
Cancer-Associated Fibroblasts in Pancreatic Ductal Adenocarcinoma: An Update on Heterogeneity and Therapeutic Targeting


Abstract
:1. Introduction
2. The PDAC Tumor Microenvironment
3. Cancer-Associated Fibroblasts (CAFs)
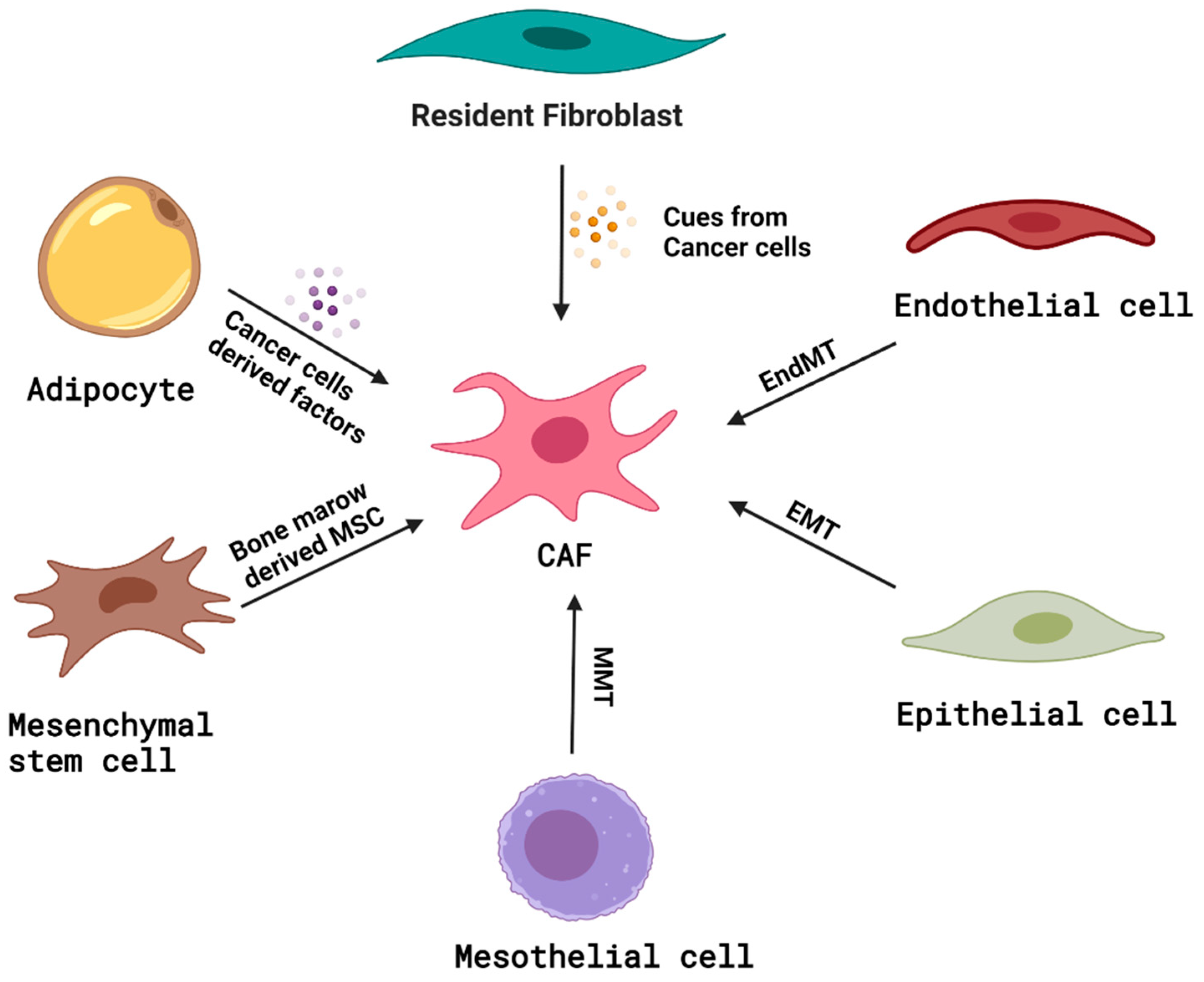
4. Origin of CAFs
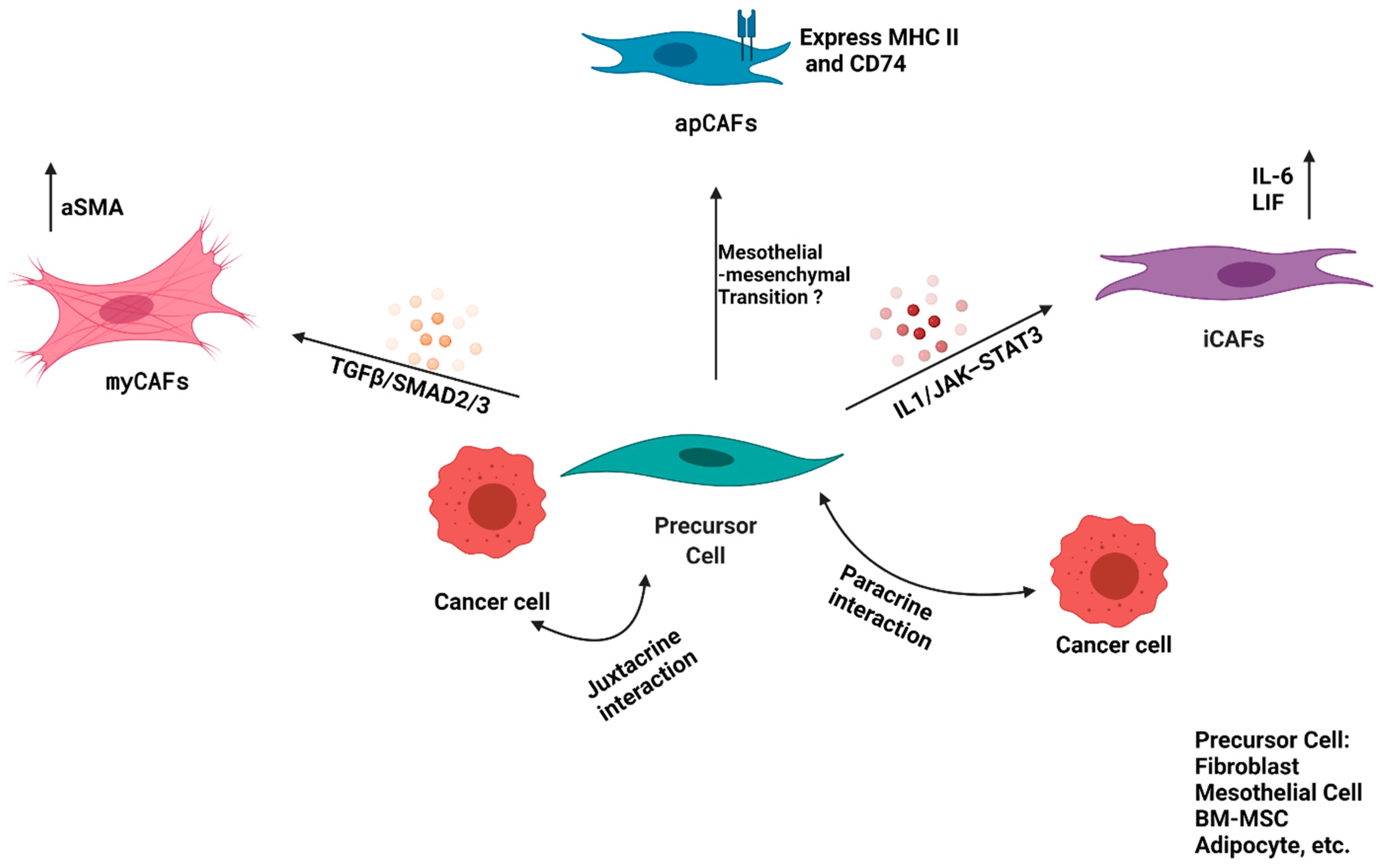
5. CAF Heterogeneity
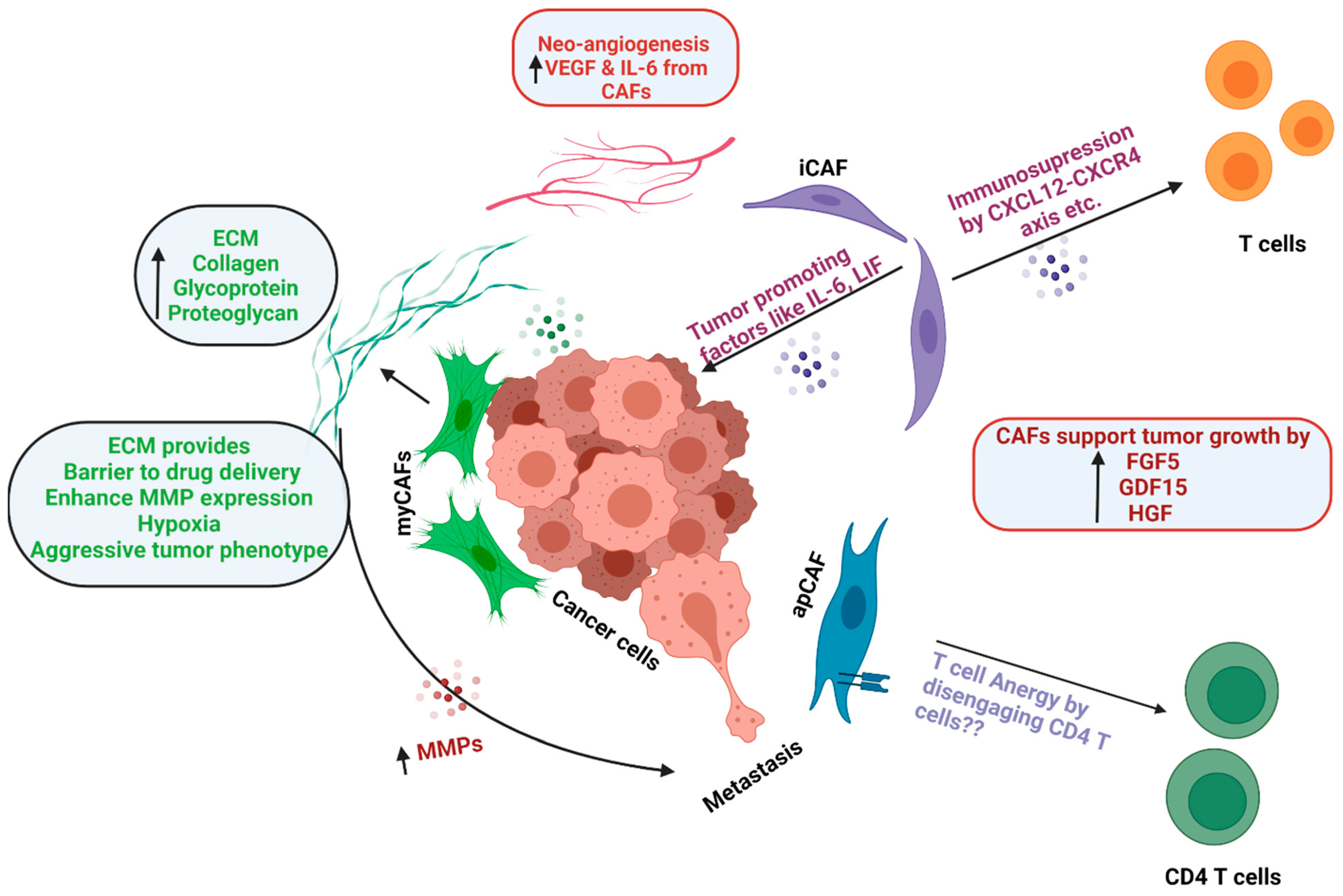
6. Functions of CAFs
7. CAFs as Therapeutic Targets
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [Green Version]
- Collisson, E.A.; Bailey, P.; Chang, D.K.; Biankin, A.V. Molecular subtypes of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Fan, Z.; Gong, Y.; Jin, K.; Yang, C.; Cheng, H.; Huang, D.; Ni, Q.; Liu, C.; Yu, X. Characteristics and Outcomes of Pancreatic Cancer by Histological Subtypes. Pancreas 2019, 48, 817–822. [Google Scholar] [CrossRef] [PubMed]
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Primers 2016, 2, 16022. [Google Scholar] [CrossRef] [PubMed]
- Mizrahi, J.D.; Surana, R.; Valle, J.W.; Shroff, R.T. Pancreatic cancer. Lancet 2020, 395, 2008–2020. [Google Scholar] [CrossRef]
- Jain, T.; Dudeja, V. The war against pancreatic cancer in 2020—advances on all fronts. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 99–100. [Google Scholar] [CrossRef] [PubMed]
- Ho, W.J.; Jaffee, E.M.; Zheng, L. The tumour microenvironment in pancreatic cancer—clinical challenges and opportunities. Nat. Rev. Clin. Oncol. 2020, 17, 527–540. [Google Scholar] [CrossRef]
- Feig, C.; Gopinathan, A.; Neesse, A.; Chan, D.S.; Cook, N.; Tuveson, D.A. The pancreas cancer microenvironment. Clin. Cancer Res. 2012, 18, 4266–4276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, J.-E.; Hajdu, C.H.; Liot, C.; Miller, G.; Dustin, M.L.; Bar-Sagi, D. Crosstalk between regulatory T cells and tumor-associated dendritic cells negates anti-tumor immunity in pancreatic cancer. Cell Rep. 2017, 20, 558–571. [Google Scholar] [CrossRef] [Green Version]
- Looi, C.-K.; Chung, F.F.-L.; Leong, C.-O.; Wong, S.-F.; Rosli, R.; Mai, C. Therapeutic challenges and current immunomodulatory strategies in targeting the immunosuppressive pancreatic tumor microenvironment. J. Exp. Clin. Cancer Res. 2019, 38, 162. [Google Scholar] [CrossRef]
- Giri, B.; Sharma, P.; Jain, T.; Ferrantella, A.; Vaish, U.; Mehra, S.; Garg, B.; Iyer, S.; Sethi, V.; Malchiodi, Z.; et al. Hsp70 modulates immune response in pancreatic cancer through dendritic cells. Oncoimmunology 2021, 10, 1976952. [Google Scholar] [CrossRef] [PubMed]
- Hegde, S.; Krisnawan, V.E.; Herzog, B.H.; Zuo, C.; Breden, M.A.; Knolhoff, B.L.; Hogg, G.D.; Tang, J.P.; Baer, J.M.; Mpoy, C.; et al. Dendritic cell paucity leads to dysfunctional immune surveillance in pancreatic cancer. Cancer Cell 2020, 37, 289–307. [Google Scholar] [CrossRef] [PubMed]
- Pergamo, M.; Miller, G. Myeloid-derived suppressor cells and their role in pancreatic cancer. Cancer Gene Ther. 2017, 24, 100–105. [Google Scholar] [CrossRef]
- Jain, T.; Sharma, P.; Are, A.C.; Vickers, S.M.; Dudeja, V. New Insights Into the Cancer–Microbiome–Immune Axis: Decrypting a Decade of Discoveries. Front. Immunol. 2021, 12, 622064. [Google Scholar] [CrossRef]
- Hay, E.D. An overview of epithelio-mesenchymal transformation. Acta Anat. 1995, 154, 8–20. [Google Scholar] [CrossRef]
- Sharpe, P.T. Neural crest and tooth morphogenesis. Adv. Dent. Res. 2001, 15, 4–7. [Google Scholar] [CrossRef]
- Gabbiani, G. The myofibroblast in wound healing and fibrocontractive diseases. J. Pathol. 2003, 200, 500–503. [Google Scholar] [CrossRef]
- Ahmadzadeh, M.; Rosenberg, S.A. TGF-beta 1 attenuates the acquisition and expression of effector function by tumor antigen-specific human memory CD8 T cells. J. Immunol. 2005, 174, 5215–5223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef] [Green Version]
- Chakravarthy, A.; Khan, L.; Bensler, N.P.; Bose, P.; De Carvalho, D.D. TGF-beta-associated extracellular matrix genes link cancer-associated fibroblasts to immune evasion and immunotherapy failure. Nat. Commun. 2018, 9, 4692. [Google Scholar] [CrossRef] [PubMed]
- Biffi, G.; Tuveson, D.A. Diversity and Biology of Cancer-Associated Fibroblasts. Physiol. Rev. 2021, 101, 147–176. [Google Scholar] [CrossRef] [PubMed]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer. 2020, 20, 174–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arina, A.; Idel, C.; Hyjek, E.M.; Alegre, M.L.; Wang, Y.; Bindokas, V.P.; Weichselbaum, R.R.; Schreiber, H. Tumor-associated fibroblasts predominantly come from local and not circulating precursors. Proc. Natl. Acad. Sci. USA 2016, 113, 7551–7556. [Google Scholar] [CrossRef] [Green Version]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 2005, 121, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Karnoub, A.E.; Dash, A.B.; Vo, A.P.; Sullivan, A.; Brooks, M.W.; Bell, G.W.; Richardson, A.L.; Polyak, K.; Tubo, R.; Weinberg, R.A. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 2007, 449, 557–563. [Google Scholar] [CrossRef]
- Mishra, P.J.; Mishra, P.J.; Humeniuk, R.; Medina, D.J.; Alexe, G.; Mesirov, J.P.; Ganesan, S.; Glod, J.W.; Banerjee, D. Carcinoma-associated fibroblast-like differentiation of human mesenchymal stem cells. Cancer Res. 2008, 68, 4331–4339. [Google Scholar] [CrossRef] [Green Version]
- Sandoval, P.; Jimenez-Heffernan, J.A.; Rynne-Vidal, A.; Perez-Lozano, M.L.; Gilsanz, A.; Ruiz-Carpio, V.; Reyes, R.; Garcia-Bordas, J.; Stamatakis, K.; Dotor, J.; et al. Carcinoma-associated fibroblasts derive from mesothelial cells via mesothelial-to-mesenchymal transition in peritoneal metastasis. J. Pathol. 2013, 231, 517–531. [Google Scholar] [CrossRef] [Green Version]
- Kidd, S.; Spaeth, E.; Watson, K.; Burks, J.; Lu, H.; Klopp, A.; Andreeff, M.; Marini, F.C. Origins of the tumor microenvironment: Quantitative assessment of adipose-derived and bone marrow-derived stroma. PLoS ONE 2012, 7, e30563. [Google Scholar] [CrossRef] [Green Version]
- McDonald, L.T.; Russell, D.L.; Kelly, R.R.; Xiong, Y.; Motamarry, A.; Patel, R.K.; Jones, J.A.; Watson, P.M.; Turner, D.P.; Watson, D.K.; et al. Hematopoietic stem cell-derived cancer-associated fibroblasts are novel contributors to the pro-tumorigenic microenvironment. Neoplasia 2015, 17, 434–448. [Google Scholar] [CrossRef] [Green Version]
- Direkze, N.C.; Forbes, S.J.; Brittan, M.; Hunt, T.; Jeffery, R.; Preston, S.L.; Poulsom, R.; Hodivala-Dilke, K.; Alison, M.R.; Wright, N.A. Multiple organ engraftment by bone-marrow-derived myofibroblasts and fibroblasts in bone-marrow-transplanted mice. Stem Cells 2003, 21, 514–520. [Google Scholar] [CrossRef] [PubMed]
- Iwano, M.; Plieth, D.; Danoff, T.M.; Xue, C.; Okada, H.; Neilson, E.G. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J. Clin. Investig. 2002, 110, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, E.M.; Potenta, S.; Xie, L.; Zeisberg, M.; Kalluri, R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res. 2007, 67, 10123–10128. [Google Scholar] [CrossRef] [Green Version]
- Quante, M.; Tu, S.P.; Tomita, H.; Gonda, T.; Wang, S.S.; Takashi, S.; Baik, G.H.; Shibata, W.; Diprete, B.; Betz, K.S.; et al. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell 2011, 19, 257–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jotzu, C.; Alt, E.; Welte, G.; Li, J.; Hennessy, B.T.; Devarajan, E.; Krishnappa, S.; Pinilla, S.; Droll, L.; Song, Y.H. Adipose tissue-derived stem cells differentiate into carcinoma-associated fibroblast-like cells under the influence of tumor-derived factors. Anal. Cell. Pathol. 2010, 33, 61–79. [Google Scholar] [CrossRef]
- Apte, M.V.; Pirola, R.C.; Wilson, J.S. Pancreatic stellate cells: A starring role in normal and diseased pancreas. Front. Physiol. 2012, 3, 344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apte, M.V.; Park, S.; Phillips, P.A.; Santucci, N.; Goldstein, D.; Kumar, R.K.; Ramm, G.A.; Buchler, M.; Friess, H.; McCarroll, J.A.; et al. Desmoplastic reaction in pancreatic cancer: Role of pancreatic stellate cells. Pancreas 2004, 29, 179–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bachem, M.G.; Schunemann, M.; Ramadani, M.; Siech, M.; Beger, H.; Buck, A.; Zhou, S.; Schmid-Kotsas, A.; Adler, G. Pancreatic carcinoma cells induce fibrosis by stimulating proliferation and matrix synthesis of stellate cells. Gastroenterology 2005, 128, 907–921. [Google Scholar] [CrossRef] [PubMed]
- Ohlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef]
- Helms, E.J.; Berry, M.W.; Chaw, R.C.; DuFort, C.C.; Sun, D.; Onate, M.K.; Oon, C.; Bhattacharyya, S.; Sanford-Crane, H.; Horton, W.; et al. Mesenchymal Lineage Heterogeneity Underlies Non-Redundant Functions of Pancreatic Cancer-Associated Fibroblasts. Cancer Discov. 2021. [Google Scholar] [CrossRef]
- Dominguez, C.X.; Muller, S.; Keerthivasan, S.; Koeppen, H.; Hung, J.; Gierke, S.; Breart, B.; Foreman, O.; Bainbridge, T.W.; Castiglioni, A.; et al. Single-Cell RNA Sequencing Reveals Stromal Evolution into LRRC15(+) Myofibroblasts as a Determinant of Patient Response to Cancer Immunotherapy. Cancer Discov. 2020, 10, 232–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, P.E.; Adoumie, M.; Kim, E.C.; Zhang, Y.; Scales, M.K.; El-Tawil, Y.S.; Shaikh, A.Z.; Wen, H.J.; Bednar, F.; Allen, B.L.; et al. Differential Contribution of Pancreatic Fibroblast Subsets to the Pancreatic Cancer Stroma. Cell. Mol. Gastroenterol. Hepatol. 2020, 10, 581–599. [Google Scholar] [CrossRef]
- Ishii, G.; Sangai, T.; Oda, T.; Aoyagi, Y.; Hasebe, T.; Kanomata, N.; Endoh, Y.; Okumura, C.; Okuhara, Y.; Magae, J.; et al. Bone-marrow-derived myofibroblasts contribute to the cancer-induced stromal reaction. Biochem. Biophys. Res. Commun. 2003, 309, 232–240. [Google Scholar] [CrossRef]
- Okumura, T.; Ohuchida, K.; Kibe, S.; Iwamoto, C.; Ando, Y.; Takesue, S.; Nakayama, H.; Abe, T.; Endo, S.; Koikawa, K.; et al. Adipose tissue-derived stromal cells are sources of cancer-associated fibroblasts and enhance tumor progression by dense collagen matrix. Int. J. Cancer 2019, 144, 1401–1413. [Google Scholar] [CrossRef] [PubMed]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutton, C.; Heider, F.; Blanco-Gomez, A.; Banyard, A.; Kononov, A.; Zhang, X.; Karim, S.; Paulus-Hock, V.; Watt, D.; Steele, N.; et al. Single-cell analysis defines a pancreatic fibroblast lineage that supports anti-tumor immunity. Cancer Cell 2021, 39, 1227–1244. [Google Scholar] [CrossRef]
- Kraman, M.; Bambrough, P.J.; Arnold, J.N.; Roberts, E.W.; Magiera, L.; Jones, J.O.; Gopinathan, A.; Tuveson, D.A.; Fearon, D.T. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science 2010, 330, 827–830. [Google Scholar] [CrossRef] [Green Version]
- Ozdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef] [Green Version]
- Vonlaufen, A.; Joshi, S.; Qu, C.; Phillips, P.A.; Xu, Z.; Parker, N.R.; Toi, C.S.; Pirola, R.C.; Wilson, J.S.; Goldstein, D.; et al. Pancreatic stellate cells: Partners in crime with pancreatic cancer cells. Cancer Rep. 2008, 68, 2085–2093. [Google Scholar] [CrossRef] [Green Version]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biffi, G.; Oni, T.E.; Spielman, B.; Hao, Y.; Elyada, E.; Park, Y.; Preall, J.; Tuveson, D.A. IL1-Induced JAK/STAT Signaling Is Antagonized by TGFbeta to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2019, 9, 282–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Lazarus, J.; Steele, N.G.; Yan, W.; Lee, H.-J.; Nwosu, Z.C.; Halbrook, C.J.; Menjivar, R.E.; Kemp, S.B.; Sirihorachai, V.; et al. Regulatory T-cell depletion alters the tumor microenvironment and accelerates pancreatic carcinogenesis. Cancer Discov. 2020, 10, 422–439. [Google Scholar] [CrossRef] [Green Version]
- Grünwald, B.T.; Devisme, A.; Andrieux, G.; Vyas, F.; Aliar, K.; McCloskey, C.W.; Macklin, A.; Jang, G.H.; Denroche, R.; Romero, J.M.; et al. Spatially confined sub-tumor microenvironments in pancreatic cancer. Cell 2021, 184, 5577–5592. [Google Scholar] [CrossRef]
- Monteran, L.; Erez, N. The Dark Side of Fibroblasts: Cancer-Associated Fibroblasts as Mediators of Immunosuppression in the Tumor Microenvironment. Front. Immunol. 2019, 10, 1835. [Google Scholar] [CrossRef] [Green Version]
- Zeltz, C.; Primac, I.; Erusappan, P.; Alam, J.; Noel, A.; Gullberg, D. Cancer-associated fibroblasts in desmoplastic tumors: Emerging role of integrins. Semin. Cancer Biol. 2020, 62, 166–181. [Google Scholar] [CrossRef]
- DuFort, C.C.; DelGiorno, K.E.; Hingorani, S.R. Mounting Pressure in the Microenvironment: Fluids, Solids, and Cells in Pancreatic Ductal Adenocarcinoma. Gastroenterology 2016, 150, 1545–1557 e1542. [Google Scholar] [CrossRef] [Green Version]
- Paszek, M.J.; Zahir, N.; Johnson, K.R.; Lakins, J.N.; Rozenberg, G.I.; Gefen, A.; Reinhart-King, C.A.; Margulies, S.S.; Dembo, M.; Boettiger, D.; et al. Tensional homeostasis and the malignant phenotype. Cancer Cell 2005, 8, 241–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Liu, J.; Huang, H.; Ye, M.; Li, X.; Wu, R.; Liu, H.; Song, Y. Metastasis-associated fibroblasts: An emerging target for metastatic cancer. Biomark. Res. 2021, 9, 47. [Google Scholar] [CrossRef] [PubMed]
- Fukumura, D.; Xavier, R.; Sugiura, T.; Chen, Y.; Park, E.C.; Lu, N.; Selig, M.; Nielsen, G.; Taksir, T.; Jain, R.K.; et al. Tumor induction of VEGF promoter activity in stromal cells. Cell 1998, 94, 715–725. [Google Scholar] [CrossRef] [Green Version]
- Nagasaki, T.; Hara, M.; Nakanishi, H.; Takahashi, H.; Sato, M.; Takeyama, H. Interleukin-6 released by colon cancer-associated fibroblasts is critical for tumour angiogenesis: Anti-interleukin-6 receptor antibody suppressed angiogenesis and inhibited tumour-stroma interaction. Br. J. Cancer 2014, 110, 469–478. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Recouvreux, M.V.; Jung, M.; Galenkamp, K.M.O.; Li, Y.; Zagnitko, O.; Scott, D.A.; Lowy, A.M.; Commisso, C. Macropinocytosis in Cancer-Associated Fibroblasts is Dependent on CaMKK2/ARHGEF2 Signaling and Functions to Support Tumor and Stromal Cell Fitness. Cancer Discov. 2021, 11. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Gao, W.; Lytle, N.K.; Huang, P.; Yuan, X.; Dann, A.M.; Ridinger-Saison, M.; DelGiorno, K.E.; Antal, C.E.; Liang, G.; et al. Targeting LIF-mediated paracrine interaction for pancreatic cancer therapy and monitoring. Nature 2019, 569, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Tape, C.J.; Ling, S.; Dimitriadi, M.; McMahon, K.M.; Worboys, J.D.; Leong, H.S.; Norrie, I.C.; Miller, C.J.; Poulogiannis, G.; Lauffenburger, D.A.; et al. Oncogenic KRAS Regulates Tumor Cell Signaling via Stromal Reciprocation. Cell 2016, 165, 1818. [Google Scholar] [CrossRef] [PubMed]
- Bruzzese, F.; Hagglof, C.; Leone, A.; Sjoberg, E.; Roca, M.S.; Kiflemariam, S.; Sjoblom, T.; Hammarsten, P.; Egevad, L.; Bergh, A.; et al. Local and systemic protumorigenic effects of cancer-associated fibroblast-derived GDF15. Cancer Res. 2014, 74, 3408–3417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fearon, D.T. The carcinoma-associated fibroblast expressing fibroblast activation protein and escape from immune surveillance. Cancer Immunol. Res. 2014, 2, 187–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salmon, H.; Franciszkiewicz, K.; Damotte, D.; Dieu-Nosjean, M.C.; Validire, P.; Trautmann, A.; Mami-Chouaib, F.; Donnadieu, E. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J. Clin. Investig. 2012, 122, 899–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flint, T.R.; Janowitz, T.; Connell, C.M.; Roberts, E.W.; Denton, A.E.; Coll, A.P.; Jodrell, D.I.; Fearon, D.T. Tumor-Induced IL-6 Reprograms Host Metabolism to Suppress Anti-tumor Immunity. Cell Metab. 2016, 24, 672–684. [Google Scholar] [CrossRef]
- Chen, Y.; Kim, J.; Yang, S.; Wang, H.; Wu, C.J.; Sugimoto, H.; LeBleu, V.S.; Kalluri, R. Type I collagen deletion in alphaSMA(+) myofibroblasts augments immune suppression and accelerates progression of pancreatic cancer. Cancer Cell 2021, 39, 548–565.e546. [Google Scholar] [CrossRef]
- Lo, A.; Li, C.P.; Buza, E.L.; Blomberg, R.; Govindaraju, P.; Avery, D.; Monslow, J.; Hsiao, M.; Pure, E. Fibroblast activation protein augments progression and metastasis of pancreatic ductal adenocarcinoma. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [Green Version]
- Hofheinz, R.D.; al-Batran, S.E.; Hartmann, F.; Hartung, G.; Jager, D.; Renner, C.; Tanswell, P.; Kunz, U.; Amelsberg, A.; Kuthan, H.; et al. Stromal antigen targeting by a humanised monoclonal antibody: An early phase II trial of sibrotuzumab in patients with metastatic colorectal cancer. Onkologie 2003, 26, 44–48. [Google Scholar] [CrossRef]
- Duperret, E.K.; Trautz, A.; Ammons, D.; Perales-Puchalt, A.; Wise, M.C.; Yan, J.; Reed, C.; Weiner, D.B. Alteration of the Tumor Stroma Using a Consensus DNA Vaccine Targeting Fibroblast Activation Protein (FAP) Synergizes with Antitumor Vaccine Therapy in Mice. Clin. Cancer Res. 2018, 24, 1190–1201. [Google Scholar] [CrossRef] [Green Version]
- Lo, A.; Wang, L.S.; Scholler, J.; Monslow, J.; Avery, D.; Newick, K.; O’Brien, S.; Evans, R.A.; Bajor, D.J.; Clendenin, C.; et al. Tumor-Promoting Desmoplasia Is Disrupted by Depleting FAP-Expressing Stromal Cells. Cancer Res. 2015, 75, 2800–2810. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.C.; Lo, A.; Scholler, J.; Sun, J.; Majumdar, R.S.; Kapoor, V.; Antzis, M.; Cotner, C.E.; Johnson, L.A.; Durham, A.C.; et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol. Res. 2014, 2, 154–166. [Google Scholar] [CrossRef] [Green Version]
- Bailey, J.M.; Swanson, B.J.; Hamada, T.; Eggers, J.P.; Singh, P.K.; Caffery, T.; Ouellette, M.M.; Hollingsworth, M.A. Sonic hedgehog promotes desmoplasia in pancreatic cancer. Clin. Cancer Res. 2008, 14, 5995–6004. [Google Scholar] [CrossRef] [Green Version]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef] [Green Version]
- Feldmann, G.; Habbe, N.; Dhara, S.; Bisht, S.; Alvarez, H.; Fendrich, V.; Beaty, R.; Mullendore, M.; Karikari, C.; Bardeesy, N.; et al. Hedgehog inhibition prolongs survival in a genetically engineered mouse model of pancreatic cancer. Gut 2008, 57, 1420–1430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.J.; Perera, R.M.; Wang, H.; Wu, D.C.; Liu, X.S.; Han, S.; Fitamant, J.; Jones, P.D.; Ghanta, K.S.; Kawano, S.; et al. Stromal response to Hedgehog signaling restrains pancreatic cancer progression. Proc. Natl. Acad. Sci. USA 2014, 111, E3091–E3100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steele, N.G.; Biffi, G.; Kemp, S.B.; Zhang, Y.; Drouillard, D.; Syu, L.; Hao, Y.; Oni, T.E.; Brosnan, E.; Elyada, E.; et al. Inhibition of Hedgehog Signaling Alters Fibroblast Composition in Pancreatic Cancer. Clin. Cancer Res. 2021, 27, 2023–2037. [Google Scholar] [CrossRef] [PubMed]
- Catenacci, D.V.; Junttila, M.R.; Karrison, T.; Bahary, N.; Horiba, M.N.; Nattam, S.R.; Marsh, R.; Wallace, J.; Kozloff, M.; Rajdev, L.; et al. Randomized Phase Ib/II Study of Gemcitabine Plus Placebo or Vismodegib, a Hedgehog Pathway Inhibitor, in Patients With Metastatic Pancreatic Cancer. J. Clin. Oncol. 2015, 33, 4284–4292. [Google Scholar] [CrossRef] [PubMed]
- Richards, D.A.; Stephenson, J.; Wolpin, B.M.; Becerra, C.; Hamm, J.T.; Messersmith, W.A.; Devens, S.; Cushing, J.; Schmalbach, T.; Fuchs, C.S.; et al. A phase Ib trial of IPI-926, a hedgehog pathway inhibitor, plus gemcitabine in patients with metastatic pancreatic cancer. J. Clin. Oncol. 2012, 30, 213. [Google Scholar] [CrossRef]
- Kim, E.J.; Sahai, V.; Abel, E.V.; Griffith, K.A.; Greenson, J.K.; Takebe, N.; Khan, G.N.; Blau, J.L.; Craig, R.; Balis, U.G.; et al. Pilot clinical trial of hedgehog pathway inhibitor GDC-0449 (vismodegib) in combination with gemcitabine in patients with metastatic pancreatic adenocarcinoma. Clin. Cancer Res. 2014, 20, 5937–5945. [Google Scholar] [CrossRef] [Green Version]
- A Study Evaluating IPI-926 in Combination with Gemcitabine in Patients with Metastatic Pancreatic Cancer. Available online: https://ClinicalTrials.gov/show/NCT01130142 (accessed on 16 October 2021).
- Vismodegib and Gemcitabine Hydrochloride in Treating Patients with Advanced Pancreatic Cancer. Available online: https://ClinicalTrials.gov/show/NCT01195415 (accessed on 16 October 2021).
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 418–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobetz, M.A.; Chan, D.S.; Neesse, A.; Bapiro, T.E.; Cook, N.; Frese, K.K.; Feig, C.; Nakagawa, T.; Caldwell, M.E.; Zecchini, H.I.; et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut 2013, 62, 112–120. [Google Scholar] [CrossRef]
- Ramanathan, R.K.; McDonough, S.L.; Philip, P.A.; Hingorani, S.R.; Lacy, J.; Kortmansky, J.S.; Thumar, J.; Chiorean, E.G.; Shields, A.F.; Behl, D.; et al. Phase IB/II Randomized Study of FOLFIRINOX Plus Pegylated Recombinant Human Hyaluronidase Versus FOLFIRINOX Alone in Patients With Metastatic Pancreatic Adenocarcinoma: SWOG S1313. J. Clin. Oncol. 2019, 37, 1062–1069. [Google Scholar] [CrossRef]
- Tempero, M.A.; Van Cutsem, E.; Sigal, D.; Oh, D.-Y.; Fazio, N.; Macarulla, T.; Hitre, E.; Hammel, P.; Hendifar, A.E.; Bates, S.E. HALO 109-301: A randomized, double-blind, placebo-controlled, phase 3 study of pegvorhyaluronidase alfa (PEGPH20)+ nab-paclitaxel/gemcitabine (AG) in patients (pts) with previously untreated hyaluronan (HA)-high metastatic pancreatic ductal adenocarcinoma (mPDA). Am. Soc. Clin. Oncol. 2020, 38, 638. [Google Scholar]
- Second-Line Study of PEGPH20 and Pembro for HA High Metastatic PDAC. Available online: https://ClinicalTrials.gov/show/NCT03634332 (accessed on 16 October 2021).
- Diop-Frimpong, B.; Chauhan, V.P.; Krane, S.; Boucher, Y.; Jain, R.K. Losartan inhibits collagen I synthesis and improves the distribution and efficacy of nanotherapeutics in tumors. Proc. Natl. Acad. Sci. USA 2011, 108, 2909–2914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chauhan, V.P.; Martin, J.D.; Liu, H.; Lacorre, D.A.; Jain, S.R.; Kozin, S.V.; Stylianopoulos, T.; Mousa, A.S.; Han, X.; Adstamongkonkul, P.; et al. Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat. Commun. 2013, 4, 2516. [Google Scholar] [CrossRef] [Green Version]
- Murphy, J.E.; Wo, J.Y.; Ryan, D.P.; Clark, J.W.; Jiang, W.; Yeap, B.Y.; Drapek, L.C.; Ly, L.; Baglini, C.V.; Blaszkowsky, L.S.; et al. Total Neoadjuvant Therapy With FOLFIRINOX in Combination With Losartan Followed by Chemoradiotherapy for Locally Advanced Pancreatic Cancer: A Phase 2 Clinical Trial. JAMA Oncol. 2019, 5, 1020–1027. [Google Scholar] [CrossRef]
- Losartan and Nivolumab in Combination with FOLFIRINOX and SBRT in Localized Pancreatic Cancer. Available online: https://ClinicalTrials.gov/show/NCT03563248 (accessed on 16 October 2021).
- Sulzmaier, F.J.; Jean, C.; Schlaepfer, D.D. FAK in cancer: Mechanistic findings and clinical applications. Nat. Rev. Cancer 2014, 14, 598–610. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Hegde, S.; Knolhoff, B.L.; Zhu, Y.; Herndon, J.M.; Meyer, M.A.; Nywening, T.M.; Hawkins, W.G.; Shapiro, I.M.; Weaver, D.T.; et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat. Med. 2016, 22, 851–860. [Google Scholar] [CrossRef]
- Whatcott, C.J.; Ng, S.; Barrett, M.T.; Hostetter, G.; Von Hoff, D.D.; Han, H. Inhibition of ROCK1 kinase modulates both tumor cells and stromal fibroblasts in pancreatic cancer. PLoS ONE 2017, 12, e0183871. [Google Scholar] [CrossRef] [Green Version]
- Rath, N.; Morton, J.P.; Julian, L.; Helbig, L.; Kadir, S.; McGhee, E.J.; Anderson, K.I.; Kalna, G.; Mullin, M.; Pinho, A.V.; et al. ROCK signaling promotes collagen remodeling to facilitate invasive pancreatic ductal adenocarcinoma tumor cell growth. EMBO Mol. Med. 2017, 9, 198–218. [Google Scholar] [CrossRef] [PubMed]
- Molnar, J.; Fong, K.S.; He, Q.P.; Hayashi, K.; Kim, Y.; Fong, S.F.; Fogelgren, B.; Szauter, K.M.; Mink, M.; Csiszar, K. Structural and functional diversity of lysyl oxidase and the LOX-like proteins. Biochim. Biophys. Acta 2003, 1647, 220–224. [Google Scholar] [CrossRef]
- Wu, L.; Zhu, Y. The function and mechanisms of action of LOXL2 in cancer (Review). Int. J. Mol. Med. 2015, 36, 1200–1204. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, S.; Saraiva, N.; Rijo, P.; Fernandes, A.S. LOXL2 Inhibitors and Breast Cancer Progression. Antioxidants 2021, 10, 312. [Google Scholar] [CrossRef]
- Benson, A.B., 3rd; Wainberg, Z.A.; Hecht, J.R.; Vyushkov, D.; Dong, H.; Bendell, J.; Kudrik, F. A Phase II Randomized, Double-Blind, Placebo-Controlled Study of Simtuzumab or Placebo in Combination with Gemcitabine for the First-Line Treatment of Pancreatic Adenocarcinoma. Oncologist 2017, 22, 241-e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- A Study to Evaluate the Efficacy and Safety of Simtuzumab Combined with Gemcitabine for Metastatic Pancreatic Adenocarcinoma. Available online: https://ClinicalTrials.gov/show/NCT01472198 (accessed on 17 October 2021).
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L.; et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Wang, X.; Contino, G.; Liesa, M.; Sahin, E.; Ying, H.; Bause, A.; Li, Y.; Stommel, J.M.; Dell’antonio, G.; et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011, 25, 717–729. [Google Scholar] [CrossRef] [Green Version]
- Commisso, C.; Davidson, S.M.; Soydaner-Azeloglu, R.G.; Parker, S.J.; Kamphorst, J.J.; Hackett, S.; Grabocka, E.; Nofal, M.; Drebin, J.A.; Thompson, C.B.; et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 2013, 497, 633–637. [Google Scholar] [CrossRef] [Green Version]
- Sousa, C.M.; Biancur, D.E.; Wang, X.; Halbrook, C.J.; Sherman, M.H.; Zhang, L.; Kremer, D.; Hwang, R.F.; Witkiewicz, A.K.; Ying, H.; et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 2016, 536, 479–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, S.J.; Amendola, C.R.; Hollinshead, K.E.R.; Yu, Q.; Yamamoto, K.; Encarnacion-Rosado, J.; Rose, R.E.; LaRue, M.M.; Sohn, A.S.W.; Biancur, D.E.; et al. Selective Alanine Transporter Utilization Creates a Targetable Metabolic Niche in Pancreatic Cancer. Cancer Discov. 2020, 10, 1018–1037. [Google Scholar] [CrossRef]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496, 101–105. [Google Scholar] [CrossRef]
- Francescone, R.; Barbosa Vendramini-Costa, D.; Franco-Barraza, J.; Wagner, J.; Muir, A.; Lau, A.N.; Gabitova, L.; Pazina, T.; Gupta, S.; Luong, T.; et al. Netrin G1 Promotes Pancreatic Tumorigenesis through Cancer-Associated Fibroblast-Driven Nutritional Support and Immunosuppression. Cancer Discov. 2021, 11, 446–479. [Google Scholar] [CrossRef]
- Auciello, F.R.; Bulusu, V.; Oon, C.; Tait-Mulder, J.; Berry, M.; Bhattacharyya, S.; Tumanov, S.; Allen-Petersen, B.L.; Link, J.; Kendsersky, N.D.; et al. A Stromal Lysolipid-Autotaxin Signaling Axis Promotes Pancreatic Tumor Progression. Cancer Discov. 2019, 9, 617–627. [Google Scholar] [CrossRef] [Green Version]
- Encarnacion-Rosado, J.; Kimmelman, A.C. Harnessing metabolic dependencies in pancreatic cancers. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 482–492. [Google Scholar] [CrossRef]
- Dvorak, H.F. Tumors: Wounds that do not heal-redux. Cancer Immunol. Res. 2015, 3, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, H.Y.; Sneddon, J.B.; Alizadeh, A.A.; Sood, R.; West, R.B.; Montgomery, K.; Chi, J.T.; van de Rijn, M.; Botstein, D.; Brown, P.O. Gene expression signature of fibroblast serum response predicts human cancer progression: Similarities between tumors and wounds. PLoS Biol. 2004, 2, e7. [Google Scholar] [CrossRef] [Green Version]
- Garg, B.; Giri, B.; Modi, S.; Sethi, V.; Castro, I.; Umland, O.; Ban, Y.; Lavania, S.; Dawra, R.; Banerjee, S.; et al. NFkappaB in Pancreatic Stellate Cells Reduces Infiltration of Tumors by Cytotoxic T Cells and Killing of Cancer Cells, via Up-regulation of CXCL12. Gastroenterology 2018, 155, 880–891.e888. [Google Scholar] [CrossRef]
- Singh, S.; Srivastava, S.K.; Bhardwaj, A.; Owen, L.B.; Singh, A.P. CXCL12-CXCR4 signalling axis confers gemcitabine resistance to pancreatic cancer cells: A novel target for therapy. Br. J. Cancer 2010, 103, 1671–1679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olaptesed (NOX-A12) Alone and in Combination with Pembrolizumab in Colorectal and Pancreatic Cancer. Available online: https://ClinicalTrials.gov/show/NCT03168139 (accessed on 16 October 2021).
- Study Assessing Safety and Efficacy of Combination of BL-8040 and Pembrolizumab in Metastatic Pancreatic Cancer Patients (COMBAT/KEYNOTE-202). Available online: https://ClinicalTrials.gov/show/NCT02826486 (accessed on 16 October 2021).
- Long, K.B.; Tooker, G.; Tooker, E.; Luque, S.L.; Lee, J.W.; Pan, X.; Beatty, G.L. IL6 Receptor Blockade Enhances Chemotherapy Efficacy in Pancreatic Ductal Adenocarcinoma. Mol. Cancer Ther. 2017, 16, 1898–1908. [Google Scholar] [CrossRef] [PubMed]
- Nagathihalli, N.S.; Castellanos, J.A.; VanSaun, M.N.; Dai, X.; Ambrose, M.; Guo, Q.; Xiong, Y.; Merchant, N.B. Pancreatic stellate cell secreted IL-6 stimulates STAT3 dependent invasiveness of pancreatic intraepithelial neoplasia and cancer cells. Oncotarget 2016, 7, 65982–65992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.W.; Stone, M.L.; Porrett, P.M.; Thomas, S.K.; Komar, C.A.; Li, J.H.; Delman, D.; Graham, K.; Gladney, W.L.; Hua, X.; et al. Hepatocytes direct the formation of a pro-metastatic niche in the liver. Nature 2019, 567, 249–252. [Google Scholar] [CrossRef] [PubMed]
- Mace, T.A.; Shakya, R.; Pitarresi, J.R.; Swanson, B.; McQuinn, C.W.; Loftus, S.; Nordquist, E.; Cruz-Monserrate, Z.; Yu, L.; Young, G.; et al. IL-6 and PD-L1 antibody blockade combination therapy reduces tumour progression in murine models of pancreatic cancer. Gut 2018, 67, 320–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kesh, K.; Garrido, V.T.; Dosch, A.; Durden, B.; Gupta, V.K.; Sharma, N.S.; Lyle, M.; Nagathihalli, N.; Merchant, N.; Saluja, A.; et al. Stroma secreted IL6 selects for “stem-like” population and alters pancreatic tumor microenvironment by reprogramming metabolic pathways. Cell Death Dis. 2020, 11, 967. [Google Scholar] [CrossRef]
- Sun, X.; He, X.; Zhang, Y.; Hosaka, K.; Andersson, P.; Wu, J.; Wu, J.; Jing, X.; Du, Q.; Hui, X.; et al. Inflammatory cell-derived CXCL3 promotes pancreatic cancer metastasis through a novel myofibroblast-hijacked cancer escape mechanism. Gut 2021. [Google Scholar] [CrossRef]
- Siltuximab and Spartalizumab in Patients with Metastatic Pancreatic Cancer. Available online: https://ClinicalTrials.gov/show/NCT04191421 (accessed on 16 October 2021).
- van Duijneveldt, G.; Griffin, M.D.W.; Putoczki, T.L. Emerging roles for the IL-6 family of cytokines in pancreatic cancer. Clin. Sci. 2020, 134, 2091–2115. [Google Scholar] [CrossRef] [PubMed]
- Sherman, M.H.; Yu, R.T.; Engle, D.D.; Ding, N.; Atkins, A.R.; Tiriac, H.; Collisson, E.A.; Connor, F.; Van Dyke, T.; Kozlov, S.; et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell 2014, 159, 80–93. [Google Scholar] [CrossRef] [Green Version]
- A Phase Ib Pharmacodynamic Study of Neoadjuvant Paricalcitol in Resectable Pancreatic Cancer A Phase Ib Pharmacodynamic Study of Neoadjuvant Paricalcitol in Resectable Pancreatic Cancer. Available online: https://ClinicalTrials.gov/show/NCT03300921 (accessed on 16 October 2021).
- High-Dose Vitamin D3 in Pancreas Cancer. Available online: https://ClinicalTrials.gov/show/NCT03472833 (accessed on 16 October 2021).
- Paricalcitol Plus Gemcitabine and Nab-Paclitaxel in Metastatic Pancreatic Cancer. Available online: https://ClinicalTrials.gov/show/NCT03520790 (accessed on 16 October 2021).
- Combination Therapy for Patients with Untreated Metastatic Pancreatic Ductal Adenocarcinoma. Available online: https://ClinicalTrials.gov/show/NCT02754726 (accessed on 16 October 2021).
- Chronopoulos, A.; Robinson, B.; Sarper, M.; Cortes, E.; Auernheimer, V.; Lachowski, D.; Attwood, S.; Garcia, R.; Ghassemi, S.; Fabry, B.; et al. ATRA mechanically reprograms pancreatic stellate cells to suppress matrix remodelling and inhibit cancer cell invasion. Nat. Commun. 2016, 7, 12630. [Google Scholar] [CrossRef] [Green Version]
- Froeling, F.E.; Feig, C.; Chelala, C.; Dobson, R.; Mein, C.E.; Tuveson, D.A.; Clevers, H.; Hart, I.R.; Kocher, H.M. Retinoic acid-induced pancreatic stellate cell quiescence reduces paracrine Wnt-β-catenin signaling to slow tumor progression. Gastroenterology 2011, 141, 1486–1497. [Google Scholar] [CrossRef]
- Kocher, H.M.; Basu, B.; Froeling, F.E.M.; Sarker, D.; Slater, S.; Carlin, D.; deSouza, N.M.; De Paepe, K.N.; Goulart, M.R.; Hughes, C.; et al. Phase I clinical trial repurposing all-trans retinoic acid as a stromal targeting agent for pancreatic cancer. Nat. Commun. 2020, 11, 4841. [Google Scholar] [CrossRef] [PubMed]
- Stromal TARgeting for PAncreatic Cancer (STAR_PAC). Available online: https://ClinicalTrials.gov/show/NCT03307148 (accessed on 16 October 2021).
- Hurwitz, H.; Van Cutsem, E.; Bendell, J.; Hidalgo, M.; Li, C.P.; Salvo, M.G.; Macarulla, T.; Sahai, V.; Sama, A.; Greeno, E.; et al. Ruxolitinib + capecitabine in advanced/metastatic pancreatic cancer after disease progression/intolerance to first-line therapy: JANUS 1 and 2 randomized phase III studies. Investig. New Drugs 2018, 36, 683–695. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Li, L.; Jiang, H.; Li, Q.; Wang-Gillam, A.; Yu, J.; Head, R.; Liu, J.; Ruzinova, M.B.; Lim, K.H. Tumor-Stroma IL1beta-IRAK4 Feedforward Circuitry Drives Tumor Fibrosis, Chemoresistance, and Poor Prognosis in Pancreatic Cancer. Cancer Res. 2018, 78, 1700–1712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gemcitabine, Nab-Paclitaxel, Cisplatin and Anakinra Treatment on Patients with Pancreatic Cancer. Available online: https://ClinicalTrials.gov/show/NCT02550327 (accessed on 16 October 2021).




{kind=link}
{kind=link}
{kind=link}
| S. No. | Target | Name of Therapeutic | Rationale Based on Pre-Clinical Studies | Current Status | ClinicalTrials.gov Identifier |
|---|---|---|---|---|---|
| 1. | Hedgehog Pathway | IPI-926 | Inhibition of the Hedgehog Pathway, leading to reduced CAF activation | Phase II was halted due to the early detection of a shorter median overall survival (OS) in the experimental arm, compared to the placebo arm. | NCT01130142 |
| 2. | Hedgehog Pathway | Vismodegib | The phase Ib/II randomized clinical trial, evaluating the addition of Vismodegib to gemcitabine, showed no treatment benefit for OS or progression free survival (PFS). | NCT01195415 | |
| 3. | Hyaluronic acid | PEGPH20 | Depletion of stroma by PEGPH20, which may synergize with immunotherapy | Clinical trials failed to show any benefit. | NCT03634332 |
| 4. | Angiotensin receptor | Losartan | Attenuation of collagen and hyaluronan deposition by CAFs through inhibition of TGF-β signaling | Encouraging results in locally advanced PDAC | NCT03563248 |
| 5. | LOXL2 | Simtuzumab | Inhibition of matrix-remodeling enzyme Lysyl oxidase-like 2, an ECM remodeling enzyme | Study completed, and addition of Simtuzumab to gemcitabine did not improve clinical outcomes | NCT01472198 |
| 6. | CXCL12-CXCR4 Axis | Olaptesed (NOX-A12) | Modulation of PDAC TME by reducing immunosuppressive factors, as CXCL12 secreted by iCAFs promotes tumorigenesis by reducing CD8 T cell infiltration | Clinical trial ongoing | NCT03168139 |
| 7. | CXCL12-CXCR4 Axis | BL-8040 CXCR4 Antagonist | Clinical trial ongoing | NCT02826486 | |
| 8. | IL-6 | Siltuximab | Combination of IL-6 and PD-L1 blockade decreases tumor growth, improves survival, and leads to increased infiltration of effector CD8+ T cells | Clinical trial ongoing | NCT04191421 |
| 9. | Vitamin D receptor (VDR) | Paricalcitol (Vitamin D Receptor Agonist) | Modulating signaling in tumor microenvironment. CAFs highly express VDR, and treating them with Vitamin D can induce a quiescent phenotype | Clinical trials ongoing | NCT03520790 |
| NCT03300921 | |||||
| NCT02754726 | |||||
| 10. | Vitamin D receptor (VDR) | Vitamin D3 | Clinical trials ongoing | NCT03472833 | |
| 11. | Stroma | All Trans Retinoic Acid (ATRA) | Inducing CAF quiescence and decreasing motility, leading to decreased tumor growth through decreased Wnt-β Catenin signaling | Clinical trials ongoing | NCT03307148 |
| 12. | IL-1R | Anakinra (IL-1R antagonist) | By switching iCAF to a myCAF phenotype | Clinical trials ongoing | NCT02550327 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vaish, U.; Jain, T.; Are, A.C.; Dudeja, V. Cancer-Associated Fibroblasts in Pancreatic Ductal Adenocarcinoma: An Update on Heterogeneity and Therapeutic Targeting. Int. J. Mol. Sci. 2021, 22, 13408. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222413408
Vaish U, Jain T, Are AC, Dudeja V. Cancer-Associated Fibroblasts in Pancreatic Ductal Adenocarcinoma: An Update on Heterogeneity and Therapeutic Targeting. International Journal of Molecular Sciences. 2021; 22(24):13408. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222413408
Chicago/Turabian StyleVaish, Utpreksha, Tejeshwar Jain, Abhi C. Are, and Vikas Dudeja. 2021. "Cancer-Associated Fibroblasts in Pancreatic Ductal Adenocarcinoma: An Update on Heterogeneity and Therapeutic Targeting" International Journal of Molecular Sciences 22, no. 24: 13408. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222413408