
Drug Discovery for Mycobacterium tuberculosis Using Structure-Based Computer-Aided Drug Design Approach

 , ,
, ,
Abstract
:1. Introduction
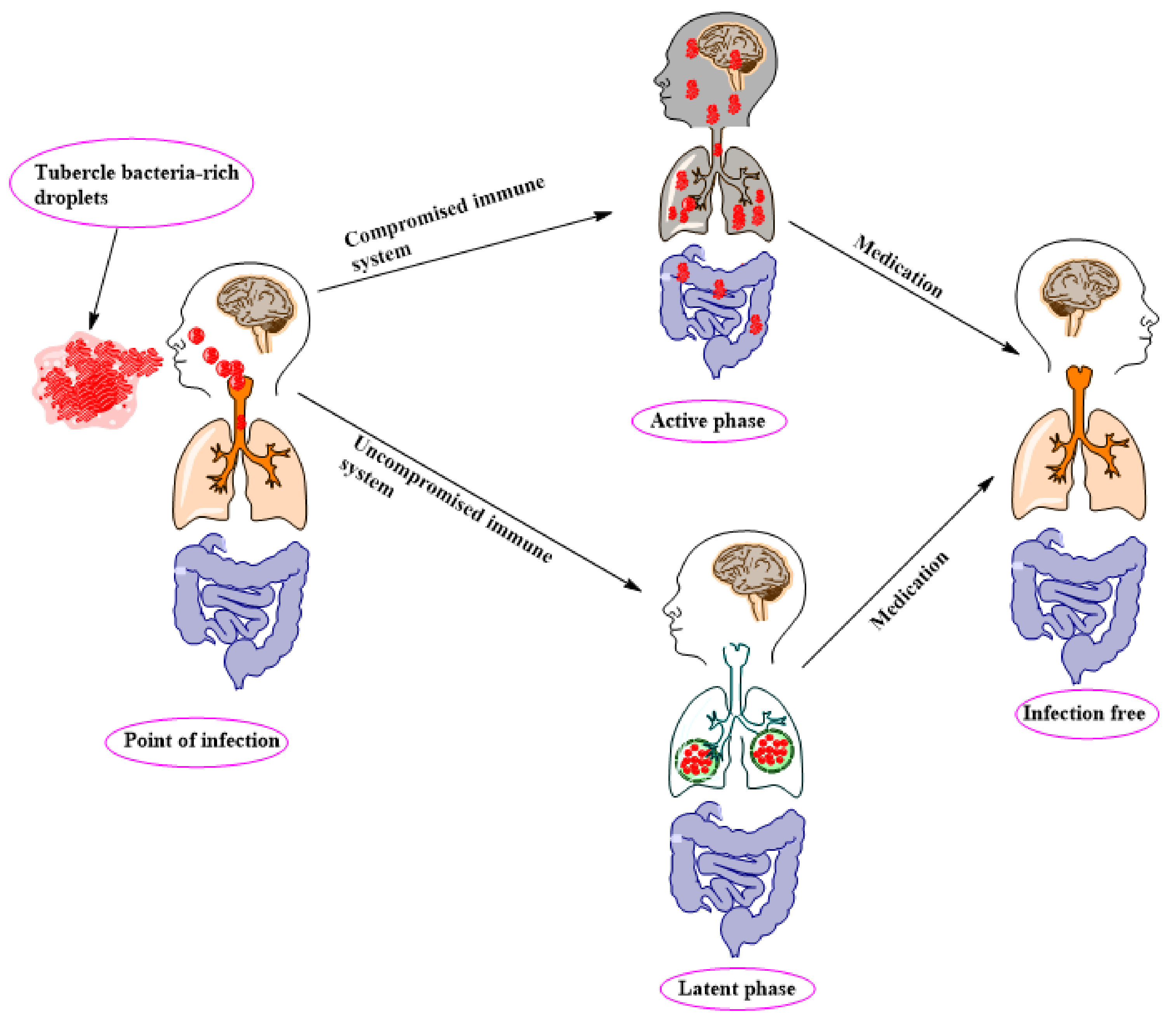
2. TB Pathology, Management, and Control
2.1. TB Drug Management and Classification
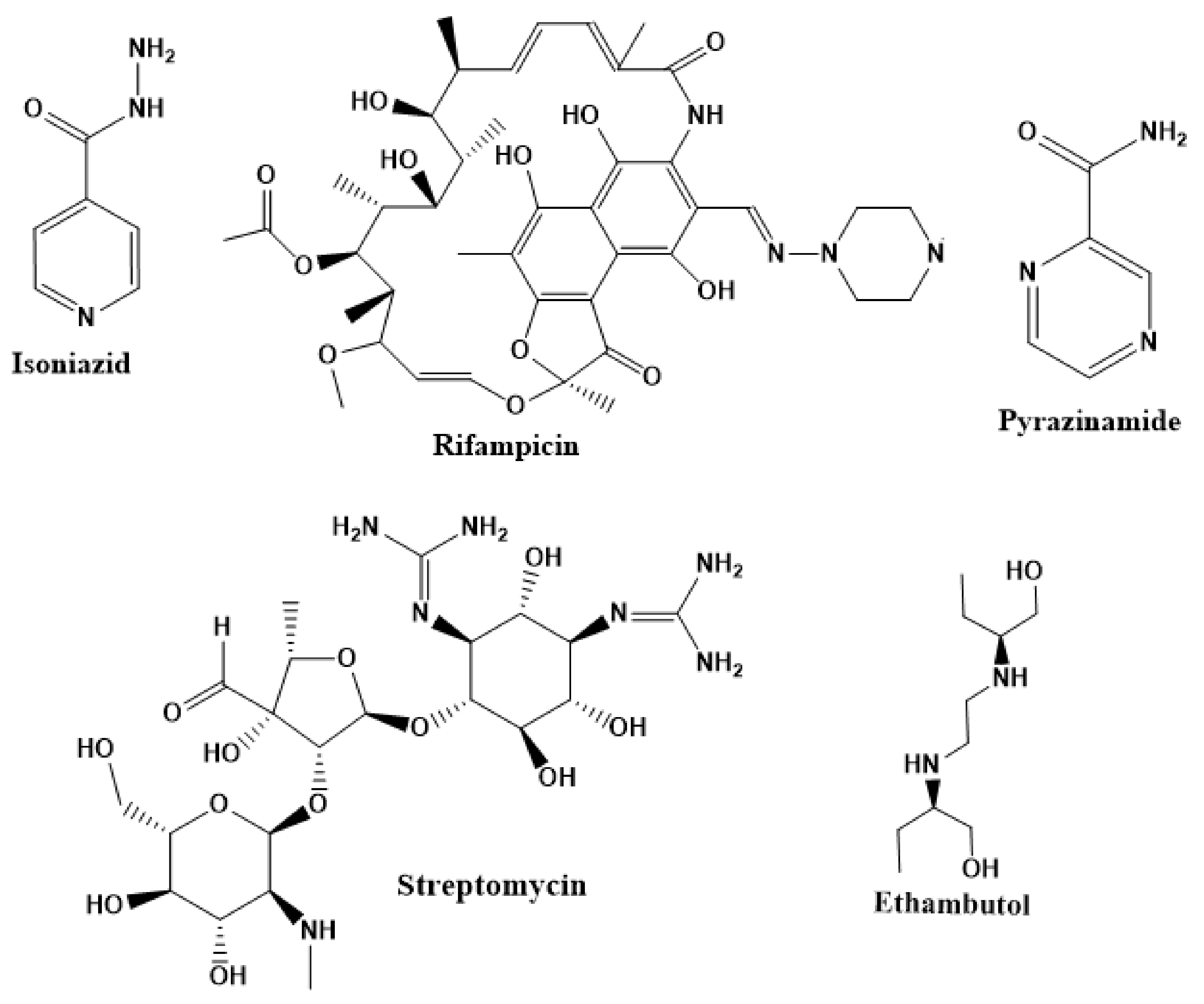
2.2. First-Line Drugs
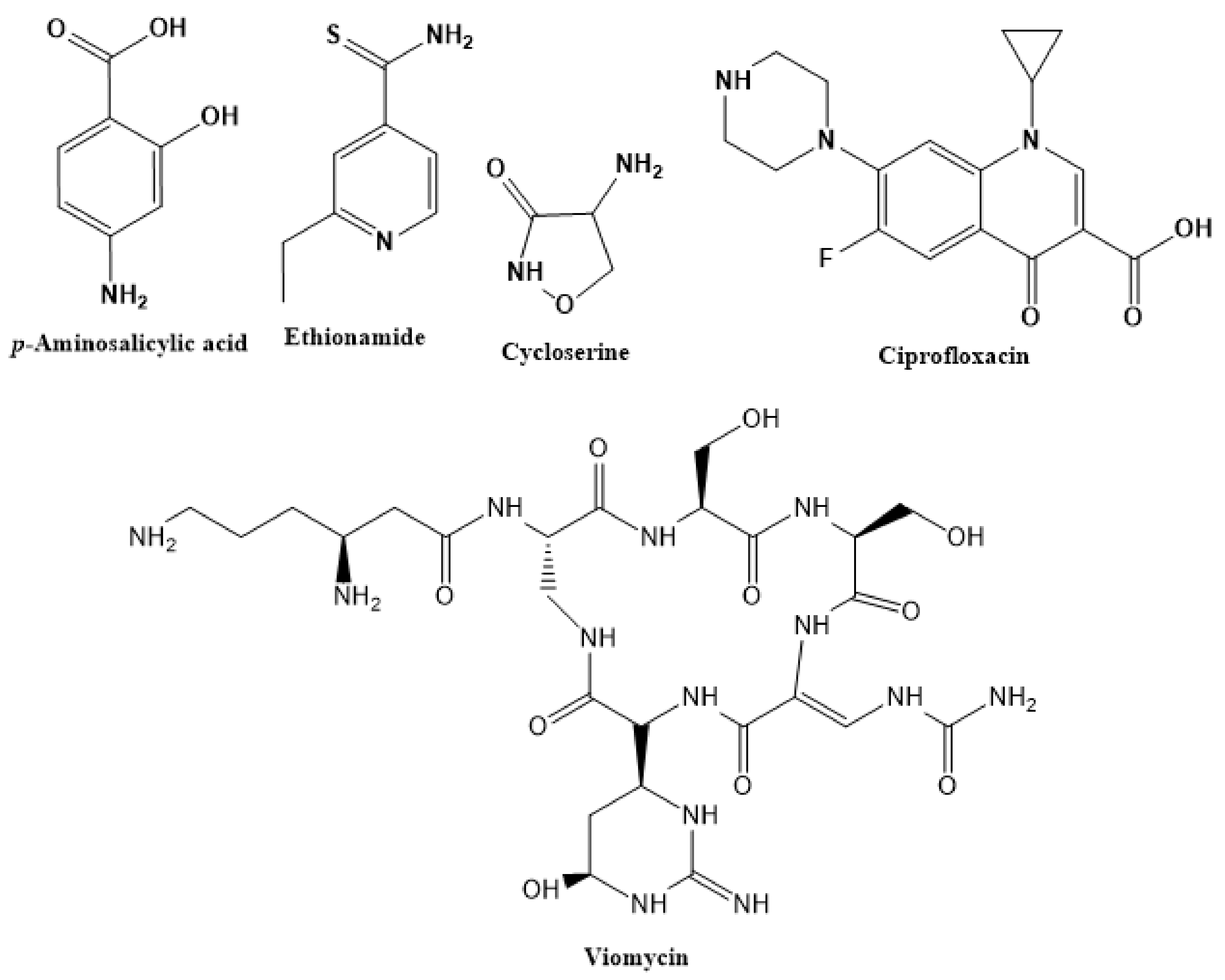
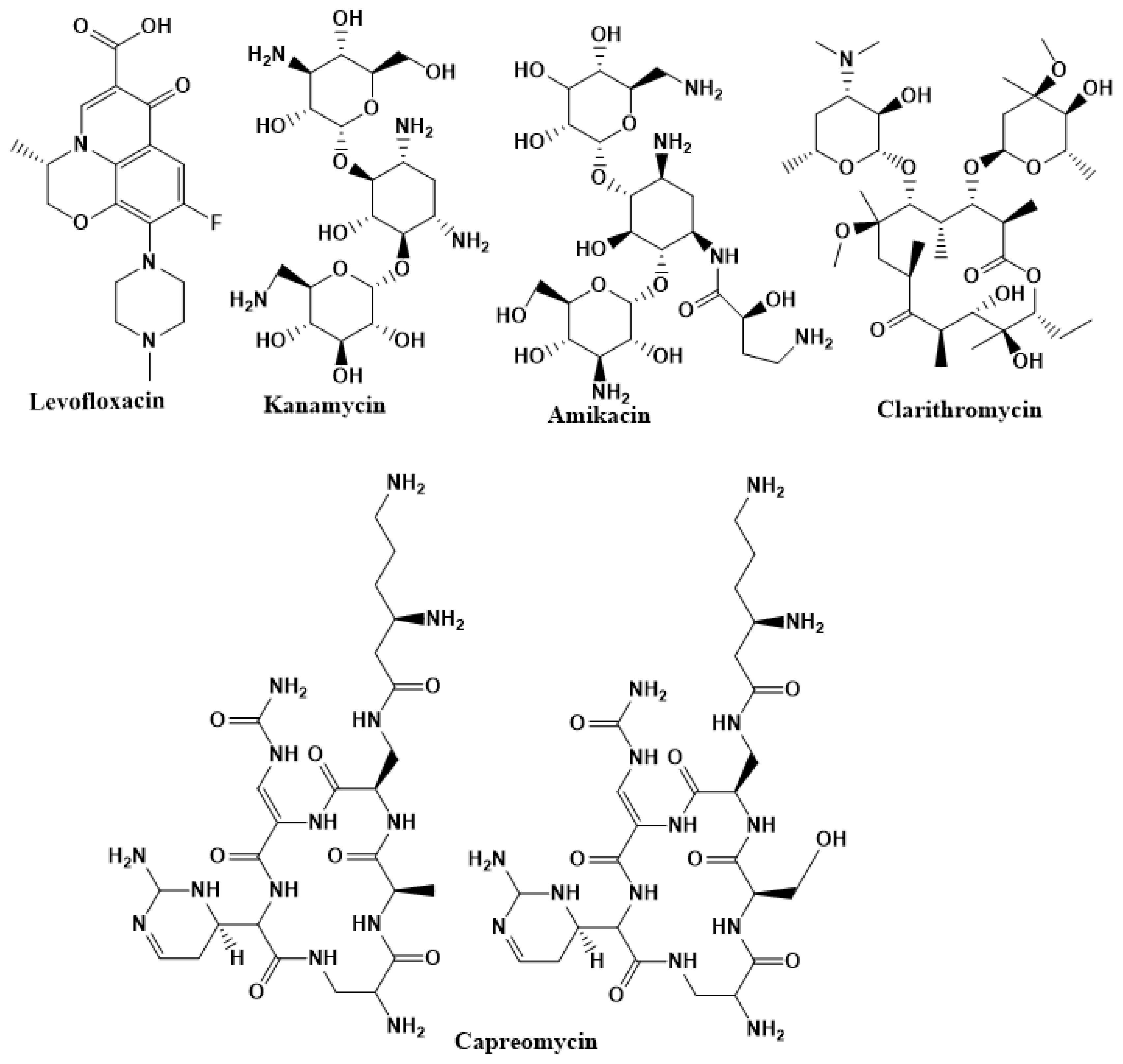
2.3. Second-Line Drugs
2.4. Emergence and Treatment of Multi-Drug Resistant TB (MDR-TB) and Extensively Drug-Resistant TB (XDR-TB)
2.5. Current TB Drugs’ Mechanism and Resistance Development
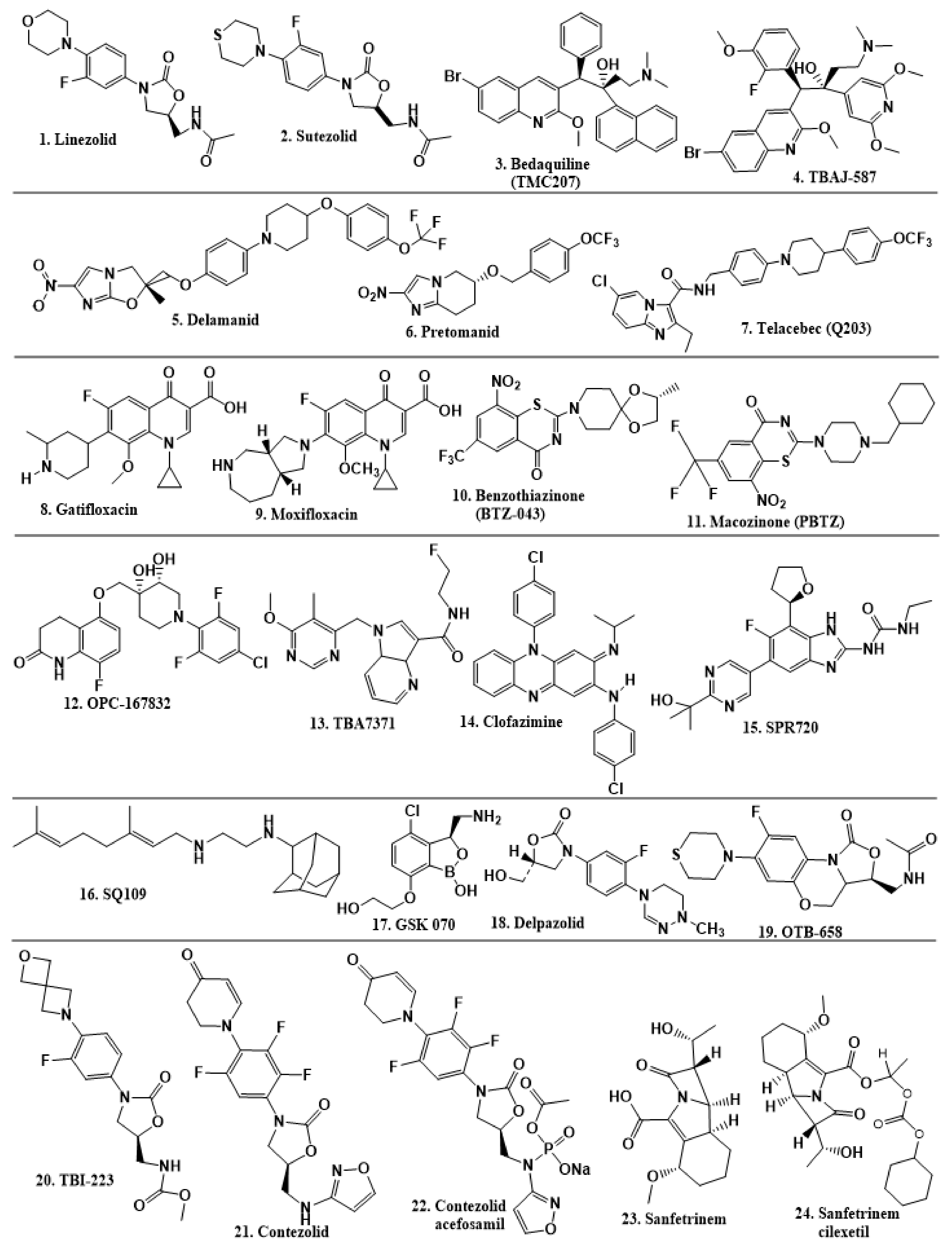
2.6. New TB Drugs Discovered through HTS and Other Approaches
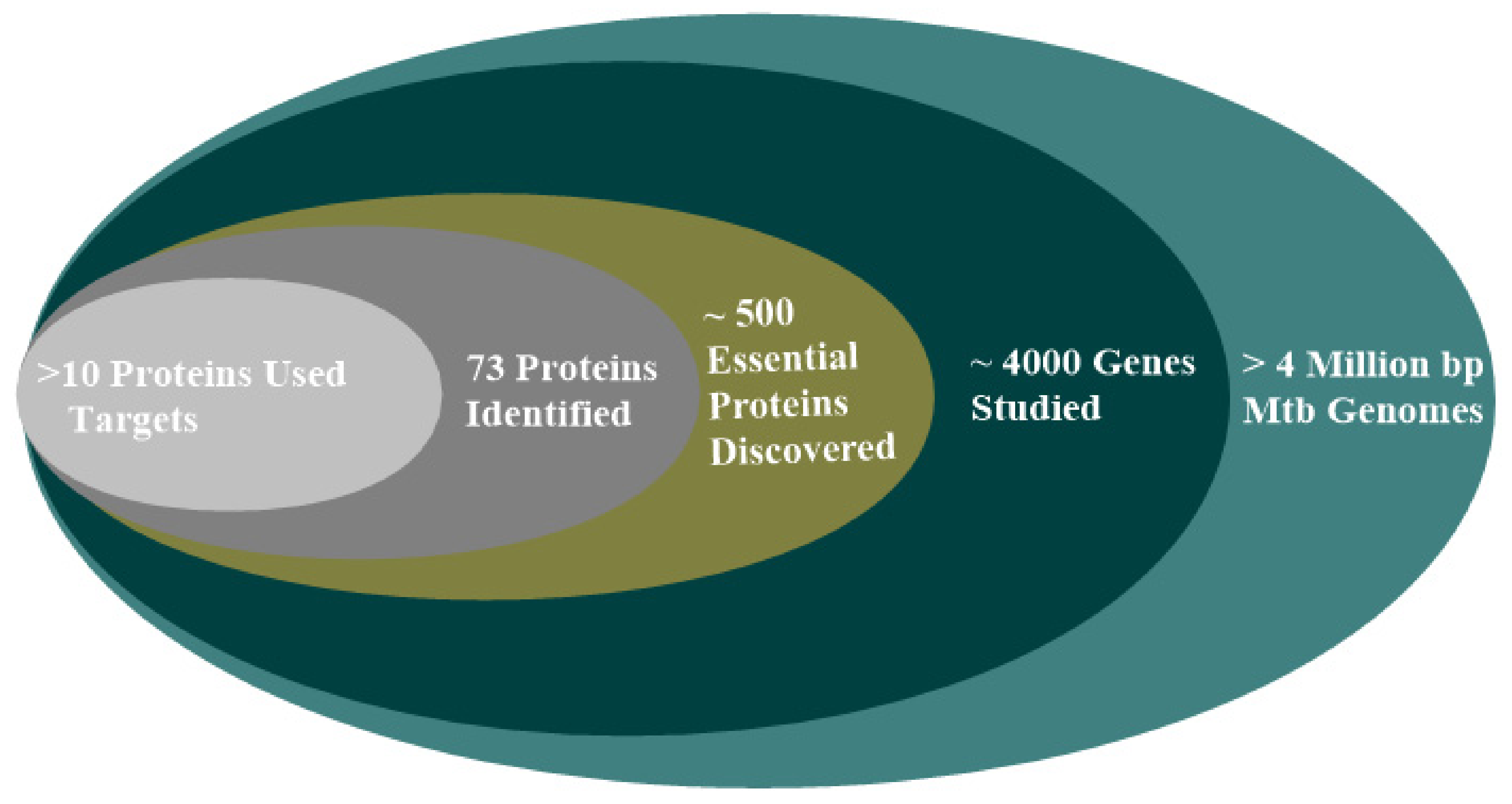
2.7. Protein Target in Mtb Drug Design
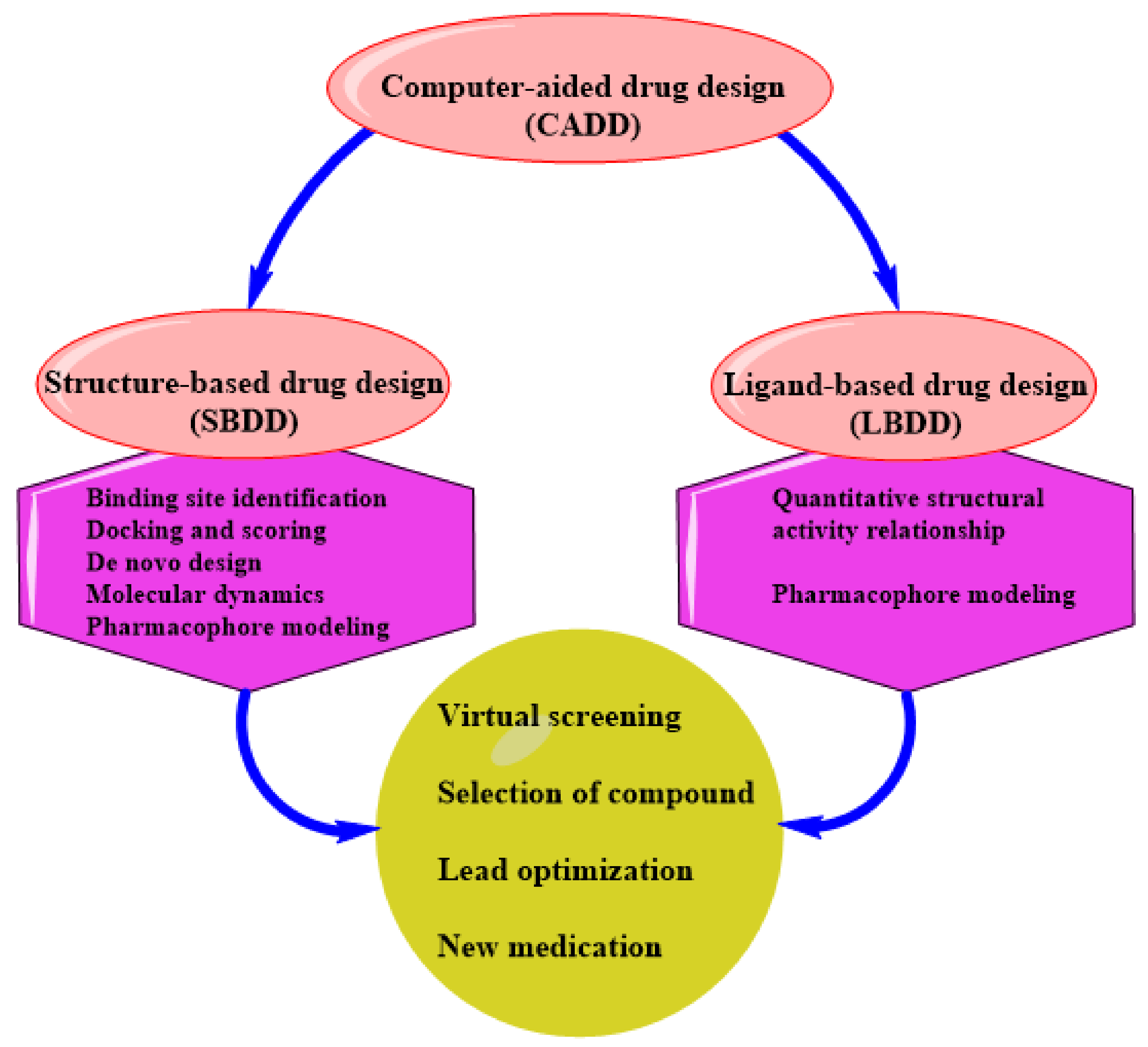
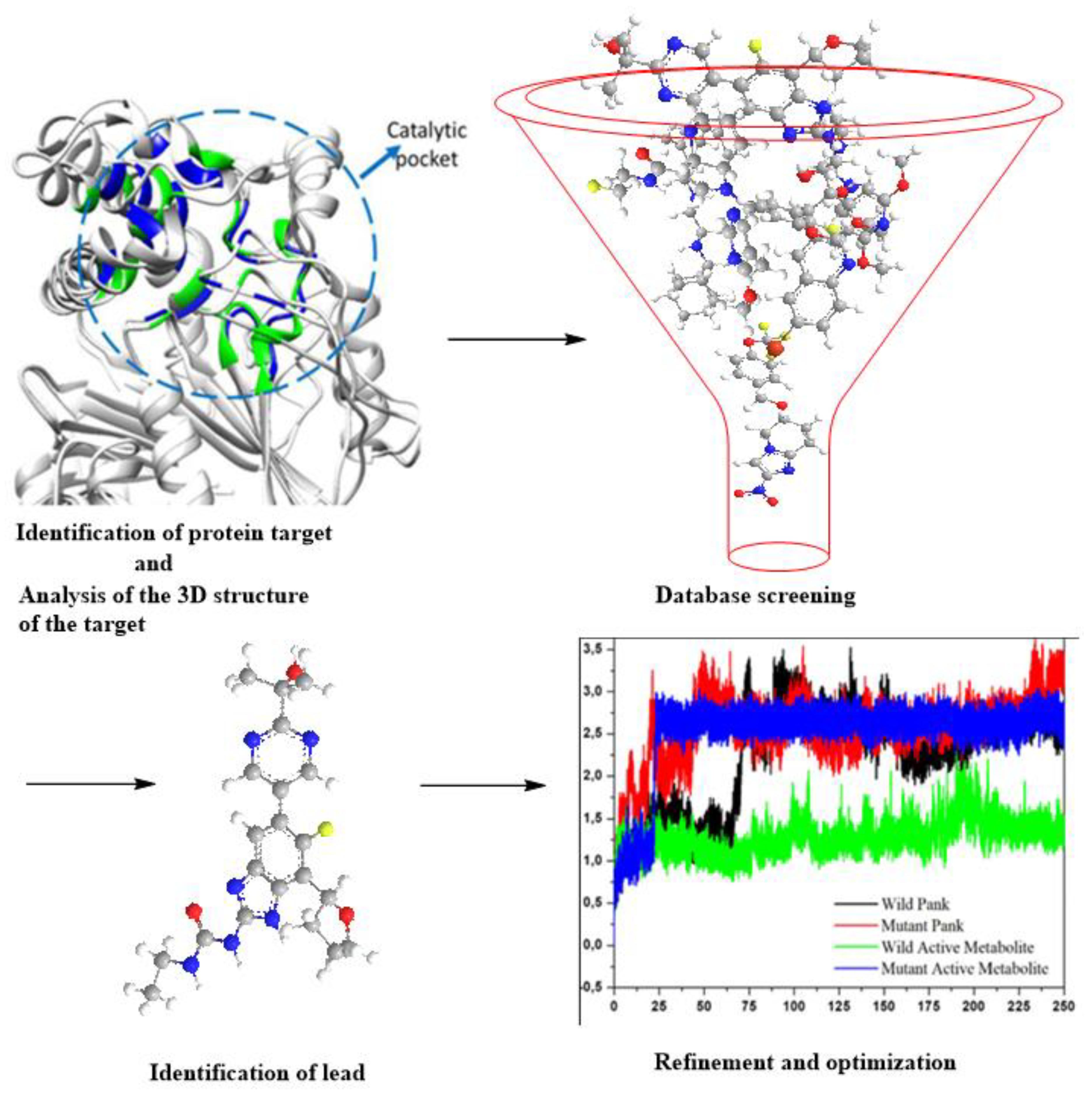
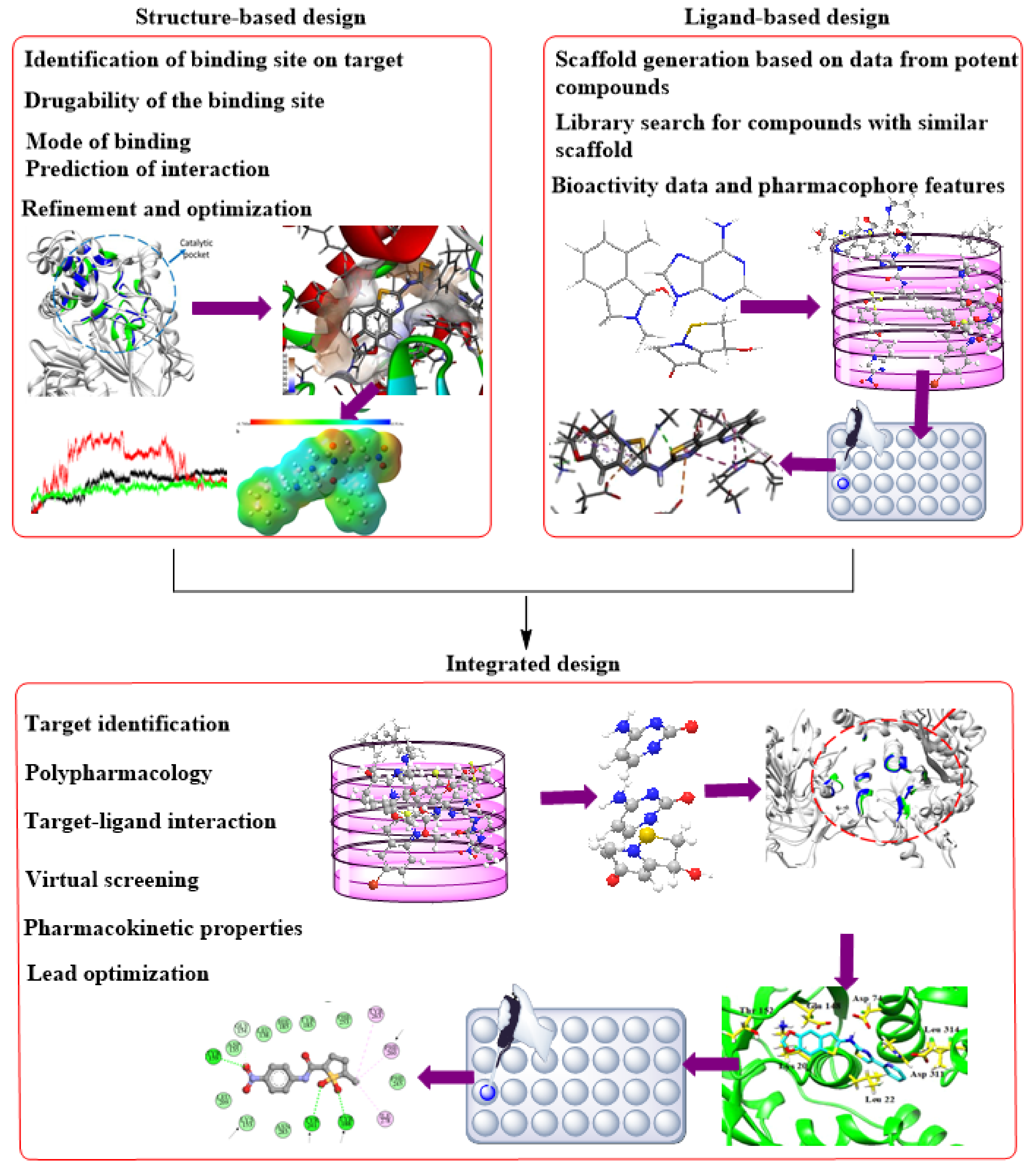
3. SBDD as an Indispensable Tool in Computational Drug Design
SBDD in Drug Discovery and Design
4. Status of Computational-Aided Drug Design and Discovery in TB
5. Data Application and Management in Tuberculosis Drug Development
5.1. SBDD Based on Mtb Proteins
5.2. Virtual Screening as a Method of Lead Identification
5.3. De Novo Drug Design—A Signature to the Drug Discovery Process
5.4. Molecular Docking and Density Functional Theory Applied to Mtb
5.5. Advantages and Drawbacks of Computational Methods
6. Conclusions and Future Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Macalino, S.J.Y.; Billones, J.B.; Organo, V.G.; Carrillo, M.; Constancia, O. In silico strategies in tuberculosis drug discovery. Molecules 2020, 25, 665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization; Stop TB Initiative (World Health Organization). Treatment of Tuberculosis: Guidelines; World Health Organization: Geneva, Switzerland, 2010. [Google Scholar]
- World Health Organization. Global Tuberculosis Report 2018; WHO: Geneva, Switzerland, 2018. [Google Scholar]
- Balganesh, T.S.; Alzari, P.M.; Cole, S.T. Rising standards for tuberculosis drug development. Trends Pharmacol. Sci. 2008, 29, 576–581. [Google Scholar] [CrossRef] [PubMed]
- Ballell, L.; Field, R.A.; Duncan, K.; Young, R.J. New small-molecule synthetic antimycobacterials. Antimicrob. Agents Chemother. 2005, 49, 2153–2163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ananthan, S.; Faaleolea, E.R.; Goldman, R.C.; Hobrath, J.V.; Kwong, C.D.; Laughon, B.E.; Maddry, J.A.; Mehta, A.; Rasmussen, L.; Reynolds, R.C.; et al. High-throughput screening for inhibitors of Mycobacterium tuberculosis H37Rv. Tuberculosis 2009, 89, 334–353. [Google Scholar] [CrossRef] [Green Version]
- Maddry, J.A.; Ananthan, S.; Goldman, R.C.; Hobrath, J.V.; Kwong, C.D.; Maddox, C.; Rasmussen, L.; Reynolds, R.C.; Secrist, J.A., III; Sosa, M.I.; et al. Antituberculosis activity of the molecular libraries screening center network library. Tuberculosis 2009, 89, 354–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynolds, R.C.; Ananthan, S.; Faaleolea, E.; Hobrath, J.V.; Kwong, C.D.; Maddox, C.; Rasmussen, L.; Sosa, M.I.; Thammasuvimol, E.; White, E.L.; et al. High throughput screening of a library based on kinase inhibitor scaffolds against Mycobacterium tuberculosis H37Rv. Tuberculosis 2012, 92, 72–83. [Google Scholar] [CrossRef] [Green Version]
- Petersen, E.; Maeurer, M.; Marais, B.; Migliori, G.B.; Mwaba, P.; Ntoumi, F.; Vilaplana, C.; Kim, K.; Schito, M.; Zumla, A. World TB day 2017: Advances, challenges and opportunities in the “end-TB” era. Int. J. Infect. Dis. 2017, 56, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Liu, N.; Cummings, J.E.; England, K.; Slayden, R.A.; Tonge, P.J. Mechanism and inhibition of the FabI enoyl-ACP reductase from Burkholderia pseudomallei. J. Antimicrob. Chemother. 2011, 66, 564–573. [Google Scholar] [CrossRef] [Green Version]
- England, K.; Am Ende, C.; Lu, H.; Sullivan, T.J.; Marlenee, N.L.; Bowen, R.A.; Knudson, S.E.; Knudson, D.L.; Tonge, P.J.; Slayden, R.A. Substituted diphenyl ethers as a broad-spectrum platform for the development of chemotherapeutics for the treatment of tularaemia. J. Antimicrob. Chemother. 2009, 64, 1052–1061. [Google Scholar] [CrossRef]
- Xu, H.; Sullivan, T.J.; Sekiguchi, J.; Kirikae, T.; Ojima, I.; Stratton, C.F.; Mao, W.; Rock, F.L.; Alley, M.R.K.; Johnson, F.; et al. Mechanism and inhibition of saFabI, the enoyl reductase from Staphylococcus aureus. Biochemistry 2008, 47, 4228–4236. [Google Scholar] [CrossRef] [Green Version]
- Tipparaju, S.K.; Mulhearn, D.C.; Klein, G.M.; Chen, Y.; Tapadar, S.; Bishop, M.H.; Yang, S.; Chen, J.; Ghassemi, M.; Santarsiero, B.D.; et al. Design and Synthesis of Aryl Ether Inhibitors of the Bacillus Anthracis Enoyl–ACP Reductase. ChemMedChem 2008, 3, 1250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lobanov, V. Using artificial neural networks to drive virtual screening of combinatorial libraries. Drug Discov. Today BIOSILICO 2004, 2, 149–156. [Google Scholar] [CrossRef]
- Song, C.M.; Lim, S.J.; Tong, J.C. Recent advances in computer-aided drug design. Brief. Bioinform. 2009, 10, 579–591. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L. The many roles of computation in drug discovery. Science 2004, 303, 1813–1818. [Google Scholar] [CrossRef]
- Xiang, M.; Cao, Y.; Fan, W.; Chen, L.; Mo, Y. Computer-aided drug design: Lead discovery and optimization. Comb. Chem. High Throughput Screen. 2012, 15, 328–337. [Google Scholar] [CrossRef]
- Zhang, S. Computer-aided drug discovery and development. Drug Des. Discov. 2011, 716, 23–38. [Google Scholar]
- Berman, H.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- RSCB-PDB. A Structural View of Biology. 2021. Available online: https://www.rcsb.org/ (accessed on 8 November 2021).
- Bruch, E.M.; Petrella, S.; Bellinzoni, M. Structure-Based Drug Design for Tuberculosis: Challenges Still Ahead. Appl. Sci. 2020, 10, 4248. [Google Scholar] [CrossRef]
- Holton, S.J.; Weiss, M.S.; Tucker, P.A.; Wilmanns, M. Structure-based approaches to drug discovery against tuberculosis. Curr. Protein Pept. Sci. 2007, 8, 365–375. [Google Scholar] [CrossRef]
- Elkington, P.T.; Friedland, J.S. Permutations of time and place in tuberculosis. Lancet Infect. Dis. 2015, 15, 1357–1360. [Google Scholar] [CrossRef] [Green Version]
- Hong, B.-Y.; Maulén, N.P.; Adami, A.J.; Granados, H.; Balcells, M.E.; Cervantes, J. Microbiome changes during tuberculosis and antituberculous therapy. Clin. Microbiol. Rev. 2016, 29, 915–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Getahun, H.; Matteelli, A.; Chaisson, R.E.; Raviglione, M. Latent Mycobacterium tuberculosis infection. N. Engl. J. Med. 2015, 372, 2127–2135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, L.G.; Asch, S.M.; Yu, E.I.; Knowles, L.; Gelberg, L.; Davidson, P. A population-based survey of tuberculosis symptoms: How atypical are atypical presentations? Clin. Infect. Dis. 2000, 30, 293–299. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Global Tuberculosis Report 2019; World Health Organization: Geneva, Switzerland, 2019. [Google Scholar]
- Green, K.; Garneau-Tsodikova, S. Resistance in tuberculosis: What do we know and where can we go? Front. Microbiol. 2013, 4, 208. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. Global Tuberculosis Report 2020; World Health Organization: Geneva, Switzerland, 2020; pp. 115–127. [Google Scholar]
- World Health Organization. Guidelines on the Management of Latent Tuberculosis Infection; World Health Organization: Geneva, Switzerland, 2015. [Google Scholar]
- Horsburgh, C.R., Jr.; Barry, C.E., III; Lange, C. Treatment of tuberculosis. N. Engl. J. Med. 2015, 373, 2149–2160. [Google Scholar] [CrossRef]
- Mitchison, D.; Davies, G. The chemotherapy of tuberculosis: Past, present and future [State of the art]. Int. J. Tuberc. Lung Dis. 2012, 16, 724–732. [Google Scholar] [CrossRef]
- Zhang, Z.; Yan, J.; Xu, K.; Ji, Z.; Li, L. Tetrandrine reverses drug resistance in isoniazid and ethambutol dual drug-resistant Mycobacterium tuberculosis clinical isolates. BMC Infect. Dis. 2015, 15, 153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rich, M.; Jaramillo, E. Guidelines for the Programmatic Management of Drug-Resistant Tuberculosis; World Health Organization: Geneva, Switzerland, 2008. [Google Scholar]
- Claiborne, A.B.; Guenther, R.S.; English, R.A.; Nicholson, A. Developing and Strengthening the Global Supply Chain for Second-Line Drugs for Multidrug-Resistant Tuberculosis: Workshop Summary; National Academies Press: Washington, DC, USA, 2013. [Google Scholar]
- Wright, A.; Zignol, M. Anti-Tuberculosis Drug Resistance in the World: Fourth Global Report: The World Health Organization/International Union against Tuberculosis and Lung Disease (WHO/Union) Global Project on Anti-Tuberculosis Drug Resistance Surveillance, 2002–2007; World Health Organization: Geneva, Switzerland, 2008. [Google Scholar]
- Mahajan, R. Bedaquiline: First FDA-approved tuberculosis drug in 40 years. Int. J. Appl. Basic Med. Res. 2013, 3, 1. [Google Scholar] [CrossRef]
- Vilchèze, C.; Jacobs, W.R., Jr. The mechanism of isoniazid killing: Clarity through the scope of genetics. Annu. Rev. Microbiol. 2007, 61, 35–50. [Google Scholar] [CrossRef]
- Khan, S.R.; Morgan, A.G.M.; Michail, K.; Srivastava, N.; Whittal, R.M.; Aljuhani, N.; Siraki, A.G. Metabolism of isoniazid by neutrophil myeloperoxidase leads to isoniazid-NAD+ adduct formation: A comparison of the reactivity of isoniazid with its known human metabolites. Biochem. Pharmacol. 2016, 106, 46–55. [Google Scholar] [CrossRef]
- Preziosi, P. Isoniazid: Metabolic aspects and toxicological correlates. Curr. Drug Metab. 2007, 8, 839–851. [Google Scholar] [CrossRef]
- Rickman, K.A.; Swancutt, K.L.; Mezyk, S.P.; Kiddle, J.J. Isoniazid: Radical-induced oxidation and reduction chemistry. Bioorganic Med. Chem. Lett. 2013, 23, 3096–3100. [Google Scholar] [CrossRef]
- Timmins, G.S.; Deretic, V. Mechanisms of action of isoniazid. Mol. Microbiol. 2006, 62, 1220–1227. [Google Scholar] [CrossRef]
- Timmins, G.S.; Master, S.; Rusnak, F.; Deretic, V. Nitric oxide generated from isoniazid activation by KatG: Source of nitric oxide and activity against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2004, 48, 3006–3009. [Google Scholar] [CrossRef] [Green Version]
- Bulatovic, V.M.; Wengenack, N.L.; Uhl, J.R.; Hall, L.; Roberts, G.D.; Cockerill, F.R., III; Rusnak, F. Oxidative stress increases susceptibility of Mycobacterium tuberculosis to isoniazid. Antimicrob. Agents Chemother. 2002, 46, 2765–2771. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Li, X.; Huang, L.; Chan, J.; Chen, Y.; Deng, H.; Mi, K. Quantitative proteomics reveals novel insights into isoniazid susceptibility in mycobacteria mediated by a universal stress protein. J. Proteome Res. 2015, 14, 1445–1454. [Google Scholar] [CrossRef]
- Jena, L.; Waghmare, P.; Kashikar, S.; Kumar, S.; Harinath, B.C. Computational approach to understanding the mechanism of action of isoniazid, an anti-TB drug. Int. J. Mycobacteriol. 2014, 3, 276–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laurenzo, D.; Mousa, S.A. Mechanisms of drug resistance in Mycobacterium tuberculosis and current status of rapid molecular diagnostic testing. Acta Trop. 2011, 119, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.-L.; Sun, Q.; Liu, H.-C.; Wu, X.-C.; Xiao, T.-Y.; Zhao, X.-Q.; Li, G.-L.; Jiang, Y.; Zeng, C.-Y.; Wan, K.-L. Analysis of embCAB mutations associated with ethambutol resistance in multidrug-resistant Mycobacterium tuberculosis isolates from China. Antimicrob. Agents Chemother. 2015, 59, 2045–2050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Safi, H.; Sayers, B.; Hazbón, M.H.; Alland, D. Transfer of embB codon 306 mutations into clinical Mycobacterium tuberculosis strains alters susceptibility to ethambutol, isoniazid, and rifampin. Antimicrob. Agents Chemother. 2008, 52, 2027–2034. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Wang, X.; Cui, P.; Jin, J.; Chen, J.; Zhang, W.; Zhang, Y. ubiA (Rv3806c) encoding DPPR synthase involved in cell wall synthesis is associated with ethambutol resistance in Mycobacterium tuberculosis. Tuberculosis 2015, 95, 149–154. [Google Scholar] [CrossRef]
- Almeida Da Silva, P.E.; Palomino, J.C. Molecular basis and mechanisms of drug resistance in Mycobacterium tuberculosis: Classical and new drugs. J. Antimicrob. Chemother. 2011, 66, 1417–1430. [Google Scholar] [CrossRef]
- Zhang, Y.; Mitchison, D. The curious characteristics of pyrazinamide: A review. Int. J. Tuberc. Lung Dis. 2003, 7, 6–21. [Google Scholar]
- Zhang, Y.; Wade, M.M.; Scorpio, A.; Zhang, H.; Sun, Z. Mode of action of pyrazinamide: Disruption of Mycobacterium tuberculosis membrane transport and energetics by pyrazinoic acid. J. Antimicrob. Chemother. 2003, 52, 790–795. [Google Scholar] [CrossRef]
- Scorpio, A.; Zhang, Y. Mutations in pncA, a gene encoding pyrazinamidase/nicotinamidase, cause resistance to the antituberculous drug pyrazinamide in tubercle bacillus. Nat. Med. 1996, 2, 662–667. [Google Scholar] [CrossRef]
- Hirano, K.; Takahashi, M.; Kazumi, Y.; Fukasawa, Y.; Abe, C. Mutation in pncA is a major mechanism of pyrazinamide resistance in Mycobacterium tuberculosis. Tuber. Lung Dis. 1998, 78, 117–122. [Google Scholar] [CrossRef]
- Zhang, Y.; Shi, W.; Zhang, W.; Mitchison, D. Mechanisms of pyrazinamide action and resistance. Microbiol. Spectr. 2014, 2, MGM2-0023-2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemaitre, N.; Callebaut, I.; Frenois, F.; Jarlier, V.; Sougakoff, W. Study of the structure–activity relationships for the pyrazinamidase (PncA) from Mycobacterium tuberculosis. Biochem. J. 2001, 353, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Sheen, P.; Ferrer, P.; Gilman, R.H.; López-Llano, J.; Fuentes, P.; Valencia, E.; Zimic, M.J. Effect of pyrazinamidase activity on pyrazinamide resistance in Mycobacterium tuberculosis. Tuberculosis 2009, 89, 109–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zumla, A.; Nahid, P.; Cole, S.T. Advances in the development of new tuberculosis drugs and treatment regimens. Nat. Rev. Drug Discov. 2013, 12, 388–404. [Google Scholar] [CrossRef]
- Gillespie, S.H. Evolution of drug resistance in Mycobacterium tuberculosis: Clinical and molecular perspective. Antimicrob. Agents Chemother. 2002, 46, 267–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamoto, S.; Tamaru, A.; Nakajima, C.; Nishimura, K.; Tanaka, Y.; Tokuyama, S.; Suzuki, Y.; Ochi, K. Loss of a conserved 7-methylguanosine modification in 16S rRNA confers low-level streptomycin resistance in bacteria. Mol. Microbiol. 2007, 63, 1096–1106. [Google Scholar] [CrossRef]
- Carette, X.; Blondiaux, N.; Willery, E.; Hoos, S.; Lecat-Guillet, N.; Lens, Z.; Wohlkönig, A.; Wintjens, R.; Soror, S.H.; Frénois, F. Structural activation of the transcriptional repressor EthR from Mycobacterium tuberculosis by single amino acid change mimicking natural and synthetic ligands. Nucleic Acids Res. 2012, 40, 3018–3030. [Google Scholar] [CrossRef] [Green Version]
- DeBarber, A.E.; Mdluli, K.; Bosman, M.; Bekker, L.-G.; Barry, C.E. Ethionamide activation and sensitivity in multidrug-resistant Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2000, 97, 9677–9682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilchèze, C.; Av-Gay, Y.; Attarian, R.; Liu, Z.; Hazbón, M.H.; Colangeli, R.; Chen, B.; Liu, W.; Alland, D.; Sacchettini, J.C.; et al. Mycothiol biosynthesis is essential for ethionamide susceptibility in Mycobacterium tuberculosis. Mol. Microbiol. 2008, 69, 1316–1329. [Google Scholar] [CrossRef] [Green Version]
- Zhao, F.; Wang, X.-D.; Erber, L.N.; Luo, M.; Guo, A.-Z.; Yang, S.-S.; Gu, J.; Turman, B.J.; Gao, Y.-R.; Li, D.-F.; et al. Binding pocket alterations in dihydrofolate synthase confer resistance to para-aminosalicylic acid in clinical isolates of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2014, 58, 1479–1487. [Google Scholar] [CrossRef] [Green Version]
- Mathys, V.; Wintjens, R.; Lefevre, P.; Bertout, J.; Singhal, A.; Kiass, M.; Kurepina, N.; Wang, X.-M.; Mathema, B.; Baulard, A.; et al. Molecular genetics of para-aminosalicylic acid resistance in clinical isolates and spontaneous mutants of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2009, 53, 2100–2109. [Google Scholar] [CrossRef] [Green Version]
- Satta, G.; Witney, A.A.; Begum, N.; Canseco, J.O.; Boa, A.N.; McHugha, T.D. Role of Whole-Genome Sequencing in Characterizing the Mechanism of Action of para-Aminosalicylic Acid and Its Resistance. Antimicrob. Agents Chemother. 2020, 64, e00675-20. [Google Scholar] [CrossRef] [PubMed]
- Fàbrega, A.; Madurga, S.; Giralt, E.; Vila, J. Mechanism of action of and resistance to quinolones. Microb. Biotechnol. 2009, 2, 40–61. [Google Scholar] [CrossRef] [Green Version]
- Cheng, A.F.; Yew, W.W.; Chan, E.W.C.; Chin, M.L.; Hui, M.M.M.; Chan, R.C.Y. Multiplex PCR amplimer conformation analysis for rapid detection of gyrA mutations in fluoroquinolone-resistant Mycobacterium tuberculosis clinical isolates. Antimicrob. Agents Chemother. 2004, 48, 596–601. [Google Scholar] [CrossRef] [Green Version]
- Gordeev, M.F.; Liu, J.; Wang, X.; Yuan, Z. Water-Soluble O-Carbonyl Phosphoramidate Prodrugs for Therapeutic Administration. U.S. Patent 9,382,276, 5 July 2016. [Google Scholar]
- Wang, W.; Voss, K.M.; Liu, J.; Gordeev, M.F. Nonclinical Evaluation of Antibacterial Oxazolidinones Contezolid and Contezolid Acefosamil with Low Serotonergic Neurotoxicity. Chem. Res. Toxicol. 2021, 34, 1348–1354. [Google Scholar] [CrossRef]
- Working Group on New TB Drugs StopTBPartnership. Clinical Pipeline. 2021. Available online: https://www.newtbdrugs.org/pipeline/clinical (accessed on 7 November 2021).
- Libardo, M.D.J.; Boshoff, H.I.; Barry, C.E., III. The present state of the tuberculosis drug development pipeline. Curr. Opin. Pharmacol. 2018, 42, 81–94. [Google Scholar] [CrossRef]
- Bandodkar, B.; Shandil, R.K.; Bhat, J.; Balganesh, T.S. Two Decades of TB Drug Discovery Efforts—What Have We Learned? Appl. Sci. 2020, 10, 5704. [Google Scholar] [CrossRef]
- Palomino, J.C.; Martin, A. Drug resistance mechanisms in Mycobacterium tuberculosis. Antibiotics 2014, 3, 317–340. [Google Scholar] [CrossRef] [Green Version]
- Grzelak, E.M.; Choules, M.P.; Gao, W.; Cai, G.; Wan, B.; Wang, Y.; McAlpine, J.B.; Cheng, J.; Jin, Y.; Lee, H.; et al. Strategies in anti-Mycobacterium tuberculosis drug discovery based on phenotypic screening. J. Antibiot. 2019, 72, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Ahamad, S.; Rahman, S.; Khan, F.I.; Dwivedi, N.; Ali, S.; Kim, J.; Hassan, M.I. QSAR based therapeutic management of M. tuberculosis. Arch. Pharmacal Res. 2017, 40, 676–694. [Google Scholar] [CrossRef] [PubMed]
- Tiberi, S.; Vjecha, M.J.; Zumla, A.; Galvin, J.; Migliori, G.B.; Zumla, A. Accelerating development of new shorter TB treatment regimens in anticipation of a resurgence of multi-drug resistant TB due to the COVID-19 pandemic. Int. J. Infect. Dis. 2021. [Google Scholar] [CrossRef]
- Koul, A.; Dendouga, N.; Vergauwen, K.; Molenberghs, B.; Vranckx, L.; Willebrords, R.; Ristic, Z.; Lill, H.; Dorange, I.; Guillemont, J.; et al. Diarylquinolines target subunit c of mycobacterial ATP synthase. Nat. Chem. Biol. 2007, 3, 323–324. [Google Scholar] [CrossRef]
- Pethe, K.; Bifani, P.; Jang, J.; Kang, S.; Park, S.; Ahn, S.; Jiricek, J.; Jung, J.; Jeon, H.K.; Cechetto, J.; et al. Discovery of Q203, a potent clinical candidate for the treatment of tuberculosis. Nat. Med. 2013, 19, 1157–1160. [Google Scholar] [CrossRef]
- Kang, S.; Kim, R.Y.; Seo, M.J.; Lee, S.; Kim, Y.M.; Seo, M.; Seo, J.J.; Ko, Y.; Choi, I.; Jang, J.; et al. Lead optimization of a novel series of imidazo [1, 2-a] pyridine amides leading to a clinical candidate (Q203) as a multi-and extensively-drug-resistant anti-tuberculosis agent. J. Med. Chem. 2014, 57, 5293–5305. [Google Scholar] [CrossRef]
- Maeda, K. A new antibiotic, azomycin. J. Antibiot. 1953, 6, 182. [Google Scholar]
- Stover, C.K.; Warrener, P.; VanDevanter, D.R.; Sherman, D.R.; Arain, T.M.; Langhorne, M.H.; Anderson, S.W.; Towell, J.A.; Yuan, Y.; McMurray, D.N.; et al. A small-molecule nitroimidazopyran drug candidate for the treatment of tuberculosis. Nature 2000, 405, 962–966. [Google Scholar] [CrossRef]
- Ginsberg, A.M.; Laurenzi, M.W.; Rouse, D.J.; Whitney, K.D.; Spigelman, M.K. Safety, tolerability, and pharmacokinetics of PA-824 in healthy subjects. Antimicrob. Agents Chemother. 2009, 53, 3720–3725. [Google Scholar] [CrossRef] [Green Version]
- Sacksteder, K.A.; Protopopova, M.; Barry, C.E.; Andries, K.; Nacy, C.A. Discovery and development of SQ109: A new antitubercular drug with a novel mechanism of action. Future Microbiol. 2012, 7, 823–837. [Google Scholar] [CrossRef] [Green Version]
- Tahlan, K.; Wilson, R.; Kastrinsky, D.B.; Arora, K.; Nair, V.; Fischer, E.; Barnes, S.W.; Walker, J.R.; Alland, D.; Barry, C.E., III; et al. SQ109 targets MmpL3, a membrane transporter of trehalose monomycolate involved in mycolic acid donation to the cell wall core of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2012, 56, 1797–1809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordeev, M.F.; Yuan, Z.Y. New potent antibacterial oxazolidinone (MRX-I) with an improved class safety profile. J. Med. Chem. 2014, 57, 4487–4497. [Google Scholar] [CrossRef]
- Shoen, C.; DeStefano, M.; Hafkin, B.; Cynamon, M. In vitro and in vivo activities of contezolid (MRX-I) against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2018, 62, e00493-18. [Google Scholar] [CrossRef] [Green Version]
- Chiliza, T.E.; Pillay, M.; Pillay, B. Identification of unique essential proteins from a Mycobacterium tuberculosis F15/Lam4/KZN phage secretome library. Pathog. Dis. 2017, 75, ftx001. [Google Scholar] [CrossRef] [PubMed]
- Lamichhane, G. Novel targets in M. tuberculosis: Search for new drugs. Trends Mol. Med. 2011, 17, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Bishi, L.Y.; Vedithi, S.C.; Blundell, T.L.; Mugumbate, G.C. Computational Deorphaning of Mycobacterium Tuberculosis Targets. In Drug Discovery and Development-New Advances; IntechOpen: London, UK, 2019. [Google Scholar]
- Cheng, T.; Li, Q.; Zhou, Z.; Wang, Y.; Bryant, S.H. Structure-based virtual screening for drug discovery: A problem-centric review. AAPS J. 2012, 14, 133–141. [Google Scholar] [CrossRef] [Green Version]
- Lavecchia, A.; di Giovanni, C. Virtual screening strategies in drug discovery: A critical review. Curr. Med. Chem. 2013, 20, 2839–2860. [Google Scholar] [CrossRef] [PubMed]
- Lavecchia, A.; Cerchia, C. In silico methods to address polypharmacology: Current status, applications and future perspectives. Drug Discov. Today 2016, 21, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Moore, T.J.; Zhang, H.; Anderson, G.; Alexander, G.C. Estimated costs of pivotal trials for novel therapeutic agents approved by the US Food and Drug Administration, 2015–2016. JAMA Intern. Med. 2018, 178, 1451–1457. [Google Scholar] [CrossRef] [Green Version]
- Swinney, D.C.; Anthony, J. How were new medicines discovered? Nat. Rev. Drug Discov. 2011, 10, 507–519. [Google Scholar] [CrossRef]
- Ferreira, L.G.; Dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular docking and structure-based drug design strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.C. The process of structure-based drug design. Chem. Biol. 2003, 10, 787–797. [Google Scholar] [CrossRef] [Green Version]
- Lionta, E.; Spyrou, G.; Vassilatis, D.K.; Cournia, Z. Structure-based virtual screening for drug discovery: Principles, applications and recent advances. Curr. Top. Med. Chem. 2014, 14, 1923–1938. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Chen, Y.-P.P. Structure-based drug design to augment hit discovery. Drug Discov. Today 2011, 16, 831–839. [Google Scholar] [CrossRef]
- Searls, D.B. Data integration: Challenges for drug discovery. Nat. Rev. Drug Discov. 2005, 4, 45–58. [Google Scholar] [CrossRef]
- Batool, M.; Choi, S. Identification of druggable genome in staphylococcus aureus multidrug resistant strain. In Proceedings of the 2017 IEEE Life Sciences Conference (LSC), Sydney, Australia, 13–15 December 2017. [Google Scholar]
- Blaney, J. A very short history of structure-based design: How did we get here and where do we need to go? J. Comput.-Aided Mol. Des. 2012, 26, 13–14. [Google Scholar] [CrossRef]
- Mandal, S.; Mandal, S.K. Rational drug design. Eur. J. Pharmacol. 2009, 625, 90–100. [Google Scholar] [CrossRef]
- Wilson, G.L.; Lill, M.A. Integrating structure-based and ligand-based approaches for computational drug design. Future Med. Chem. 2011, 3, 735–750. [Google Scholar] [CrossRef]
- Urwyler, S. Allosteric modulation of family C G-protein-coupled receptors: From molecular insights to therapeutic perspectives. Pharmacol. Rev. 2011, 63, 59–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, Y. Ligand–receptor interaction platforms and their applications for drug discovery. Expert Opin. Drug Discov. 2012, 7, 969–988. [Google Scholar] [CrossRef] [PubMed]
- Wlodawer, A.; Vondrasek, J. Inhibitors of HIV-1 protease: A major success of structure-assisted drug design. Annu. Rev. Biophys. Biomol. Struct. 1998, 27, 249–284. [Google Scholar] [CrossRef] [Green Version]
- Clark, D.E. What has computer-aided molecular design ever done for drug discovery? Expert Opin. Drug Discov. 2006, 1, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Rutenber, E.E.; Stroud, R.M. Binding of the anticancer drug ZD1694 to E. coli thymidylate synthase: Assessing specificity and affinity. Structure 1996, 4, 1317–1324. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Gu, Q.; Zheng, X.; Ye, J.; Liu, Z.; Li, J.; Hu, X.; Hagler, A.; Xu, J. Discovery of new selective human aldose reductase inhibitors through virtual screening multiple binding pocket conformations. J. Chem. Inf. Modeling 2013, 53, 2409–2422. [Google Scholar] [CrossRef]
- Grover, S.; Apushkin, M.A.; Fishman, G.A. Topical dorzolamide for the treatment of cystoid macular edema in patients with retinitis pigmentosa. Am. J. Ophthalmol. 2006, 141, 850–858. [Google Scholar] [CrossRef]
- Dadashpour, S.; Tuylu Kucukkilinc, T.; Unsal Tan, O.; Ozadali, K.; Irannejad, H.; Emami, S. Design, Synthesis and In Vitro Study of 5, 6-Diaryl-1, 2, 4-triazine-3-ylthioacetate Derivatives as COX-2 and β-Amyloid Aggregation Inhibitors. Arch. Pharm. 2015, 348, 179–187. [Google Scholar] [CrossRef]
- Miller, Z.; Kim, K.-S.; Lee, D.-M.; Kasam, V.; Baek, S.E.; Lee, K.H.; Zhang, Y.-Y.; Ao, L.; Carmony, K.; Lee, N.-R.; et al. Proteasome inhibitors with pyrazole scaffolds from structure-based virtual screening. J. Med. Chem. 2015, 58, 2036–2041. [Google Scholar] [CrossRef] [PubMed]
- Marrakchi, H.; Lanéelle, G.; Quémard, A. InhA a target of the antituberculous drug isoniazid, is involved in a mycobacterial fatty acid elongation system, FAS-II. Microbiology 2000, 146, 289–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, J.-X.; Li, L.-L.; Zheng, R.-L.; Xie, H.-Z.; Cao, Z.-X.; Feng, S.; Pan, Y.-L.; Chen, X.; Wei, Y.-Q.; Yang, S.-Y. Discovery of novel Pim-1 kinase inhibitors by a hierarchical multistage virtual screening approach based on SVM model, pharmacophore, and molecular docking. J. Chem. Inf. Modeling 2011, 51, 1364–1375. [Google Scholar] [CrossRef] [PubMed]
- Matsuno, K.; Masuda, Y.; Uehara, Y.; Sato, H.; Muroya, A.; Takahashi, O.; Yokotagawa, T.; Furuya, T.; Okawara, T.; Otsuka, M.; et al. Identification of a new series of STAT3 inhibitors by virtual screening. ACS Med. Chem. Lett. 2010, 1, 371–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallerand, A.H.; Sanoski, C.A. Davis’s Canadian Drug Guide for Nurses; FA Davis: Philadelphia, PA, USA, 2020. [Google Scholar]
- Tripathi, K. Essentials of Medical Pharmacology; JP Medical Ltd.: New Delhi, India, 2013. [Google Scholar]
- Maia, E.H.B.; Assis, L.C.; de Oliveira, T.A.; da Silva, A.M.; Taranto, A.G. Structure-based virtual screening: From classical to artificial intelligence. Front. Chem. 2020, 8, 343. [Google Scholar] [CrossRef]
- Qaseem, A.; Kansagara, D.; Forciea, M.A.; Cooke, M.; Denberg, T.D. Management of chronic insomnia disorder in adults: A clinical practice guideline from the American College of Physicians. Ann. Intern. Med. 2016, 165, 125–133. [Google Scholar] [CrossRef]
- Gambacorti-Passerini, C.; Antolini, L.; Mahon, F.-X.; Guilhot, F.; Deininger, M.; Fava, C.; Nagler, A.; della Casa, C.M.; Morra, E.; Abruzzese, E.; et al. Multicenter independent assessment of outcomes in chronic myeloid leukemia patients treated with imatinib. J. Natl. Cancer Inst. 2011, 103, 553–561. [Google Scholar] [CrossRef]
- Markowitz, M.; Nguyen, B.-Y.; Gotuzzo, E.; Mendo, F.; Ratanasuwan, W.; Kovacs, C.; Prada, G.; Morales-Ramirez, J.O.; Crumpacker, C.S.; Isaacs, R.D.; et al. Rapid and durable antiretroviral effect of the HIV-1 integrase inhibitor raltegravir as part of combination therapy in treatment-naive patients with HIV-1 infection: Results of a 48-week controlled study. JAIDS J. Acquir. Immune Defic. Syndr. 2007, 46, 125–133. [Google Scholar] [CrossRef] [Green Version]
- Reymond, J.-L.; van Deursen, R.; Blum, L.C.; Ruddigkeit, L. Chemical space as a source for new drugs. MedChemComm 2010, 1, 30–38. [Google Scholar] [CrossRef]
- Ekins, S.; Freundlich, J.S.; Choi, I.; Sarker, M.; Talcott, C. Computational databases, pathway and cheminformatics tools for tuberculosis drug discovery. Trends Microbiol. 2011, 19, 65–74. [Google Scholar] [CrossRef] [Green Version]
- Macalino, S.J.Y.; Gosu, V.; Hong, S.; Choi, S. Role of computer-aided drug design in modern drug discovery. Arch. Pharmacal Res. 2015, 38, 1686–1701. [Google Scholar] [CrossRef]
- Saxena, S.; Devi, P.B.; Soni, V.; Yogeeswari, P.; Sriram, D. Identification of novel inhibitors against Mycobacterium tuberculosis L-alanine dehydrogenase (MTB-AlaDH) through structure-based virtual screening. J. Mol. Graph. Model. 2014, 47, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Saxena, S.; Samala, G.; Sridevi, J.P.; Devi, P.B.; Yogeeswari, P.; Sriram, D. Design and development of novel Mycobacterium tuberculosis l-alanine dehydrogenase inhibitors. Eur. J. Med. Chem. 2015, 92, 401–414. [Google Scholar] [CrossRef]
- Reshma, R.S.; Saxena, S.; Bobesh, K.A.; Jeankumar, V.U.; Gunda, S.; Yogeeswari, P.; Sriram, D. Design and development of new class of Mycobacterium tuberculosis L-alanine dehydrogenase inhibitors. Bioorganic Med. Chem. 2016, 24, 4499–4508. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Khare, G.; Bahal, R.K.; Ghosh, P.C.; Tyagi, A.K. Identification of Mycobacterium tuberculosis BioA inhibitors by using structure-based virtual screening. Drug Des. Dev. Ther. 2018, 12, 1065. [Google Scholar] [CrossRef] [Green Version]
- Taira, J.; Ito, T.; Nakatani, H.; Umei, T.; Baba, H.; Kawashima, S.; Maruoka, T.; Komatsu, H.; Sakamoto, H.; Aoki, S. In silico structure-based drug screening of novel antimycobacterial pharmacophores by DOCK-GOLD tandem screening. Int. J. Mycobacteriol. 2017, 6, 142. [Google Scholar]
- Billones, J.B.; Carrillo, M.C.O.; Organo, V.G.; Macalino, S.J.Y.; Sy, J.B.A.; Emnacen, I.A.; Clavio, N.A.B.; Concepcion, G.P. Toward antituberculosis drugs: In silico screening of synthetic compounds against Mycobacterium tuberculosis l, d-transpeptidase 2. Drug Des. Dev. Ther. 2016, 10, 1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korycka-Machala, M.; Nowosielski, M.; Kuron, A.; Rykowski, S.; Olejniczak, A.; Hoffmann, M.; Dziadek, J. Naphthalimides selectively inhibit the activity of bacterial, replicative DNA ligases and display bactericidal effects against tubercle bacilli. Molecules 2017, 22, 154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djaout, K.; Singh, V.; Boum, Y.; Katawera, V.; Becker, H.F.; Bush, N.G.; Hearnshaw, S.J.; Pritchard, J.E.; Bourbon, P.; Madrid, P.B.; et al. Predictive modeling targets thymidylate synthase ThyX in Mycobacterium tuberculosis. Sci. Rep. 2016, 6, 27792. [Google Scholar] [CrossRef] [Green Version]
- Luciani, R.; Saxena, P.; Surade, S.; Santucci, M.; Venturelli, A.; Borsari, C.; Marverti, G.; Ponterini, G.; Ferrari, S.; Blundell, T.L.; et al. Virtual screening and X-ray crystallography identify non-substrate analog inhibitors of flavin-dependent thymidylate synthase. J. Med. Chem. 2016, 59, 9269–9275. [Google Scholar] [CrossRef]
- Jeankumar, V.U.; Reshma, R.S.; Vats, R.; Janupally, R.; Saxena, S.; Yogeeswari, P.; Sriram, D. Engineering another class of anti-tubercular lead: Hit to lead optimization of an intriguing class of gyrase ATPase inhibitors. Eur. J. Med. Chem. 2016, 122, 216–231. [Google Scholar] [CrossRef]
- Sharma, K.; Tanwar, O.; Sharma, S.; Ali, S.; Alam, M.M.; Zaman, M.S.; Akhter, M. Structural comparison of Mtb-DHFR and h-DHFR for design, synthesis and evaluation of selective non-pteridine analogues as antitubercular agents. Bioorganic Chem. 2018, 80, 319–333. [Google Scholar] [CrossRef] [PubMed]
- Chiarelli, L.R.; Mori, M.; Barlocco, D.; Beretta, G.; Gelain, A.; Pini, E.; Porcino, M.; Mori, G.; Stelitano, G.; Costantino, L.; et al. Discovery and development of novel salicylate synthase (MbtI) furanic inhibitors as antitubercular agents. Eur. J. Med. Chem. 2018, 155, 754–763. [Google Scholar] [CrossRef] [PubMed]
- Rohilla, A.; Khare, G.; Tyagi, A.K. Virtual Screening, pharmacophore development and structure based similarity search to identify inhibitors against IdeR, a transcription factor of Mycobacterium tuberculosis. Sci. Rep. 2017, 7, 4653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dharra, R.; Talwar, S.; Singh, Y.; Gupta, R.; Cirillo, J.D.; Pandey, A.K.; Kulharia, M.; Mehta, P.K. Rational design of drug-like compounds targeting Mycobacterium marinum MelF protein. PLoS ONE 2017, 12, e0183060. [Google Scholar] [CrossRef] [Green Version]
- Gudzera, O.I.; Golub, A.G.; Bdzhola, V.G.; Volynets, G.P.; Lukashov, S.S.; Kovalenko, O.P.; Kriklivyi, I.A.; Yaremchuk, A.D.; Starosyla, S.A.; Yarmoluk, S.M.; et al. Discovery of potent anti-tuberculosis agents targeting leucyl-tRNA synthetase. Bioorganic Med. Chem. 2016, 24, 1023–1031. [Google Scholar] [CrossRef] [PubMed]
- Gudzera, O.I.; Golub, A.G.; Bdzhola, V.G.; Volynets, G.P.; Kovalenko, O.P.; Boyarshin, K.S.; Yaremchuk, A.D.; Protopopov, M.V.; Yarmoluk, S.M.; Tukalo, M.A. Identification of Mycobacterium tuberculosis leucyl-tRNA synthetase (LeuRS) inhibitors among the derivatives of 5-phenylamino-2H-[1, 2, 4] triazin-3-one. J. Enzym. Inhib. Med. Chem. 2016, 31 (Suppl. 2), 201–207. [Google Scholar] [CrossRef] [Green Version]
- Petersen, G.O.; Saxena, S.; Renuka, J.; Soni, V.; Yogeeswari, P.; Santos, D.S.; Bizarro, C.V.; Sriram, D. Structure-based virtual screening as a tool for the identification of novel inhibitors against Mycobacterium tuberculosis 3-dehydroquinate dehydratase. J. Mol. Graph. Model. 2015, 60, 124–131. [Google Scholar] [CrossRef]
- Lone, M.Y.; Athar, M.; Gupta, V.K.; Jha, P.C. Prioritization of natural compounds against mycobacterium tuberculosis 3-dehydroquinate dehydratase: A combined in-silico and in-vitro study. Biochem. Biophys. Res. Commun. 2017, 491, 1105–1111. [Google Scholar] [CrossRef]
- Buryska, T.; Daniel, L.; Kunka, A.; Brezovsky, J.; Damborsky, J.; Prokop, Z. Discovery of novel haloalkane dehalogenase inhibitors. Appl. Environ. Microbiol. 2016, 82, 1958–1965. [Google Scholar] [CrossRef] [Green Version]
- Koes, D.R.; Camacho, C.J. Pharmer: Efficient and exact pharmacophore search. J. Chem. Inf. Modeling 2011, 51, 1307–1314. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem substance and compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef]
- Reddy, T.; Riley, R.; Wymore, F.; Montgomery, P.; DeCaprio, D.; Engels, R.; Gellesch, M.; Hubble, J.; Jen, D.; Jin, H.; et al. TB database: An integrated platform for tuberculosis research. Nucleic Acids Res. 2009, 37 (Suppl. 1), D499–D508. [Google Scholar] [CrossRef]
- Galagan, J.E.; Sisk, P.; Stolte, C.; Weiner, B.; Koehrsen, M.; Wymore, F.; Reddy, T.B.K.; Zucker, J.D.; Engels, R.; Gellesch, M.; et al. TB database 2010: Overview and update. Tuberculosis 2010, 90, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Kapopoulou, A.; Lew, J.M.; Cole, S.T. The MycoBrowser portal: A comprehensive and manually annotated resource for mycobacterial genomes. Tuberculosis 2011, 91, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Patni, K.; Agarwal, P.; Kumar, A.; Meena, L.S. Computational evaluation of anticipated PE_PGRS39 protein involvement in host–pathogen interplay and its integration into vaccine development. 3 Biotech 2021, 11, 204. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, A.; Gabrielian, A.; Engle, E.; Hurt, D.E.; Alexandru, S.; Crudu, V.; Sergueev, E.; Kirichenko, V.; Lapitskii, V.; Snezhko, E.; et al. The TB portals: An open-access, web-based platform for global drug-resistant-tuberculosis data sharing and analysis. J. Clin. Microbiol. 2017, 55, 3267–3282. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, R.A.; Jabłońska, J.; Pravda, L.; Vařeková, R.S.; Thornton, J.M. PDBsum: Structural summaries of PDB entries. Protein Sci. 2018, 27, 129–134. [Google Scholar] [CrossRef]
- Liu, R.; Li, X.; Lam, K.S. Combinatorial chemistry in drug discovery. Curr. Opin. Chem. Biol. 2017, 38, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Shivanyuk, A.; Ryabukhin, S.V.; Tolmachev, A.; Bogolyubsky, A.V.; Mykytenko, D.M.; Chupryna, A.A.; Heilman, W.; Kostyuk, A.N. Enamine real database: Making chemical diversity real. Chem. Today 2007, 25, 58–59. [Google Scholar]
- Sterling, T.; Irwin, J.J. ZINC 15–ligand discovery for everyone. J. Chem. Inf. Modeling 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Ruddigkeit, L.; Van Deursen, R.; Blum, L.C.; Reymond, J.-L. Enumeration of 166 billion organic small molecules in the chemical universe database GDB-17. J. Chem. Inf. Modeling 2012, 52, 2864–2875. [Google Scholar] [CrossRef] [PubMed]
- Williams, A. ChemSpider: Integrating Structure-Based Resources Distributed Across the Internet. Enhancing Learning with Online Resources, Social Networking, and Digital Libraries. In ACS Symposium Series; American Chemical Society: Washington, DC, USA, 2010; Volume 1060, pp. 23–29. [Google Scholar]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrián-Uhalte, E.; et al. The ChEMBL database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef]
- Voigt, J.H.; Bienfait, B.; Wang, S.; Nicklaus, M.C. Comparison of the NCI open database with seven large chemical structural databases. J. Chem. Inf. Comput. Sci. 2001, 41, 702–712. [Google Scholar] [CrossRef] [PubMed]
- Schmidtke, P.; Le Guilloux, V.; Maupetit, J.; Tuffery, P. Fpocket: Online tools for protein ensemble pocket detection and tracking. Nucleic Acids Res. 2010, 38 (Suppl. 2), W582–W589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Guilloux, V.; Schmidtke, P.; Tuffery, P. Fpocket: An open source platform for ligand pocket detection. BMC Bioinform. 2009, 10, 168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussein, H.A.; Borrel, A.; Geneix, C.; Petitjean, M.; Regad, L.; Camproux, A.-C. PockDrug-Server: A new web server for predicting pocket druggability on holo and apo proteins. Nucleic Acids Res. 2015, 43, W436–W442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koes, D.R.; Camacho, C.J. PocketQuery: Protein–protein interaction inhibitor starting points from protein–protein interaction structure. Nucleic Acids Res. 2012, 40, W387–W392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brady, G.P.; Stouten, P.F. Fast prediction and visualization of protein binding pockets with PASS. J. Comput.-Aided Mol. Des. 2000, 14, 383–401. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein–ligand docking using GOLD. Proteins Struct. Funct. Bioinform. 2003, 52, 609–623. [Google Scholar] [CrossRef]
- Schrödinger, G. Glide: A Complete Solution for Ligand-Receptor Docking. 2021. Available online: https://www.schrodinger.com/products/glide (accessed on 5 September 2021).
- Rarey, M.; Kramer, B.; Lengauer, T.; Klebe, G. A fast flexible docking method using an incremental construction algorithm. J. Mol. Biol. 1996, 261, 470–489. [Google Scholar] [CrossRef] [Green Version]
- BioSolveIT. SeeSAR: The Drug Design Dashboard. 2021. Available online: https://www.biosolveit.de/SeeSAR (accessed on 5 September 2021).
- Tosco, P.; Balle, T. Open3DQSAR: A new open-source software aimed at high-throughput chemometric analysis of molecular interaction fields. J. Mol. Modeling 2011, 17, 201–208. [Google Scholar] [CrossRef]
- Dong, J.; Yao, Z.-J.; Zhu, M.-F.; Wang, N.-N.; Lu, B.; Chen, A.F.; Lu, A.-P.; Miao, H.; Zeng, W.-B.; Cao, D.-S. ChemSAR: An online pipelining platform for molecular SAR modeling. J. Cheminformat. 2017, 9, 27. [Google Scholar] [CrossRef] [Green Version]
- Schrödinger, Q. QikProp Rapid ADME Predictions of Drug Candidates. 2021. Available online: https://www.schrodinger.com/products/qikprop (accessed on 5 September 2021).
- SimulationsPlus. ADMET Predictor® Flagship Machine Learning Platform for ADMET Modeling. 2021. Available online: https://www.simulations-plus.com/software/admetpredictor/ (accessed on 5 September 2021).
- Potapov, V.; Cohen, M.; Inbar, Y.; Schreiber, G. Protein structure modelling and evaluation based on a 4-distance description of side-chain interactions. BMC Bioinform. 2010, 11, 374. [Google Scholar] [CrossRef] [Green Version]
- Laurie, A.T.; Jackson, R.M. Q-SiteFinder: An energy-based method for the prediction of protein–ligand binding sites. Bioinformatics 2005, 21, 1908–1916. [Google Scholar] [CrossRef] [PubMed]
- Wunberg, T.; Hendrix, M.; Hillisch, A.; Lobell, M.; Meier, H.; Schmeck, C.; Wild, H.; Hinzen, B. Improving the hit-to-lead process: Data-driven assessment of drug-like and lead-like screening hits. Drug Discov. Today 2006, 11, 175–180. [Google Scholar] [CrossRef]
- Smieško, M.; Vedani, A. VirtualToxLab: Exploring the toxic potential of rejuvenating substances found in traditional medicines. In In Silico Methods for Predicting Drug Toxicity; Springer: Berlin/Heidelberg, Germany, 2016; pp. 121–137. [Google Scholar]
- Krieger, E.; Joo, K.; Lee, J.; Lee, J.; Raman, S.; Thompson, J.; Tyka, M.; Baker, D.; Karplus, K. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins Struct. Funct. Bioinform. 2009, 77, 114–122. [Google Scholar] [CrossRef] [Green Version]
- Bordoli, L.; Kiefer, F.; Arnold, K.; Benkert, P.; Battey, J.; Schwede, T. Protein structure homology modeling using SWISS-MODEL workspace. Nat. Protoc. 2009, 4, 1–13. [Google Scholar] [CrossRef]
- Shi, Y.; Colombo, C.; Kuttiyatveetil, J.R.A.; Zalatar, N.; van Straaten, K.E.; Mohan, S.; Sanders, D.A.R.; Pinto, B.M. A Second, Druggable Binding Site in UDP-Galactopyranose Mutase from Mycobacterium tuberculosis? ChemBioChem 2016, 17, 2264–2273. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Vina, O.A.A. improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading Oleg Public Access. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar]
- Case, D.A.; Cheatham, T.E., III; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [Green Version]
- Shoichet, B.K. Virtual screening of chemical libraries. Nature 2004, 432, 862–865. [Google Scholar] [CrossRef] [PubMed]
- Pedretti, A.; Mazzolari, A.; Gervasoni, S.; Vistoli, G. Rescoring and linearly combining: A highly effective consensus strategy for virtual screening campaigns. Int. J. Mol. Sci. 2019, 20, 2060. [Google Scholar] [CrossRef] [Green Version]
- Vidal, D.; Thormann, M.; Pons, M. A novel search engine for virtual screening of very large databases. J. Chem. Inf. Modeling 2006, 46, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.S.; Pati, S.P.; Kumar, P.P.; Pradeep, H.N.; Sastry, G.N. Virtual screening in drug discovery-a computational perspective. Curr. Protein Pept. Sci. 2007, 8, 329–351. [Google Scholar] [CrossRef] [PubMed]
- Bissantz, C.; Folkers, G.; Rognan, D. Protein-based virtual screening of chemical databases. 1. Evaluation of different docking/scoring combinations. J. Med. Chem. 2000, 43, 4759–4767. [Google Scholar] [CrossRef]
- Willett, P.; Barnard, J.M.; Downs, G.M. Chemical similarity searching. J. Chem. Inf. Comput. Sci. 1998, 38, 983–996. [Google Scholar] [CrossRef] [Green Version]
- Bender, A.; Glen, R.C. A discussion of measures of enrichment in virtual screening: Comparing the information content of descriptors with increasing levels of sophistication. J. Chem. Inf. Modeling 2005, 45, 1369–1375. [Google Scholar] [CrossRef] [PubMed]
- Batool, M.; Ahmad, B.; Choi, S. A structure-based drug discovery paradigm. Int. J. Mol. Sci. 2019, 20, 2783. [Google Scholar] [CrossRef] [Green Version]
- Ejalonibu, M.A.; Elrashedy, A.A.; Lawal, M.M.; Soliman, M.E.; Sosibo, S.C.; Kumalo, H.M.; Mhlongo, N.N. Dual targeting approach for Mycobacterium tuberculosis drug discovery: Insights from DFT calculations and molecular dynamics simulations. Struct. Chem. 2020, 31, 557–571. [Google Scholar] [CrossRef]
- Hartenfeller, M.; Schneider, G. De novo drug design. Chemoinformat. Comput. Chem. Biol. 2010, 672, 299–323. [Google Scholar]
- Richardson, J.S.; Richardson, D.C. The de novo design of protein structures. Trends Biochem. Sci. 1989, 14, 304–309. [Google Scholar] [CrossRef]
- Lameijer, E.-W.; Tromp, R.A.; Spanjersberg, R.F.; Brussee, J.; IJzerman, A.P. Designing active template molecules by combining computational de novo design and human chemist’s expertise. J. Med. Chem. 2007, 50, 1925–1932. [Google Scholar] [CrossRef]
- Gillet, V.J. New directions in library design and analysis. Curr. Opin. Chem. Biol. 2008, 12, 372–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, G.; Fechner, U. Computer-based de novo design of drug-like molecules. Nat. Rev. Drug Discov. 2005, 4, 649–663. [Google Scholar] [CrossRef] [PubMed]
- Keserű, G.M.; Makara, G.M. Hit discovery and hit-to-lead approaches. Drug Discov. Today 2006, 11, 741–748. [Google Scholar] [CrossRef]
- Tang, Y.; Zhu, W.; Chen, K.; Jiang, H. New technologies in computer-aided drug design: Toward target identification and new chemical entity discovery. Drug Discov. Today Technol. 2006, 3, 307–313. [Google Scholar] [CrossRef]
- DeJesus, M.A.; Gerrick, E.R.; Xu, W.; Park, S.W.; Long, J.E.; Boutte, C.C.; Rubin, E.J.; Schnappinger, D.; Ehrt, S.; Fortune, S.M.; et al. Comprehensive essentiality analysis of the Mycobacterium tuberculosis genome via saturating transposon mutagenesis. MBio 2017, 8, e02133-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiménez-Luna, J.; Cuzzolin, A.; Bolcato, G.; Sturlese, M.; Moro, S. A deep-learning approach toward rational molecular docking protocol selection. Molecules 2020, 25, 2487. [Google Scholar] [CrossRef] [PubMed]
- Abrahams, K.A.; Besra, G.S. Mycobacterial drug discovery. RSC Med. Chem. 2020, 11, 1354–1365. [Google Scholar] [CrossRef]
- Billones, J.B.; Carrillo, M.C.O.; Organo, V.G.; Sy, J.B.A.; Clavio, N.A.B.; Macalino, S.J.Y.; Emnacen, I.A.; Lee, A.P.; Ko, P.K.L.; Concepcion, G.P. In silico discovery and in vitro activity of inhibitors against Mycobacterium tuberculosis 7, 8-diaminopelargonic acid synthase (Mtb BioA). Drug Des. Dev. Ther. 2017, 11, 563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, N.; Srivastava, R.; Prakash, A.; Lynn, A.M. Structure-based virtual screening, molecular dynamics simulation and MM-PBSA toward identifying the inhibitors for two-component regulatory system protein NarL of Mycobacterium Tuberculosis. J. Biomol. Struct. Dyn. 2020, 38, 3396–3410. [Google Scholar] [CrossRef]
- Tatum, N.J.; Liebeschuetz, J.W.; Cole, J.C.; Frita, R.; Herledan, A.; Baulard, A.R.; Willand, N.; Pohl, E. New active leads for tuberculosis booster drugs by structure-based drug discovery. Org. Biomol. Chem. 2017, 15, 10245–10255. [Google Scholar] [CrossRef] [PubMed]
- Kingdon, A.D.; Alderwick, L.J. Structure-based in silico approaches for drug discovery against Mycobacterium tuberculosis. Comput. Struct. Biotechnol. J. 2021, 19, 3708. [Google Scholar] [CrossRef] [PubMed]
- Rani, J.; Silla, Y.; Borah, K.; Ramachandran, S.; Bajpai, U. Repurposing of FDA-approved drugs to target MurB and MurE enzymes in Mycobacterium tuberculosis. J. Biomol. Struct. Dyn. 2020, 38, 2521–2532. [Google Scholar] [CrossRef]
- Zhang, G.; Guo, S.; Cui, H.; Qi, J. Virtual screening of small molecular inhibitors against DprE1. Molecules 2018, 23, 524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, M.T.; Blicharska, N.; Shilpi, J.A.; Seidel, V. Investigation of the anti-TB potential of selected propolis constituents using a molecular docking approach. Sci. Rep. 2018, 8, 12238. [Google Scholar] [CrossRef]
- Ravichandran, R.; Ridzwan, N.F.W.; Mohamad, S.B. Ensemble-based high-throughput virtual screening of natural ligands using the Super Natural-II database against cell-wall protein dTDP-4-dehydrorhamnose reductase (RmlD) in Mycobacterium tuberculosis. J. Biomol. Struct. Dyn. 2020, 1–10. [Google Scholar] [CrossRef]
- Scheich, C.; Szabadka, Z.; Vértessy, B.; Pütter, V.; Grolmusz, V.; Schade, M. Discovery of novel MDR-Mycobacterium tuberculosis inhibitor by new FRIGATE computational screen. PLoS ONE 2011, 6, e28428. [Google Scholar] [CrossRef]
- Kaur, G.; Pandey, B.; Kumar, A.; Garewal, N.; Grover, A.; Kaur, J. Drug targeted virtual screening and molecular dynamics of LipU protein of Mycobacterium tuberculosis and Mycobacterium leprae. J. Biomol. Struct. Dyn. 2019, 37, 1254–1269. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Srivastava, R.; Prakash, A.; Lynn, A.M. Virtual screening and free energy estimation for identifying Mycobacterium tuberculosis flavoenzyme DprE1 inhibitors. J. Mol. Graph. Model. 2021, 102, 107770. [Google Scholar] [CrossRef]
- Sundar, S.; Thangamani, L.; Manivel, G.; Kumar, P.; Piramanayagam, S. Molecular docking, molecular dynamics and MM/PBSA studies of FDA approved drugs for protein kinase a of Mycobacterium tuberculosis; application insights of drug repurposing. Inform. Med. Unlocked 2019, 16, 100210. [Google Scholar] [CrossRef]
- Kuldeep, J.; Sharma, S.K.; Sharma, T.; Singh, B.N.; Siddiqi, M.I. Targeting Mycobacterium Tuberculosis Enoyl-Acyl Carrier Protein Reductase Using Computational Tools for Identification of Potential Inhibitor and their Biological Activity. Mol. Inform. 2021, 40, 2000211. [Google Scholar] [CrossRef]
- Sivaranjani, P.; Naik, V.U.; Madhulitha, N.R.; Kumar, K.S.; Chiranjeevi, P.; Alex, S.P.; Umamaheswari, A. Design of Novel Antimycobacterial Molecule Targeting Shikimate Pathway of Mycobacterium tuberculosis. Indian J. Pharm. Sci. 2019, 81, 438–447. [Google Scholar] [CrossRef] [Green Version]
- Ballester, P.J.; Mangold, M.; Howard, N.I.; Robinson, R.L.M.; Abell, C.; Blumberger, J.; Mitchell, J.B.O. Hierarchical virtual screening for the discovery of new molecular scaffolds in antibacterial hit identification. J. R. Soc. Interface 2012, 9, 3196–3207. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, S.; Rhee, K.Y. Tuberculosis drug development: History and evolution of the mechanism-based paradigm. Cold Spring Harb. Perspect. Med. 2015, 5, a021147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, G.; Chiarelli, L.R.; Esposito, M.; Makarov, V.; Bellinzoni, M.; Hartkoorn, R.C.; Degiacomi, G.; Boldrin, F.; Ekins, S.; de Jesus Lopes Ribeiro, A.L.; et al. Thiophenecarboxamide derivatives activated by EthA kill Mycobacterium tuberculosis by inhibiting the CTP synthetase PyrG. Chem. Biol. 2015, 22, 917–927. [Google Scholar] [CrossRef]
- Rabi, S.; Patel, A.H.G.; Burger, S.K.; Verstraelen, T.; Ayers, P.W. Exploring the substrate selectivity of human sEH and M. tuberculosis EHB Using QM/MM. Struct. Chem. 2017, 28, 1501–1511. [Google Scholar] [CrossRef]
- Ramalho, T.C.; Caetano, M.S.; Josa, D.; Luz, G.P.; Freitas, E.A.; da Cunha, E.F.F. Molecular modeling of Mycobacterium tuberculosis dUTpase: Docking and catalytic mechanism studies. J. Biomol. Struct. Dyn. 2011, 28, 907–917. [Google Scholar] [CrossRef]
- Oliveira, C.G.; Maia, P.S.; Souza, P.C.; Pavan, F.R.; Leite, C.Q.F.; Viana, R.B.; Batista, A.A.; Nascimento, O.R.; Deflon, V.M. Manganese (II) complexes with thiosemicarbazones as potential anti-Mycobacterium tuberculosis agents. J. Inorg. Biochem. 2014, 132, 21–29. [Google Scholar] [CrossRef]
- Chi, G.; Manos-Turvey, A.; O’Connor, P.D.; Johnston, J.M.; Evans, G.L.; Baker, E.N.; Payne, R.J.; Lott, J.S.; Bulloch, E.M.M. Implications of binding mode and active site flexibility for inhibitor potency against the salicylate synthase from Mycobacterium tuberculosis. Biochemistry 2012, 51, 4868–4879. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision d. 01; Gaussian Inc.: Wallingford, CT, USA, 2009; Volume 201. [Google Scholar]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Beck, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648-6. [Google Scholar] [CrossRef] [Green Version]
- Indarto, A. Theoretical Modelling and Mechanistic Study of the Formation and Atmospheric Transformations of Polycyclic Aromatic Compounds and Carbonaceous Particles; Universal-Publishers: Gulf, CA, USA, 2010. [Google Scholar]
- Hamada, I. van der Waals density functional made accurate. Phys. Rev. B 2014, 89, 121103. [Google Scholar] [CrossRef]
- Grimme, S. Accurate description of van der Waals complexes by density functional theory including empirical corrections. J. Comput. Chem. 2004, 25, 1463–1473. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.J.; Mori-Sánchez, P.; Yang, W. Insights into current limitations of density functional theory. Science 2008, 321, 792–794. [Google Scholar] [CrossRef] [Green Version]
- Villoutreix, B.O.; Eudes, R.; Miteva, M.A. Structure-based virtual ligand screening: Recent success stories. Comb. Chem. High Throughput Screen. 2009, 12, 1000–1016. [Google Scholar] [CrossRef]
- Talele, T.T.; Khedkar, S.A.; Rigby, A.C. Successful applications of computer aided drug discovery: Moving drugs from concept to the clinic. Curr. Top. Med. Chem. 2010, 10, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Clark, D.E. What has virtual screening ever done for drug discovery? Expert Opin. Drug Discov. 2008, 3, 841–851. [Google Scholar] [CrossRef]
- Scior, T.; Bender, A.; Tresadern, G.; Medina-Franco, J.L.; Martínez-Mayorga, K.; Langer, T.; Cuanalo-Contreras, K.; Agrafiotis, D.K. Recognizing pitfalls in virtual screening: A critical review. J. Chem. Inf. Modeling 2012, 52, 867–881. [Google Scholar] [CrossRef] [PubMed]
- Hassan Baig, M.; Ahmad, K.; Roy, S.; Mohammad Ashraf, J.; Adil, M.; Haris Siddiqui, M.; Khan, S.; Amjad Kamal, M.; Provazník, I.; Choi, I. Computer aided drug design: Success and limitations. Curr. Pharm. Des. 2016, 22, 572–581. [Google Scholar] [CrossRef] [PubMed]
- Coupez, B.; Lewis, R. Docking and scoring-Theoretically easy, practically impossible? Curr. Med. Chem. 2006, 13, 2995–3003. [Google Scholar]
- Fujita, T. Recent success stories leading to commercializable bioactive compounds with the aid of traditional QSAR procedures. Quant. Struct.-Act. Relatsh. 1997, 16, 107–112. [Google Scholar] [CrossRef]
- Gao, Q.; Yang, L.; Zhu, Y. Pharmacophore based drug design approach as a practical process in drug discovery. Curr. Comput.-Aided Drug Des. 2010, 6, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Sardari, S.; Dezfulian, M. Cheminformatics in anti-infective agents discovery. Mini Rev. Med. Chem. 2007, 7, 181–189. [Google Scholar] [CrossRef]
- Boström, J.; Norrby, P.-O.; Liljefors, T. Conformational energy penalties of protein-bound ligands. J. Comput.-Aided Mol. Des. 1998, 12, 383. [Google Scholar] [CrossRef]
- Perola, E.; Charifson, P.S. Conformational analysis of drug-like molecules bound to proteins: An extensive study of ligand reorganization upon binding. J. Med. Chem. 2004, 47, 2499–2510. [Google Scholar] [CrossRef]
- Verma, J.; Khedkar, V.M.; Coutinho, E.C. 3D-QSAR in drug design-a review. Curr. Top. Med. Chem. 2010, 10, 95–115. [Google Scholar] [CrossRef]
- Hu, Y.; Bajorath, J. Extending the activity cliff concept: Structural categorization of activity cliffs and systematic identification of different types of cliffs in the ChEMBL database. J. Chem. Inf. Modeling 2012, 52, 1806–1811. [Google Scholar] [CrossRef]
- Hu, Y.; Stumpfe, D.; Bajorath, J. Advancing the activity cliff concept. F1000Research 2013, 2, 199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stumpfe, D.; de León, A.V.; Dimova, D.; Bajorath, J. Follow up: Advancing the activity cliff concept, part II. F1000Research 2014, 3, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geppert, H.; Vogt, M.; Bajorath, J. Current trends in ligand-based virtual screening: Molecular representations, data mining methods, new application areas, and performance evaluation. J. Chem. Inf. Modeling 2010, 50, 205–216. [Google Scholar] [CrossRef]
- Jain, A.N.; Nicholls, A. Recommendations for evaluation of computational methods. J. Comput.-Aided Mol. Des. 2008, 22, 133–139. [Google Scholar] [CrossRef] [Green Version]
- Lindorff-Larsen, K.; Maragakis, P.; Piana, S.; Shaw, D.E. Picosecond to millisecond structural dynamics in human ubiquitin. J. Phys. Chem. B 2016, 120, 8313–8320. [Google Scholar] [CrossRef]
- Noé, F. Beating the millisecond barrier in molecular dynamics simulations. Biophys. J. 2015, 108, 228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, J.; Nobrega, R.P.; Schwantes, C.; Kathuria, S.V.; Bilsel, O.; Matthews, C.R.; Lane, T.J.; Pande, V.S. Atomistic structural ensemble refinement reveals non-native structure stabilizes a sub-millisecond folding intermediate of CheY. Sci. Rep. 2017, 7, 44116. [Google Scholar] [CrossRef]
- Park, C.-W.; Seo, S.W.; Kang, N.; Ko, B.; Choi, B.W.; Park, C.M.; Chang, D.K.; Kim, H.; Kim, H.; Lee, H.; et al. Artificial intelligence in health care: Current applications and issues. J. Korean Med. Sci. 2020, 35, e379. [Google Scholar] [CrossRef]
- Al-Attar, S.; Westra, E.R.; Van Der Oost, J.; Brouns, S.J. Clustered regularly interspaced short palindromic repeats (CRISPRs): The hallmark of an ingenious antiviral defense mechanism in prokaryotes. Biol. Chem. 2011, 392, 277–289. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| The Traditional Method of Drug Development | CADD |
|---|---|
| It involves more trial-and-error processes | It is more logical |
| It involves blind screening | It is specific and mostly target-based |
| It is a more expensive approach to drug development | It minimizes the cost of drug development |
| It is a relatively more laborious and time-consuming approach | It reduces the duration required in the development of new drugs |
| It involves sequential steps | It entails steps that are not only sequential but are also parallel and straightforward. |
| It involves separate interdisciplinary drug development with more difficult processes | It coordinates interdisciplinary drug development with easier processes. |
| Drug | Class of Compound | Target | Approach | Clinical Trial Phase |
|---|---|---|---|---|
| Linezolid | Oxazolidinone | 50S ribosomal subunit | Revisiting established targets (repurposing) | Phase 2 |
| Sutezolid | Oxazolidinone | 50S ribosomal subunit | Revisiting established targets (repurposing) | Phase 1 |
| Bedaquiline (TMC207) | Diarylquinoline | ATP synthase | Phenotypic-HTS | Approved |
| TBAJ-587 | Diaryquinoline | ATPsynthase | Revisiting novel target | Preclinical |
| Delamanid | Nitroimidazoles | Cell wall biosynthesis | HTS; modification of drug scaffold | Approved |
| Pretomanid | Nitroimidazoles | Cell wall biosynthesis | HTS; modification of drug scaffold | Approved |
| Telacebec (Q203) | Imidazopyridine amides | Cytochrome bc1 complex | HTS | Phase 2 |
| Gatifloxacin | Quinolones | DNA gyrase; gyrA, gyrB | Revisiting established targets (repurposing) | Phase 3/4 |
| Moxifloxacin | Quinolones | DNA gyrase; gyrA, gyrB | Revisiting established targets (repurposing) | Phase 3/4 |
| Benzothiazinone (BTZ-043) | Benzothiazole | Decaprenylphosphoryl-β-D-ribose-2′-oxidase (DprE1) | HTS | Phase 2 |
| Macozinone (PBTZ) | Benzothiazole | DprE1 | HTS | Phase 2 |
| OPC-167832 | Carbostyril | DprE1 | HTS | Phase 2 |
| TBA7371 | Azaindoles | DprE1 | HTS; modification of drug scaffold | Phase 2A |
| Clofazimine | Riminophenazine | Electrogenic pathway, reduced by NADH dehydrogenase II | Revisiting established targets (repurposing) | Approved |
| SPR720 | Benzimidazole class | GyrB ATPase | Revisiting established target (repurposing) | Phase 2 |
| SQ109 | Ethylenediamine | Inhibition of MmpL3, MenA, and MenG and ATP | HTS; modification of drug scaffold | Phase 2 |
| GSK 070 | Oxaborole | Leucine tRNA synthase | Revisiting established target (repurposing) | Phase 2 |
| Delpazolid | Oxazolidinones | Ribosomal subunit | Revisiting established targets (repurposing) | Phase 2 |
| OTB-658 | Oxazolidinones | Ribosomal subunit | Revisiting established targets (repurposing) | Preclinical |
| TBI-223 | Oxazolidinones | Ribosomal subunit | Revisiting established targets (repurposing) | Phase 1 |
| Contezolid | Oxazolidinones | Ribosomal subunit | Modification of drug scaffold | Phase 3 |
| Contezolid acefosamil (prodrug) | Oxazolidinones | Ribosomal subunit | Modification of drug scaffold | Phase 3 |
| Sanfetrinem | Carbapenem | Cell wall biosynthesis | Revisiting established target | Phase 2 |
| Sanfetrinem cilexetil (prodrug) | Carbapenem | Cell wall biosynthesis | Revisiting established target | Phase 2 |
| Drug | Target | Target Disease | Computational Methods | Refs. |
|---|---|---|---|---|
Epalrestat | Aldose reductase | Diabetic neuropathy | MD and SBVS | [111] |
Amprenavir | Antiretroviral protease | HIV | Protein modeling and molecular dynamics (MD) | [108,109] |
Dorzolamide | Carbonic anhydrase | Glaucoma, cystoid macular edema | Fragment-based screening | [112] |
Flurbiprofen | Cyclooxygenase-2 | Rheumatoid arthritis, osteoarthritis | Molecular docking | [113,114] |
Isoniazid | InhA | TB | SBVS and pharmacophore modeling | [115] |
| Pim-1 kinase inhibitors (E)-5-(4-hydroxybenzylidene)-2-iminothiazolidin-4-one  3-fluoro-4-((4-(isopropylamino)-5-nitropyrimidin-2-yl)amino)benzoic acid  4-(benzofuran-2-yl)-6-ethyl-2H-chromen-2-one  | Pim-1 kinase | Cancer | Hierarchical multistage VS | [116] |
STX-0119 | STAT3 | Lymphoma | SBVS | [117] |
Raltitrexed | Thymidylate synthase | HIV | SBDD | [98] |
Norfloxacin | Topoisomerase II, IV | Urinary tract infection | SBVS | [118] |
Cimetidine | Histamine H2 receptor antagonist | Gastrointestinal disorder (ulcer) | SBVS | [119] |
Zanamivir | Neuraminidase inhibitor | Influenza | SBVS | [120] |
Zolpidem | GABAA receptor agonist | Insomnia | SBVS | [121] |
Imatinib | Bcr-Abi tyrosine-kinase inhibitor | Cancer | SBVS | [122] |
Raltegravir | HIV integrase strand transfer inhibitor | HIV/AIDS | SBVS | [123] |
| System | PDB Structures | Function | Anti-Mtb Activity | Ref. |
|---|---|---|---|---|
| L-alanine dehydrogenase | 2VHW | Biosynthesis of l-alanine | IC50/35.5 μM b | [127] |
| L-alanine dehydrogenase | 4LMP | Biosynthesis of l-alanine | MIC/1.53 μM | [128] |
| L-alanine dehydrogenase | 2VOJ | Biosynthesis of l-alanine | MIC/11.81 µM | [129] |
| 7,8-diaminopelargonic acid synthase | 3TFU | Biotin biosynthesis pathway | MIC/25 μM | [129] |
| 7,8-diaminopelargonic acid synthase | 3TFU | Biotin biosynthesis pathway | MIC/7.86 μM | [130] |
| Cyclopropane mycolic acid synthase 1 | 1KPH | Cell wall | MIC50/5.1 μM | [131] |
| l,d-transpeptidase 2 | 3TUR | Cell wall | MIC94/25.0 μM MIC89/0.2 μM | [132] |
| GlmU protein [58] | 3ST8 a | Cell wall | IC50/9.0 μM b | |
| NAD⁺-dependent DNA ligase A | 1ZAU/1TAE | DNA metabolism | MIC50/15 µM | [133] |
| Flavin-dependent thymidylate synthase | 2AF6 a | DNA metabolism | MIC90/125 μM | [134] |
| Flavin-dependent thymidylate synthase | 2AF6 | DNA metabolism | IC29/100 μM b | [135] |
| DNA gyrase | 4BAE | DNA topology | MIC/7.8 µM | [136] |
| Dihydrofolate reductase | Mtb: 1DF7; human: 1OHJ | Folate pathway | MIC/25 μM | [137] |
| Salicylate synthase | 3VEH | Iron acquisition | MIC99/156 μM | [138] |
| Transcription factor IdeR | 1U8R | Iron acquisition control | MIC90/17.5 μg/ml | [139] |
| Flavin-dependent oxidoreductase MelF | 2WGK | Needed to withstand ROS-and RNS-induced stress | MIC/13.5 μM | [140] |
| Leucyl-tRNA synthetase | 2V0C | Protein synthesis | MIC/25 µM | [141,142] |
| 3-dehydroquinate dehydratase | 2Y71 | Shikimate pathway | MIC/6.25 µg/mL | [143] |
| 3-dehydroquinate dehydratase | 15 PDB structures | Shikimate pathway | MIC/100 mg/ml | [144] |
| Haloalkane dehalogenase | 2QVB | Unknown | Kd/3.37 µM b | [145] |
| Structure | IUPAC Name | Enzymatic Inhibition |
|---|---|---|
 | (2S,2′S,3S,3′S,4R,4′R,5R,5′R,6S,6′S)-6,6′-([1,1′-biphenyl]-4,4′-diylbis(azanediyl))bis(2-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol) | Biosynthesis of l-alanine [127] |
 | tert-butyl 2-(4-(benzyloxy)benzamido)-3-carbamoyl-4,7-dihydrothieno [2,3-c]pyridine-6(5H)-carboxylate | Biosynthesis of l-alanine [128] |
 | N1, N3-bis(benzo[d]thiazol-2-yl)-2-(isonicotinamido)cyclobutane-1,3-dicarboxamide | Biosynthesis of l-alanine [129] |
 | (Z)-N-(2-isopropoxyphenyl)-2-oxo-2-((3-(trifluoromethyl)cyclohexyl)amino)acetimidic acid | Biotin biosynthesis pathway [129] |
 | (E)-4-((2-(1-naphthoyl)hydrazono)methyl) benzoic acid | Biotin biosynthesis pathway [130] |
 | N-(2,5-diethoxy-4-(3-(4-nitro-1,3-dioxoisoindolin-2-yl)propanamido)phenyl) benzamide | Cell wall [131] |
 | (Z)-N-(2-(5-methyl-1H-1,2,4-triazol-3-yl) phenyl)-4-(methylsulfonamido)benzimidic acid | Cell wall [132] |
 | (Z)-5-(furan-3-ylmethylene)-6-hydroxy-3-(4-methoxyphenyl)-2-thioxo-2,5-dihydropyrimidin-4(3H)-one | Cell wall [133] |
 | N-(1,3-dioxo-2-(2-(pyrrolidin-1-yl)ethyl)-2,3-dihydro-1H-benzo[de]isoquinolin-5-yl)-N-oxohydroxylammonium | DNA metabolism [134] |
 | 2-(10-hydroxydecyl)-5,6-dimethoxy-3-methylcyclohexa-2,5-diene-1,4-dione | DNA metabolism [135] |
 | 7-chloro-3,5-dihydro-4H-imidazo [4, 5-d]pyridazin-4-one | DNA metabolism [136] |
 | 4-(7-chloroquinolin-4-yl)-N-(4-fluorophenyl)piperazine-1-carbothioamide | DNA topology [137] |
 | 4-((3-acetyl-1-benzyl-2-methyl-1H-indol-5-yl)oxy)butanoic acid | Folate pathway [138] |
 | 5-(4-nitrophenyl)furan-2-carboxylic acid | Iron acquisition [139] |
 | 1-(3-chloro-4-methylphenyl)-3-tosylpyrrolidine-2,5-dione | Iron acquisition control [140] |
 | (E)-N-(4-(2-(4-((5-(diethylamino)pentan-2-yl)amino)-6-methoxyquinolin-2-yl)vinyl)phenyl)-N-oxohydroxylammonium | Needed to withstand ROS- and RNS-induced stress [141] |
 | (Z)-4-((2-(4-(4-bromophenyl)thiazol-2-yl)hydrazono)methyl)-2-methoxy-6-nitrophenol | Protein synthesis [142] |
 | 3-(((Z)-5-((E)-4-(benzyloxy)benzylidene)-3-methyl-4-oxothiazolidin-2-ylidene)amino)benzoic acid | Shikimate pathway [143] |
 | 7-((4,5-dihydroxy-6-(hydroxymethyl)-3-((3,4,5-trihydroxy-6-methyltetrahydro-2H-pyran-2-yl)oxy)tetrahydro-2H-pyran-2-yl)oxy)-5-hydroxy-2-(4-hydroxyphenyl)chroman-4-one | Shikimate pathway [144] |
 | 2-phenyl-5-(4H-1,2,4-triazol-4-yl)benzo[d]oxazole | Unknown [145] |
| Database | Number of Compounds | Website * | Ref. |
|---|---|---|---|
| ** Enamine REAL | 700 million | https://enamine.net/ | [155] |
| ** ZINC | 230 million | http://zinc.docking.org/ | [156] |
| ** GDB-17 | 166 billion | http://gdb.unibe.ch/ | [157] |
| ** PubChem | 97 million | https://pubchem.ncbi.nlm.nih.gov/ | [147] |
| ** ChemSpider [142] | 77 million | http://www.chemspider.com/ | [158] |
| *** eMolecules | 24.6 million | http://www.emolecules.com | |
| ** ChEMBL | 1.9 million | https://www.ebi.ac.uk/chembl/ | [159] |
| *** ASINEX | 600,000 | http://www.asinex.com | |
| ** NCI | 460,000 | https://cactus.nci.nih.gov/download/roadma/ | [160] |
| Purpose | Program | Website * | Refs. |
|---|---|---|---|
| Prediction of binding sites and drugability | ** fpocket | https://github.com/Discngine/fpocket | [161,162] |
| ** PockDrug | http://pockdrug.rpbs.univ-paris-diderot.fr/cgi-bin/index.py?page=home | [163] | |
| ** PocketQuery | http://pocketquery.csb.pitt.edu/ | [164] | |
| ** PASS | http://www.ccl.net/cca/software/UNIX/pass/overview.html | [165] | |
| Docking | ** Autodock | http://autodock.scripps.edu/ | [166] |
| *** GOLD | https://www.ccdc.cam.ac.uk/solutions/csddiscovery/components/gold/ | [167] | |
| *** Glide | https://www.schrodinger.com/glide/ | [168] | |
| *** FlexX | https://www.biosolveit.de/flexx/index.html | [169] | |
| QSAR | *** SeeSAR | https://www.biosolveit.de/SeeSAR/ | [170] |
| ** Open3DQSAR | http://open3dqsar.sourceforge.net/?Home | [171] | |
| ** ChemSAR | http://chemsar.scbdd.com/ | [172] | |
| ADMET | *** QikProp | https://www.schrodinger.com/qikprop | [173] |
| *** ADMET Predictor | https://www.simulations-plus.com/software/overview/ | [174] | |
| ** admetSAR | http://lmmd.ecust.edu.cn/admetsar1/home/ | [175,176,177] | |
| ** VirtualToxLab | http://www.biograf.ch/index.php?id=home | [15,178,179,180] |
| Program | Library of Compounds Screened | Enzyme (Function) | Ref. |
|---|---|---|---|
| AutoDock Vina | FDA-approved: DrugBank (1932); eLEA3D (1852) | MurB and MurE (peptidoglycan biosynthesis) | [207] |
| ChemDiv dataset (135,755) | DprE1 (arabinogalactan biosynthesis) | [208] | |
| NCI; Enamine; Asinex; ChemBridge; Vitas-M Lab (total: 5.6 million) | InhA (mycolic acid biosynthesis) | [209] | |
| AutoDock 4.0 | Super Natural II database (570) | RmlD (carbohydrate biosynthesis) | [210] |
| CDOCKER | Enamine REAL database (4.5 million) | BioA (biotin biosynthesis) | [203] |
| Frigate | ZINC database (2 million) | Antigen 85c (lipid metabolism) | [211] |
| Glide | FDA-approved (6282) | LipU (lipid hydrolysis) | [212] |
| ChEMBL antimycobacterial (30,789) | DprE1 (arabinogalactan biosynthesis) | [213] | |
| FDA-approved (3176) | PknA (protein kinase) | [214] | |
| Preselected from Maybridge database (1026) | InhA (mycolic acid biosynthesis) | [215] | |
| Preselected from DrugBank database (1082) | AroB (shikimate pathway) | [216] | |
| GOLD | Drugs Now subset of ZINC database (409, 201) | EthR (transcriptional regulator) | [205] |
| GOLD and Plants | Preselected from Enamine database (2050) | MbtI (mycobactin synthesis) | [138] |
| GOLD and RFScore | Selection from 9 million compounds (4379) | AroQ (Shikimate pathway) | [217] |
| UCSF Chimera | CDD-823953; GSK-735826A | PyrG and PanK (siosynthesis of DNA and RNA) | [192] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ejalonibu, M.A.; Ogundare, S.A.; Elrashedy, A.A.; Ejalonibu, M.A.; Lawal, M.M.; Mhlongo, N.N.; Kumalo, H.M. Drug Discovery for Mycobacterium tuberculosis Using Structure-Based Computer-Aided Drug Design Approach. Int. J. Mol. Sci. 2021, 22, 13259. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222413259
Ejalonibu MA, Ogundare SA, Elrashedy AA, Ejalonibu MA, Lawal MM, Mhlongo NN, Kumalo HM. Drug Discovery for Mycobacterium tuberculosis Using Structure-Based Computer-Aided Drug Design Approach. International Journal of Molecular Sciences. 2021; 22(24):13259. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222413259
Chicago/Turabian StyleEjalonibu, Murtala A., Segun A. Ogundare, Ahmed A. Elrashedy, Morufat A. Ejalonibu, Monsurat M. Lawal, Ndumiso N. Mhlongo, and Hezekiel M. Kumalo. 2021. "Drug Discovery for Mycobacterium tuberculosis Using Structure-Based Computer-Aided Drug Design Approach" International Journal of Molecular Sciences 22, no. 24: 13259. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222413259