underlying inflammation-induced synaptic alterations. Acta Neuropathol. 2018, 135, 529–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.; Zhu, L.; Ma, R.; Ren, J.; Wang, J.; Gao, S.; Yang, D.; Ning, K.; Ling, B.; Lu, B. Astrocytic miR-324-5p is essential for synaptic formation by suppressing the secretion of CCL5 from astrocytes. Cell Death Dis. 2019, 10, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Tsai, Y.-W.; Sung, H.-H.; Li, J.-C.; Yeh, C.-Y.; Chen, P.-Y.; Cheng, Y.-J.; Chen, C.-H.; Tsai, Y.-C.; Chien, C.-T. Glia-derived exosomal miR-274 targets Sprouty in trachea and synaptic boutons to modulate growth and responses to hypoxia. Proc. Natl. Acad. Sci. USA 2019, 116, 24651–24661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mannironi, C.; Biundo, A.; Rajendran, S.; De Vito, F.; Saba, L.; Caioli, S.; Zona, C.; Ciotti, T.; Caristi, S.; Perlas, E. miR-135a regulates synaptic transmission and anxiety-like behavior in amygdala. Mol. Neurobiol. 2018, 55, 3301–3315. [Google Scholar] [CrossRef] [PubMed]
- Pons-Espinal, M.; Gasperini, C.; Marzi, M.J.; Braccia, C.; Armirotti, A.; Pötzsch, A.; Walker, T.L.; Fabel, K.; Nicassio, F.; Kempermann, G. MiR-135a-5p is critical for exercise-induced adult neurogenesis. Stem Cell Rep. 2019, 12, 1298–1312. [Google Scholar] [CrossRef] [Green Version]
- Smrt, R.D.; Szulwach, K.E.; Pfeiffer, R.L.; Li, X.; Guo, W.; Pathania, M.; Teng, Z.Q.; Luo, Y.; Peng, J.; Bordey, A. MicroRNA miR-137 regulates neuronal maturation by targeting ubiquitin ligase mind bomb-1. Stem Cells 2010, 28, 1060–1070. [Google Scholar] [CrossRef] [Green Version]
- Peng, H.; Harvey, B.T.; Richards, C.I.; Nixon, K. Neuron-Derived Extracellular Vesicles Modulate Microglia Activation and Function. Biology 2021, 10, 948. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Cao, L.-L.; Wang, X.-P.; Guo, W.; Guo, R.-B.; Sun, Y.-Q.; Xue, T.-F.; Cai, Z.-Y.; Ji, J.; Cheng, H. Neuronal extracellular vesicle derived miR-98 prevents salvageable neurons from microglial phagocytosis in acute ischemic stroke. Cell Death Dis. 2021, 12, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.T.; Dorman, L.C.; Pan, S.; Vainchtein, I.D.; Han, R.T.; Nakao-Inoue, H.; Taloma, S.E.; Barron, J.J.; Molofsky, A.B.; Kheirbek, M.A. Microglial remodeling of the extracellular matrix promotes synapse plasticity. Cell 2020, 182, 388–403. [Google Scholar] [CrossRef] [PubMed]
- Tønnesen, J.; Inavalli, V.K.; Nägerl, U.V. Super-resolution imaging of the extracellular space in living brain tissue. Cell 2018, 172, 1108–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswas, S.; Bachay, G.; Chu, J.; Hunter, D.D.; Brunken, W.J. Laminin-dependent interaction between astrocytes and microglia: A role in retinal angiogenesis. Am. J. Pathol. 2017, 187, 2112–2127. [Google Scholar] [CrossRef] [Green Version]
- Stoyanov, S.; Sun, W.; Düsedau, H.P.; Cangalaya, C.; Choi, I.; Mirzapourdelavar, H.; Baidoe-Ansah, D.; Kaushik, R.; Neumann, J.; Dunay, I.R. Attenuation of the extracellular matrix restores microglial activity during the early stage of amyloidosis. Glia 2021, 69, 182–200. [Google Scholar] [CrossRef] [PubMed]
- Crapser, J.D.; Spangenberg, E.E.; Barahona, R.A.; Arreola, M.A.; Hohsfield, L.A.; Green, K.N. Microglia facilitate loss of perineuronal nets in the Alzheimer’s disease brain. EBioMedicine 2020, 58, 102919. [Google Scholar] [CrossRef] [PubMed]
- Strackeljan, L.; Baczynska, E.; Cangalaya, C.; Baidoe-Ansah, D.; Wlodarczyk, J.; Kaushik, R.; Dityatev, A. Microglia Depletion-Induced Remodeling of Extracellular Matrix and Excitatory Synapses in the Hippocampus of Adult Mice. Cells 2021, 10, 1862. [Google Scholar] [CrossRef] [PubMed]
- Alberini, C.M.; Cruz, E.; Descalzi, G.; Bessières, B.; Gao, V. Astrocyte glycogen and lactate: New insights into learning and memory mechanisms. Glia 2018, 66, 1244–1262. [Google Scholar] [CrossRef] [PubMed]
- Charvériat, M.; Guiard, B.P. Serotonergic neurons in the treatment of mood disorders: The dialogue with astrocytes. Prog. Brain Res. 2021, 259, 197–228. [Google Scholar] [PubMed]
- Pfeiffer, T.; Avignone, E.; Nägerl, U.V. Induction of hippocampal long-term potentiation increases the morphological dynamics of microglial processes and prolongs their contacts with dendritic spines. Sci. Rep. 2016, 6, 32422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stellwagen, D.; Malenka, R.C. Synaptic scaling mediated by glial TNF-α. Nature 2006, 440, 1054–1059. [Google Scholar] [CrossRef] [PubMed]
- Saw, G.; Krishna, K.; Gupta, N.; Soong, T.W.; Mallilankaraman, K.; Sajikumar, S.; Dheen, S.T. Epigenetic regulation of microglial phosphatidylinositol 3-kinase pathway involved in long-term potentiation and synaptic plasticity in rats. Glia 2020, 68, 656–669. [Google Scholar] [CrossRef] [PubMed]
- Sa de Almeida, J.; Vargas, M.; Fonseca-Gomes, J.; Tanqueiro, S.R.; Belo, R.F.; Miranda-Lourenço, C.; Sebastião, A.M.; Diógenes, M.J.; Pais, T.F. Microglial Sirtuin 2 Shapes Long-Term Potentiation in Hippocampal Slices. Front. Neurosci. 2020, 14, 614. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, K.; Hayakawa, M.; Morimoto, Y. Minocycline prevents the impairment of hippocampal long-term potentiation in the septic mouse. Shock. Inj. Inflamm. Sepsis Lab. Clin. Approaches 2017, 48, 209–214. [Google Scholar] [CrossRef] [Green Version]
- Pribiag, H.; Stellwagen, D. TNF-α downregulates inhibitory neurotransmission through protein phosphatase 1-dependent trafficking of GABAA receptors. J. Neurosci. 2013, 33, 15879–15893. [Google Scholar] [CrossRef]
- Yu, T.; Zhang, X.; Shi, H.; Tian, J.; Sun, L.; Hu, X.; Cui, W.; Du, D. P2Y12 regulates microglia activation and excitatory synaptic transmission in spinal lamina II neurons during neuropathic pain in rodents. Cell Death Dis. 2019, 10, 165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reischer, G.; Heinke, B.; Sandkühler, J. Interferon-γ facilitates the synaptic transmission between primary afferent C-fibres and lamina I neurons in the rat spinal dorsal horn via microglia activation. Mol. Pain 2020, 16, 1744806920917249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neniskyte, U.; Vilalta, A.; Brown, G.C. Tumour necrosis factor alpha-induced neuronal loss is mediated by microglial phagocytosis. FEBS Lett. 2014, 588, 2952–2956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bassett, B.; Subramaniyam, S.; Fan, Y.; Varney, S.; Pan, H.; Carneiro, A.M.; Chung, C.Y. Minocycline alleviates depression-like symptoms by rescuing decrease in neurogenesis in dorsal hippocampus via blocking microglia activation/phagocytosis. Brain Behav. Immun. 2021, 91, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Hornik, T.C.; Vilalta, A.; Brown, G.C. Activated microglia cause reversible apoptosis of pheochromocytoma cells, inducing their cell death by phagocytosis. J. Cell Sci. 2016, 129, 65–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knafo, S.; Alonso-Nanclares, L.; Gonzalez-Soriano, J.; Merino-Serrais, P.; Fernaud-Espinosa, I.; Ferrer, I.; DeFelipe, J. Widespread changes in dendritic spines in a model of Alzheimer’s disease. Cereb. Cortex 2009, 19, 586–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fatemi, S.H.; Reutiman, T.J.; Folsom, T.D.; Thuras, P.D. GABA A receptor downregulation in brains of subjects with autism. J. Autism Dev. Disord. 2009, 39, 223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, R.C.; McCollum, L.A.; Schoonover, K.E.; Mabry, S.J.; Roche, J.K.; Lahti, A.C. Ultrastructural evidence for glutamatergic dysregulation in schizophrenia. Schizophr. Res. 2020, 31, S0920–S9964. [Google Scholar] [CrossRef] [PubMed]
- Mabry, S.J.; McCollum, L.A.; Farmer, C.B.; Bloom, E.S.; Roberts, R.C. Evidence for altered excitatory and inhibitory tone in the post-mortem substantia nigra in schizophrenia. World J. Biol. Psychiatry 2019, 21, 339–356. [Google Scholar] [CrossRef] [PubMed]
- Mitew, S.; Kirkcaldie, M.T.; Dickson, T.C.; Vickers, J.C. Altered synapses and gliotransmission in Alzheimer’s disease and AD model mice. Neurobiol. Aging 2013, 34, 2341–2351. [Google Scholar] [CrossRef] [PubMed]
- Kurucu, H.; Colom-Cadena, M.; Davies, C.; Wilkins, L.; King, D.; Rose, J.; Tzioras, M.; Tulloch, J.H.; Smith, C.; Spires-Jones, T.L. Inhibitory synapse loss and accumulation of amyloid beta in inhibitory presynaptic terminals in Alzheimer’s disease. Eur. J. Neurol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Petrache, A.L.; Rajulawalla, A.; Shi, A.; Wetzel, A.; Saito, T.; Saido, T.C.; Harvey, K.; Ali, A.B. Aberrant excitatory–inhibitory synaptic mechanisms in entorhinal cortex microcircuits during the pathogenesis of Alzheimer’s disease. Cereb. Cortex 2019, 29, 1834–1850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davenport, E.C.; Szulc, B.R.; Drew, J.; Taylor, J.; Morgan, T.; Higgs, N.F.; López-Doménech, G.; Kittler, J.T. Autism and schizophrenia-associated CYFIP1 regulates the balance of synaptic excitation and inhibition. Cell Rep. 2019, 26, 2037–2051. [Google Scholar] [CrossRef] [Green Version]
- Howell, B.W.; Smith, K.M. Synaptic structural protein dysfunction leads to altered excitation inhibition ratios in models of autism spectrum disorder. Pharmacol. Res. 2019, 139, 207–214. [Google Scholar] [CrossRef]
- Yang, J.; Yang, H.; Liu, Y.; Li, X.; Qin, L.; Lou, H.; Duan, S.; Wang, H. Astrocytes contribute to synapse elimination via type 2 inositol 1,4,5-trisphosphate receptor-dependent release of ATP. Elife 2016, 5, e15043. [Google Scholar] [CrossRef] [Green Version]
- Chung, W.S.; Clarke, L.E.; Wang, G.X.; Stafford, B.K.; Sher, A.; Chakraborty, C.; Joung, J.; Foo, L.C.; Thompson, A.; Chen, C.; et al. Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature 2013, 504, 394–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Yue, H.; Hu, Z.; Shen, Y.; Ma, J.; Li, J.; Wang, X.-D.; Wang, L.; Sun, B.; Shi, P. Microglia mediate forgetting via complement-dependent synaptic elimination. Science 2020, 367, 688–694. [Google Scholar] [CrossRef]
- Choudhury, M.E.; Miyanishi, K.; Takeda, H.; Islam, A.; Matsuoka, N.; Kubo, M.; Matsumoto, S.; Kunieda, T.; Nomoto, M.; Yano, H. Phagocytic elimination of synapses by microglia during sleep. Glia 2020, 68, 44–59. [Google Scholar] [CrossRef] [Green Version]
- Miyanishi, K.; Sato, A.; Kihara, N.; Utsunomiya, R.; Tanaka, J. Synaptic elimination by microglia and disturbed higher brain functions. Neurochem. Int. 2021, 142, 104901. [Google Scholar] [CrossRef]
- Henstridge, C.M.; Tzioras, M.; Paolicelli, R.C. Glial contribution to excitatory and inhibitory synapse loss in neurodegeneration. Front. Cell Neurosci. 2019, 13, 63. [Google Scholar] [CrossRef] [Green Version]
- Um, J.W. Roles of glial cells in sculpting inhibitory synapses and neural circuits. Front. Mol. Neurosci. 2017, 10, 381. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, H.; Abe, M.; Morimoto, C.; Iida, T.; Okabe, S.; Sakimura, K.; Hashimoto, K. Microglia permit climbing fiber elimination by promoting GABAergic inhibition in the developing cerebellum. Nat. Commun. 2018, 9, 1–15. [Google Scholar] [CrossRef]
- Cantaut-Belarif, Y.; Antri, M.; Pizzarelli, R.; Colasse, S.; Vaccari, I.; Soares, S.; Renner, M.; Dallel, R.; Triller, A.; Bessis, A. Microglia control the glycinergic but not the GABAergic synapses via prostaglandin E2 in the spinal cord. J. Cell Biol. 2017, 216, 2979–2989. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.; Yao, W.; Tambini, M.D.; Yin, T.; Norris, K.A.; D’Adamio, L. Microglia TREM2R47H alzheimer-linked variant enhances excitatory transmission and reduces LTP via increased TNF-α levels. Elife 2020, 9, e57513. [Google Scholar] [CrossRef] [PubMed]
- Semyanov, A.; Verkhratsky, A. Astrocytic processes: From tripartite synapses to the active milieu. Trends Neurosci. 2021, 44, 781–792. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Neuronal gp130 signalling via IL-6R activates STAT3, which causes miR-21-5p upregulation, inhibition of PTEN, and decreased Caspase-3 and -9, thus reducing the likelihood of apoptosis; miR-21-5p upregulation also results in the release of miR-21-5p exosomes.
Figure 1.
Neuronal gp130 signalling via IL-6R activates STAT3, which causes miR-21-5p upregulation, inhibition of PTEN, and decreased Caspase-3 and -9, thus reducing the likelihood of apoptosis; miR-21-5p upregulation also results in the release of miR-21-5p exosomes.

Figure 2.
Schematic illustration of the penta-partite synapse consisting of the pre- and post-synaptic neuronal membranes, astrocyte processes (green), extracellular matrix components (dots) and a microglial process (orange) that may be transiently present.
Figure 2.
Schematic illustration of the penta-partite synapse consisting of the pre- and post-synaptic neuronal membranes, astrocyte processes (green), extracellular matrix components (dots) and a microglial process (orange) that may be transiently present.


{kind=link}
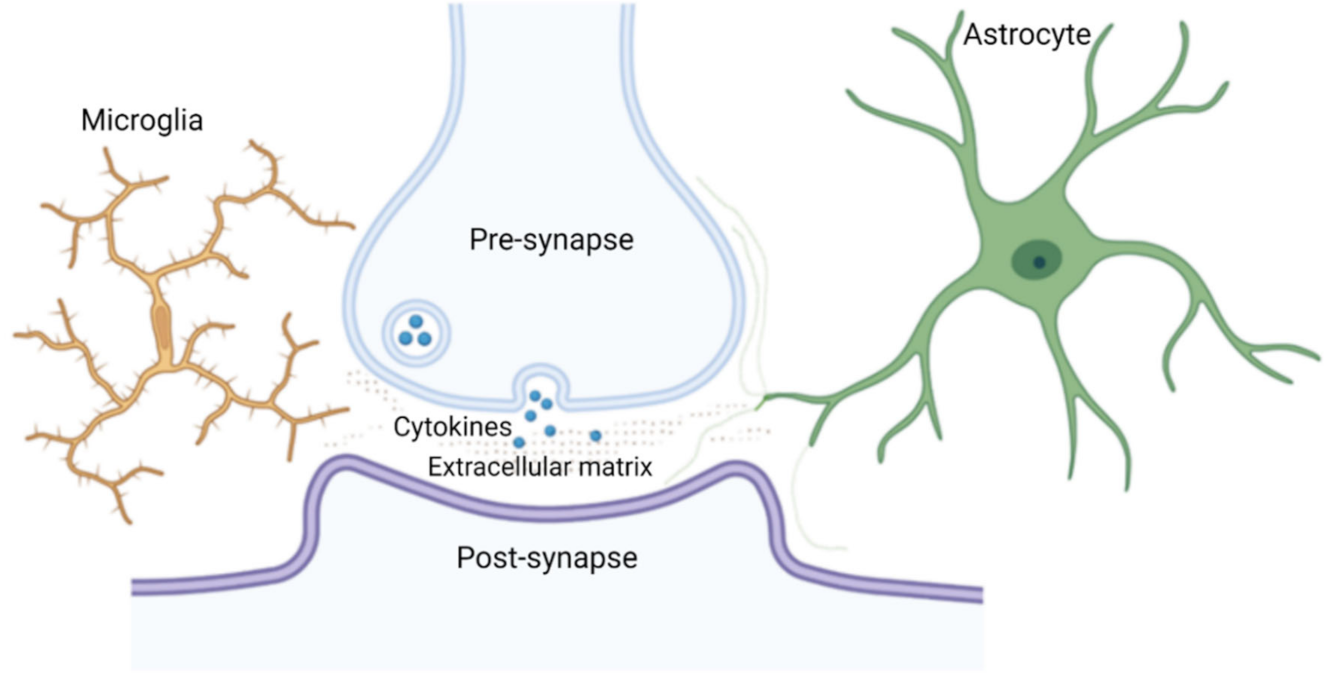{kind=link}
Table 1.
Cytokines and mRNA transcripts for cytokine receptors identified in unstimulated human, rat and mice microglia and neurons.
Table 1.
Cytokines and mRNA transcripts for cytokine receptors identified in unstimulated human, rat and mice microglia and neurons.
| Cytokine Family | Cytokines | mRNA Transcripts for Cytokine Receptors | Cell-Type |
|---|---|---|---|
| Interleukins | IL-1β IL-6 IL-8 IL-10 IL-12 IL-15 | IL1-RI IL1-RII IL-5R IL-6R IL-8R IL-9R IL-10R IL-12R IL-13R IL-15R gp130 | Human microglia [66] |
| TNF | TNF-α | TNFRI TNFRII | Human microglia [66] |
| Macrophage Inflammatory Proteins | MIP-1α MIP-1β MCP-1 | Human microglia [66] | |
| CXC chemokines | CX3CL1 SDF-1α CXCL12 | CX3CR1 CXCR4/CXCR7 | Human, mice microglia [67,68] Rat, mice neuron [69,70,71,72,73] |
| CC chemokines | MCP-1 (CCL2) MIP-1α (CCL3) MIP-1β (CCL4) RANTES (CCL5) C10/MRP-1 (CCL6) | Rat, mice neurons [69,70,71,72,73] and mice microglia [74] |
Table 2.
Common microglial mediators and their receptors involved in synaptic plasticity.
| Mediators | Receptor | Receptor Family |
|---|---|---|
| ATP, ADP | P2X4R, P2X7R | Purinergic |
| P2Y2R, P2Y4R, P2Y6R, P2Y12R, P2Y14R | ||
| A1R, A2AR, A2BR, A3R | Adenosine | |
| CX3CL1 | CX3CR1 | CX3 chemokine |
| CXCL10 | CXCR3 | CXC chemokine |
| CXCL12 | CXCR4 | |
| CCL2 CCL3 CCL5 | CCR2 CCR3 CCR1, CCR3, CCR5 | C-C chemokine |
| Glutamate | NMDAR | Ionotropic glutamate |
| IL-1β, TNF-α | AMPAR | |
| GABA | GABAAR GABABR | GABA |
| IL-1β | IL-1R1 | Interleukin-1 |
| C3, C1q | CR3 | Complement |
| CD200 | CD200R | OX-2 membrane glycoprotein |
| TGF-β1 | TGF-β1R | Transforming growth factor |
| NA | β2-AR | Adrenoceptor family of the 7-transmembrane superfamily |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Aramideh, J.A.; Vidal-Itriago, A.; Morsch, M.; Graeber, M.B. Cytokine Signalling at the Microglial Penta-Partite Synapse. Int. J. Mol. Sci. 2021, 22, 13186. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222413186
AMA Style
Aramideh JA, Vidal-Itriago A, Morsch M, Graeber MB. Cytokine Signalling at the Microglial Penta-Partite Synapse. International Journal of Molecular Sciences. 2021; 22(24):13186. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222413186
Chicago/Turabian StyleAramideh, Jason Abbas, Andres Vidal-Itriago, Marco Morsch, and Manuel B. Graeber. 2021. "Cytokine Signalling at the Microglial Penta-Partite Synapse" International Journal of Molecular Sciences 22, no. 24: 13186. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222413186
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.