
Mechanism of Blood–Heart-Barrier Leakage: Implications for COVID-19 Induced Cardiovascular Injury

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
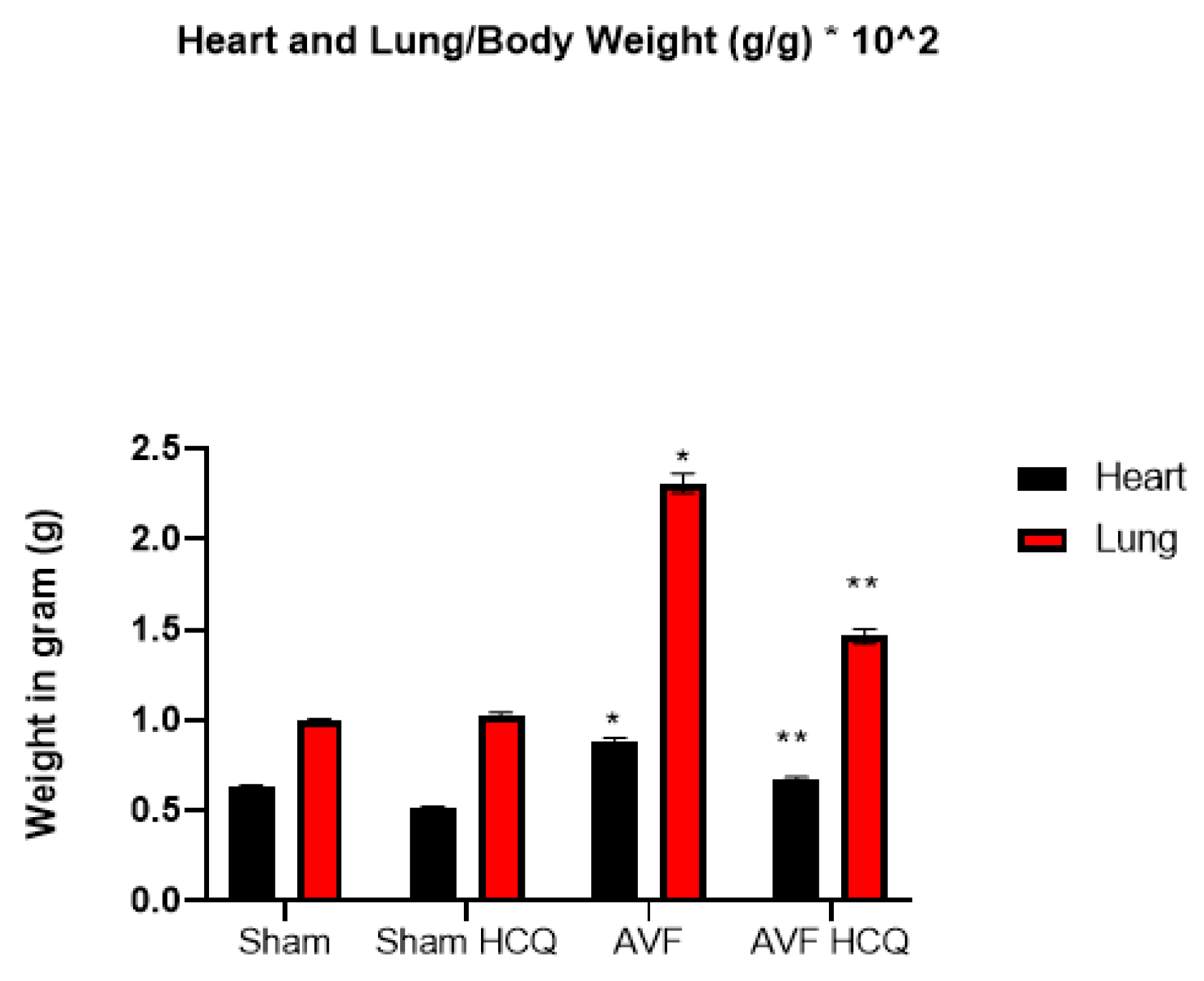
2.1. Effects of HCQ on Heart and Lung Weights
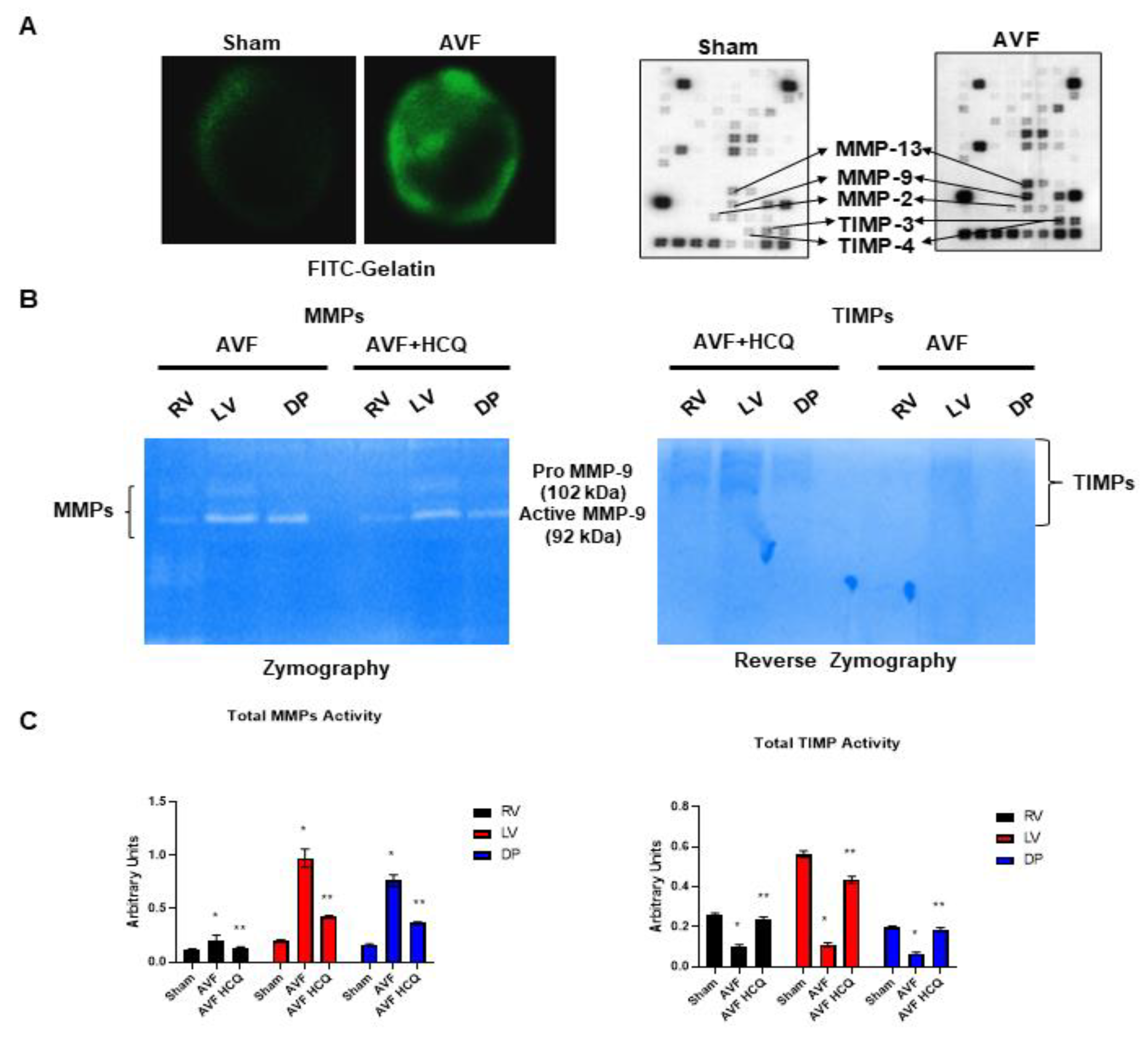
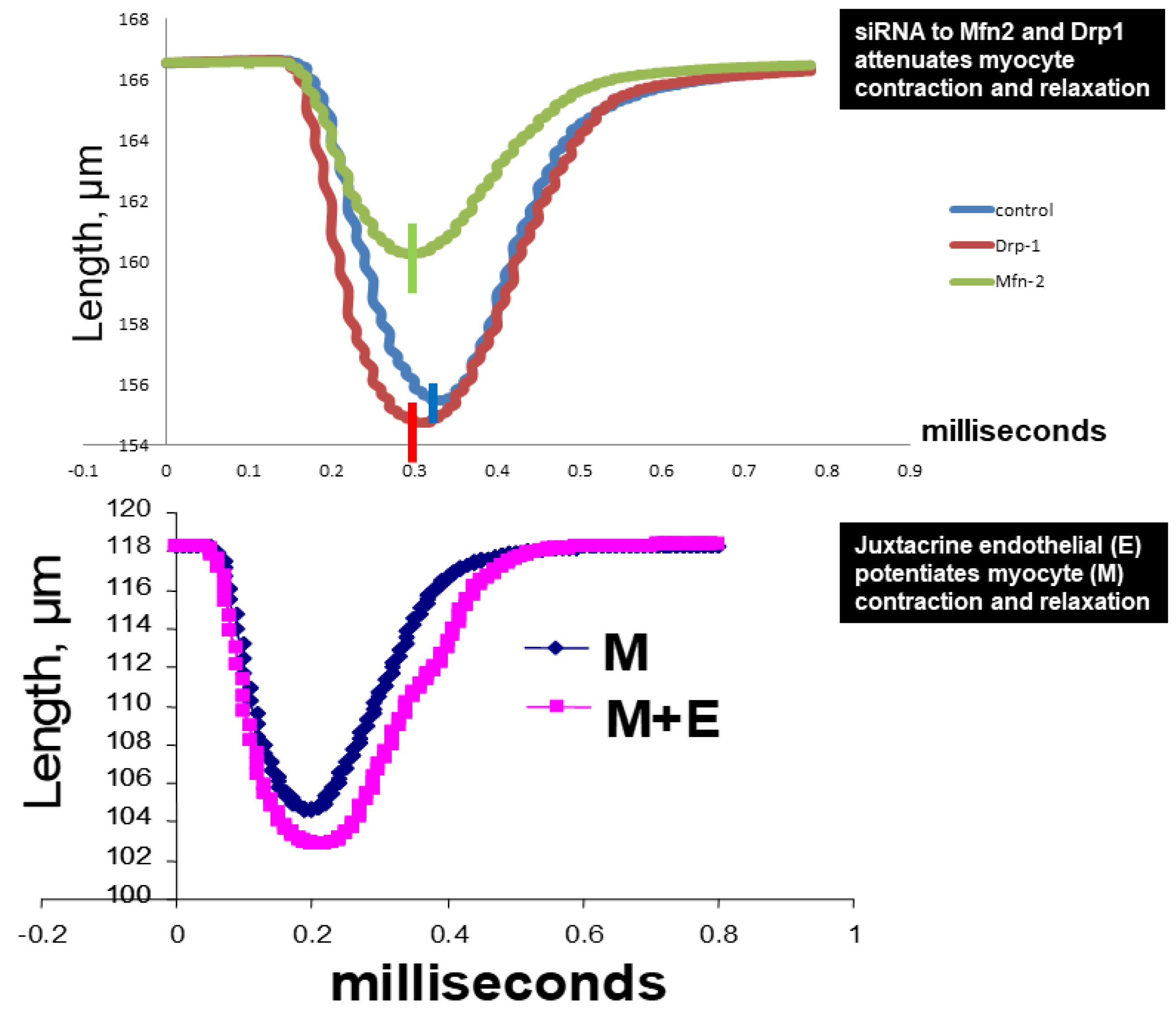
2.2. Assessment of CK Isoforms, MMPs, and TIMPs Activities, and Myocytes Contractions
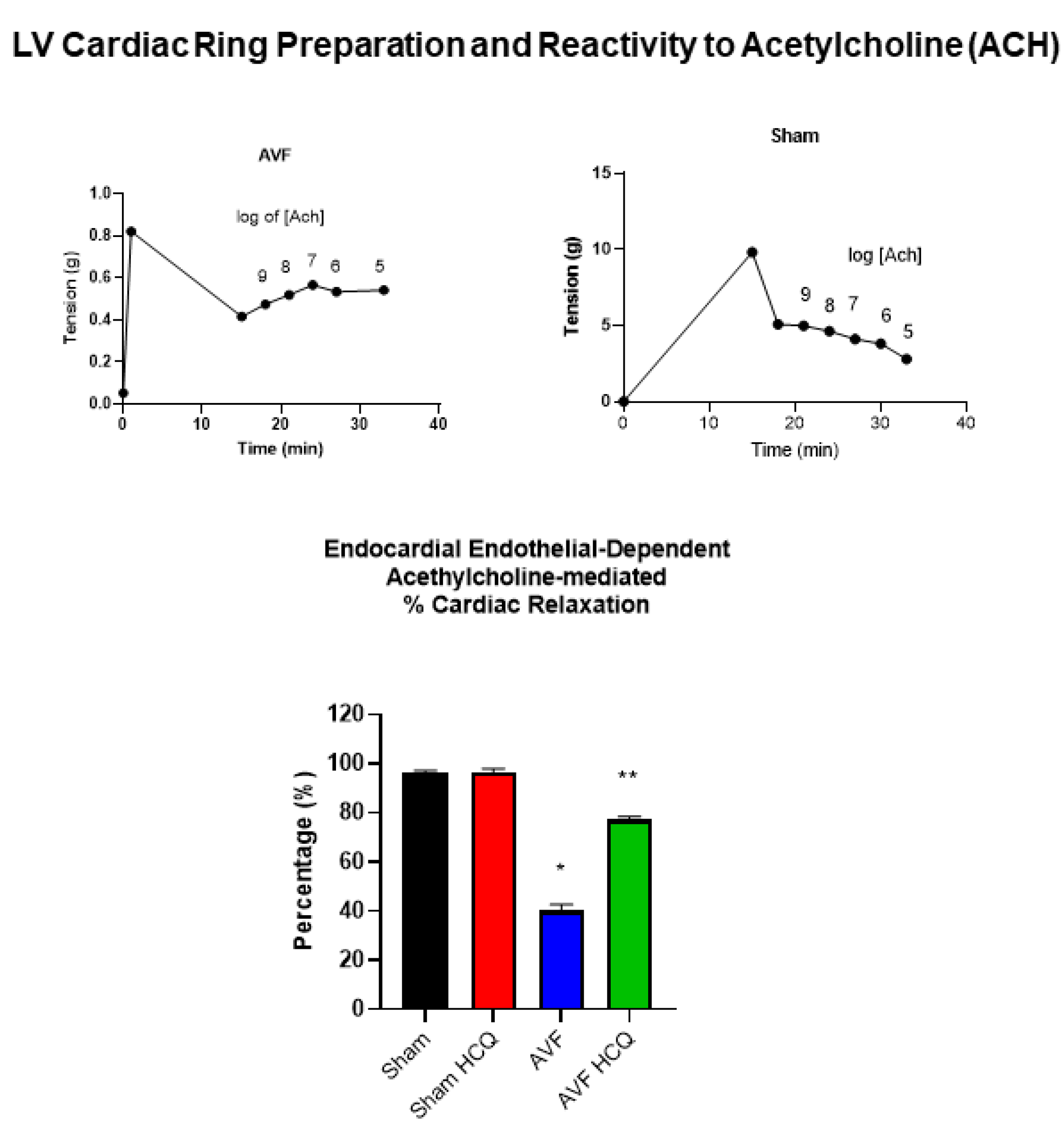
2.3. Measurement of Contraction, and Relaxation of Ventricle Rings, and Microvascular Permeability
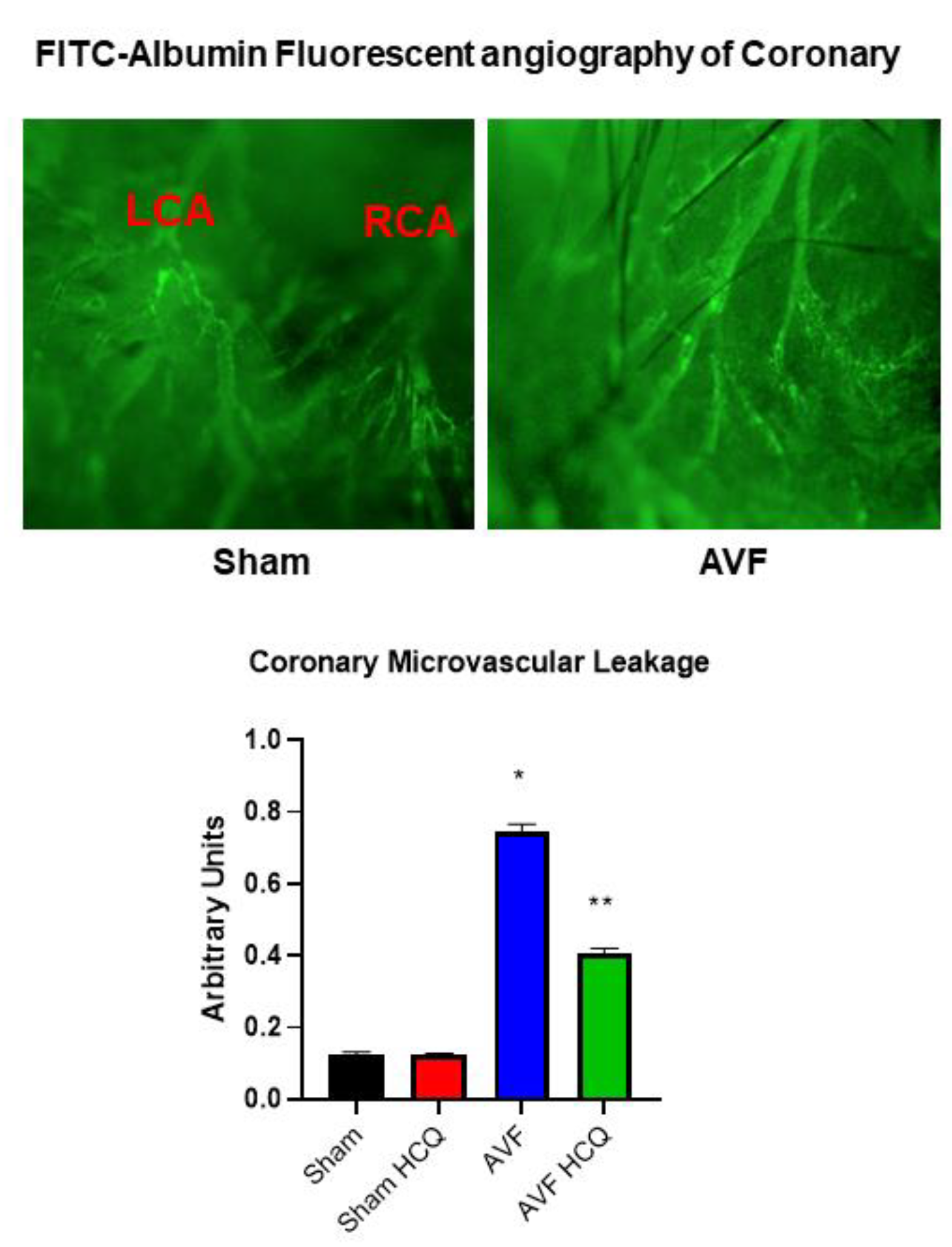
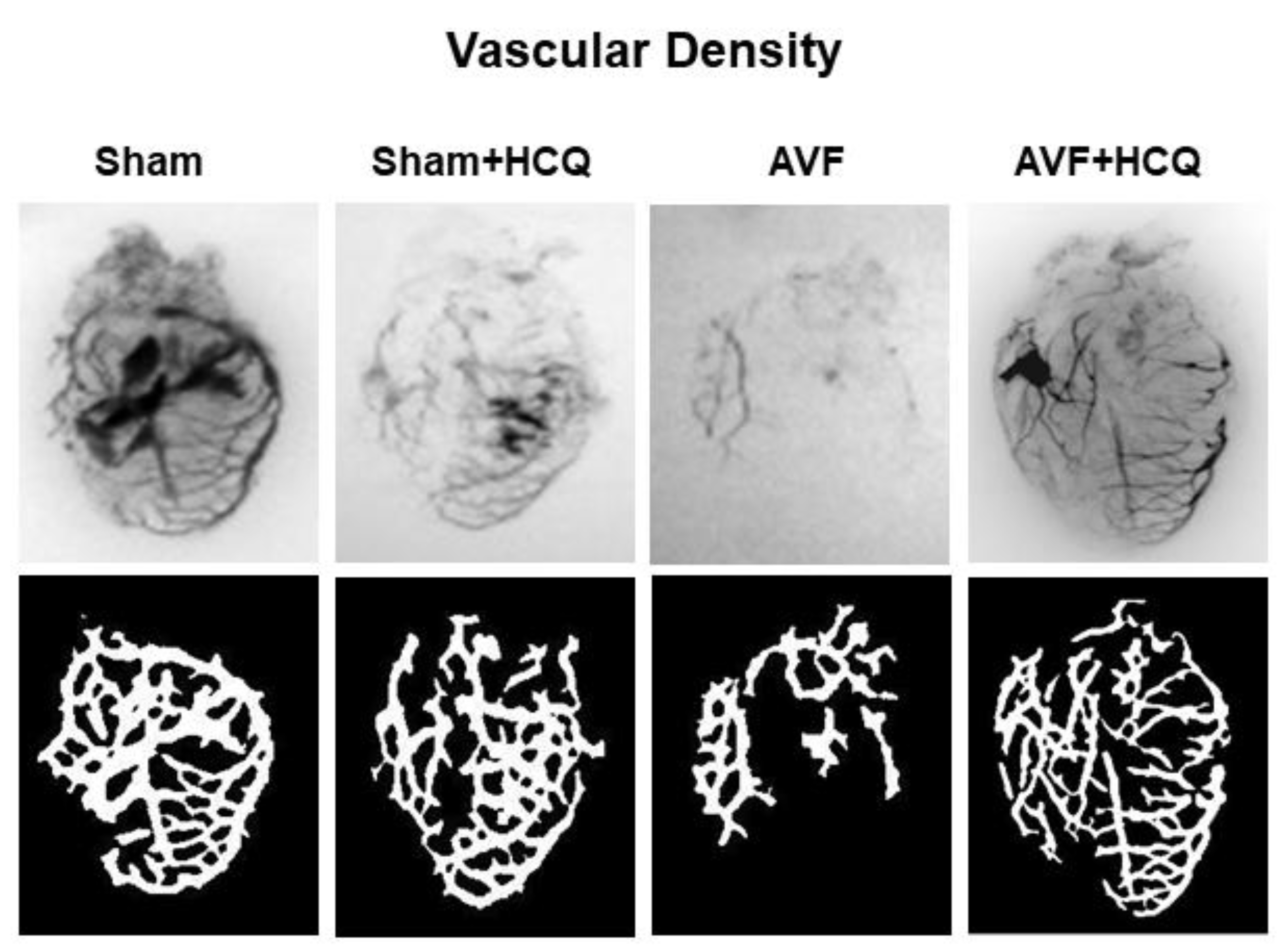
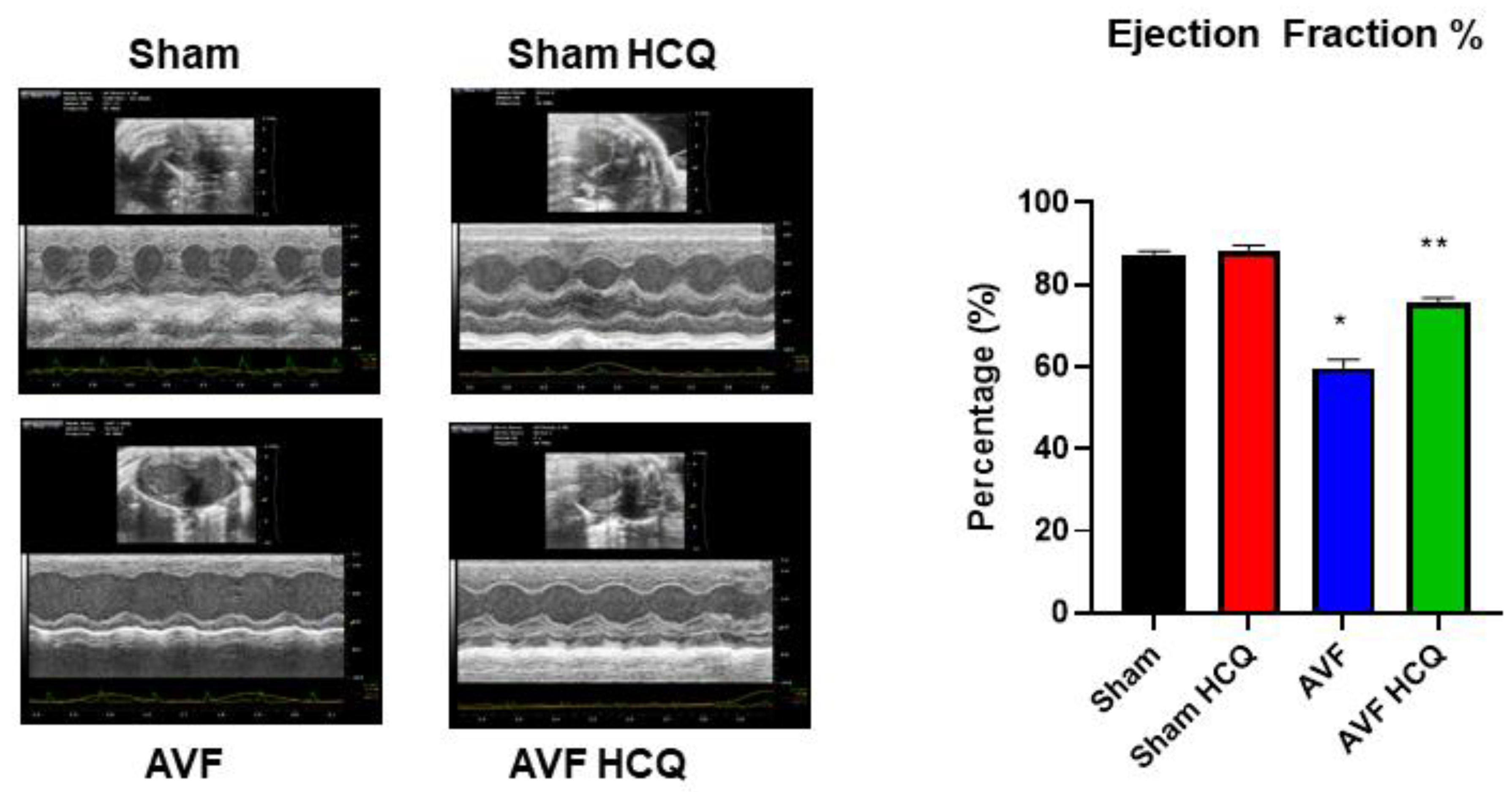
2.4. X-Ray Angiography, Ex Vivo Coronary Vascular Leakage, and Echocardiography
3. Discussion
4. Materials and Methods
4.1. Animal Protocol, and Treatment
4.2. Plasma Creatine Phosphokinase (CK) Activity
4.3. Zymography and Reverse Zymography
4.4. Myocyte Contraction
4.5. Cardiac Ring Preparation, and Endothelial Myocyte Coupling
4.6. FITC-Albumin Perfusion, and Vascular Leakage
4.7. Barium Sulfate Contrast X-ray Angiography
4.8. Coronary Leakage Due to Endothelial Dysfunction
4.9. Echocardiography, and Cardiac Function
4.10. Statistical Analysis
4.11. Ethics Approval
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tyagi, S.C.; Singh, M. Multi-organ damage by COVID-19: Congestive (cardio-pulmonary) heart failure, and blood-heart barrier leakage. Mol. Cell. Biochem. 2021, 476, 1891–1895. [Google Scholar] [CrossRef] [PubMed]
- Brutsaert, D.L.; Fransen, P.; Andries, L.J.; De Keulenaer, G.; Sys, S.U. Cardiac endothelium and myocardial function. Cardiovasc. Res. 1998, 38, 281–290. [Google Scholar] [CrossRef] [Green Version]
- Brutsaert, D.L. Cardiac Endothelial-Myocardial Signaling: Its Role in Cardiac Growth, Contractile Performance, and Rhythmicity. Physiol. Rev. 2003, 83, 59–115. [Google Scholar] [CrossRef] [PubMed]
- Smiljic, S. The clinical significance of endocardial endothelial dysfunction. Medicina 2017, 53, 295–302. [Google Scholar] [CrossRef]
- Dbouk, H.A.; Mroue, R.M.; El-Sabban, M.E.; Talhouk, R.S. Connexins: A myriad of functions ex-tending beyond assembly of gap junction channels. Cell Commun. Signal. CCS 2009, 7, 4. [Google Scholar] [CrossRef] [Green Version]
- Boengler, K.; Schulz, R. Connexin 43 and Mitochondria in Cardiovascular Health and Disease. Adv. Exp. Med. Biol. 2017, 982, 227–246. [Google Scholar] [CrossRef]
- Veeranki, S.; Givvimani, S.; Kundu, S.; Metreveli, N.; Pushpakumar, S.; Tyagi, S.C. Moderate intensity exercise prevents diabetic cardiomyopathy associated contractile dysfunction through restoration of mitochondrial function and connexin 43 levels in db/db mice. J. Mol. Cell. Cardiol. 2016, 92, 163–173. [Google Scholar] [CrossRef] [Green Version]
- Cruz Rodriguez, J.B.; Lange, R.A.; Mukherjee, D. Gamut of cardiac manifestations and complica-tions of COVID-19: A contemporary review. J. Investig. Med. Off. Publ. Am. Fed. Clin. Res. 2020, 68, 1334–1340. [Google Scholar]
- Adeghate, E.A.; Eid, N.; Singh, J. Mechanisms of COVID-19-induced heart failure: A short review. Heart Fail. Rev. 2021, 26, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Bader, F.; Manla, Y.; Atallah, B.; Starling, R.C. Heart failure and COVID-19. Heart Fail. Rev. 2021, 26, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Deswal, A.; Khalid, U. COVID-19 myocarditis and long-term heart failure sequelae. Curr. Opin. Cardiol. 2021, 36, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Mehra, M.R.; Ruschitzka, F. COVID-19 Illness and Heart Failure: A Missing Link? JACC Heart Fail. 2020, 8, 512–514. [Google Scholar] [CrossRef] [PubMed]
- Shchendrygina, A.; Nagel, E.; Puntmann, V.O.; Valbuena-Lopez, S. COVID-19 myocarditis and prospective heart failure burden. Expert Rev. Cardiovasc. 2021, 19, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Abbasi, J. Researchers Investigate What COVID-19 Does to the Heart. JAMA 2021, 325, 808. [Google Scholar] [CrossRef]
- Toschi, E.; Barillari, G.; Sgadari, C.; Bacigalupo, I.; Cereseto, A.; Carlei, D.; Palladino, C.; Zietz, C.; Leone, P.; Stürzl, M.; et al. Activation of matrix-metalloproteinase-2 and membrane-type-1-matrix-metalloproteinase in endothelial cells and induction of vascular permeability in vivo by human immunodeficiency virus-1 Tat protein and basic fibroblast growth factor. Mol. Biol. Cell 2001, 12, 2934–2946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, A.T.; Bürgers, H.F.; Rabie, T.; Marti, H.H. Matrix metalloproteinase-9 mediates hypoxia-induced vascular leakage in the brain via tight junction rearrangement. J. Cerebral Blood Flow Metab. Off. J. Int. Soc. Cerebral Blood Flow Metab. 2010, 30, 837–848. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, G.A.; Yang, Y. Vasogenic edema due to tight junction disruption by matrix metallo-proteinases in cerebral ischemia. Neurosurg. Focus 2007, 22, E4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jadhav, V.; Yamaguchi, M.; Obenaus, A.; Zhang, J.H. Matrix metalloproteinase inhibition attenuates brain edema after surgical brain injury. Improv. Results Peripher. Nerve Surg. 2008, 102, 357–361. [Google Scholar] [CrossRef]
- DeLeon-Pennell, K.Y.; Meschiari, C.A.; Jung, M.; Lindsey, M.L. Matrix Metalloproteinases in Myocardial Infarction and Heart Failure. Matrix metalloproteinases in myocardial infarction and heart failure. Prog. Mol. Biol. Transl. Sci. 2017, 147, 75–100. [Google Scholar]
- Hunt, M.J.; Aru, G.M.; Hayden, M.R.; Moore, C.K.; Hoit, B.D.; Tyagi, S.C. Induction of oxidative stress and disintegrin metalloproteinase in human heart end-stage failure. Am. J. Physiol. Cell. Mol. Physiol. 2002, 283, L239–L245. [Google Scholar] [CrossRef]
- Zima, A.V.; Pabbidi, M.R.; Lipsius, S.L.; Blatter, L.A. Effects of mitochondrial uncoupling on Ca2+ signaling during excitation-contraction coupling in atrial myocytes. Am. J. Physiol. Circ. Physiol. 2013, 304, H983–H993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, W.E.; Sen, U.; Tyagi, N.; Kumar, M.; Carneal, G.; Aggrawal, D.; Newsome, J.; Tyagi, S.C. PPAR gamma agonist normalizes glomerular filtration rate, tissue levels of homocysteine, and attenuates endothelial-myocyte uncoupling in alloxan induced diabetic mice. Int. J. Biol. Sci. 2008, 4, 236–244. [Google Scholar] [CrossRef] [Green Version]
- Sen, U.; Tyagi, N.; Moshal, K.S.; Kartha, G.K.; Rosenberger, R.; Henderson, B.C.; Joshua, I.G.; Tyagi, S.C. Cardiac Synchronous and Dys-synchronous Remodeling in Diabetes Mellitus. Antioxid. Redox Signal. 2007, 9, 971–978. [Google Scholar] [CrossRef] [PubMed]
- Rosenberger, D.; Moshal, K.S.; Kartha, G.K.; Tyagi, N.; Sen, U.; Lominadze, D.; Maldonado, C.; Roberts, A.M.; Tyagi, S.C. Arrhythmia and neuronal/endothelial myocyte uncoupling in hyper-homocysteinemia. Arch. Physiol. Biochem. 2006, 112, 219–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyagi, S.C.; Rodriguez, W.; Patel, A.M.; Roberts, A.M.; Falcone, J.C.; Passmore, J.C.; Fleming, J.T.; Joshua, I.G. Hyperhomocysteinemic Diabetic Cardiomyopathy: Oxidative Stress, Remodeling, and Endothelial-Myocyte Uncoupling. J. Cardiovasc. Pharm. Ther. 2005, 10, 1–10. [Google Scholar] [CrossRef]
- Segers, V.F.M.; Brutsaert, D.L.; De Keulenaer, G.W. Cardiac Remodeling: Endothelial Cells Have More to Say Than Just NO. Front. Physiol. 2018, 9, 382. [Google Scholar] [CrossRef] [PubMed]
- Givvimani, S.; Qipshidze, N.; Tyagi, N.; Mishra, P.K.; Sen, U.; Tyagi, S.C. Synergism between arrhythmia and hyperhomo-cysteinemia in structural heart disease. Int. J. Physiol. Pathophysiol. Pharmacol. 2011, 3, 107–119. [Google Scholar]
- Tyagi, N.; Vacek, J.C.; Givvimani, S.; Sen, U.; Tyagi, S.C. Cardiac specific deletion of N-methyl-d-aspartate receptor 1 ameliorates mtMMP-9 mediated autophagy/mitophagy in hyper-homocysteinemia. J. Recept. Signal Transduct. Res. 2010, 30, 78–87. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Brodsky, S.; Kumari, S.; Valiunas, V.; Brink, P.; Kaide, J.-I.; Nasjletti, A.; Goligorsky, M.S. Paradoxical overexpression and translocation of connexin43 in homocysteine-treated endothelial cells. Am. J. Physiol. Circ. Physiol. 2002, 282, H2124–H2133. [Google Scholar] [CrossRef] [Green Version]
- Moshal, K.S.; Tyagi, N.; Henderson, B.; Ovechkin, A.; Tyagi, S.C. Protease-activated receptor and endothelial-myocyte uncoupling in chronic heart failure. Am. J. Physiol. Circ. Physiol. 2005, 288, H2770–H2777. [Google Scholar] [CrossRef]
- Ovechkin, A.; Tyagi, N.; Rodriguez, W.E.; Hayden, M.R.; Moshal, K.S.; Tyagi, S.C. Role of matrix metalloproteinase-9 in endothelial apoptosis in chronic heart failure in mice. J. Appl. Physiol. 2005, 99, 2398–2405. [Google Scholar] [CrossRef] [Green Version]
- Yazdany, J.; Kim, A. Use of Hydroxychloroquine and Chloroquine During the COVID-19 Pandemic: What Every Clinician Should Know. Ann. Intern. Med. 2020, 172, 754–755. [Google Scholar] [CrossRef] [Green Version]
- Al-Bari, A.A. Facts and Myths: Efficacies of Repurposing Chloroquine and Hydroxychloroquine for the Treatment of COVID-19. Curr. Drug Targets 2020, 21, 1703–1721. [Google Scholar] [CrossRef]
- Kamat, S.; Kumari, M. Repurposing Chloroquine against Multiple Diseases with Special Attention to SARS-CoV-2 and Associated Toxicity. Front. Pharmacol. 2021, 12, 576093. [Google Scholar] [CrossRef]
- Naghipour, S.; Ghodousi, M.; Rahsepar, S.; Elyasi, S. Repurposing of well-known medications as antivirals: Hydroxychloroquine and chloroquine—From HIV-1 infection to COVID-19. Expert Rev. Anti-Infect. Ther. 2020, 18, 1119–1133. [Google Scholar] [CrossRef]
- Shionoya, K.; Yamasaki, M.; Iwanami, S.; Ito, Y.; Fukushi, S.; Ohashi, H.; Saso, W.; Tanaka, T.; Aoki, S.; Kuramochi, K.; et al. Mefloquine, a Potent Anti-severe Acute Respiratory Syndrome-Related Coronavirus 2 (SARS-CoV-2) Drug as an Entry Inhibitor in vitro. Front. Microbiol. 2021, 12, 651403. [Google Scholar] [CrossRef] [PubMed]
- Deckert, A.; Anders, S.; de Allegri, M.; Nguyen, H.T.; Souares, A.; McMahon, S.; Boerner, K.; Meurer, M.; Herbst, K.; Sand, M.; et al. Effectiveness and cost-effectiveness of four different strategies for SARS-CoV-2 surveillance in the general population (CoV-Surv Study): A structured summary of a study protocol for a cluster-randomised, two-factorial controlled trial. Trials 2021, 22, 39. [Google Scholar] [CrossRef]
- Villanustre, F.; Chala, A.; Dev, R.; Xu, L.; LexisNexis, J.S.; Furht, B.; Khoshgoftaar, T. Modeling and tracking COVID-19 cases using Big Data analytics on HPCC system platformm. J. Big Data 2021, 8, 33. [Google Scholar] [CrossRef] [PubMed]
- Zohner, Y.E.; Morris, J.S. COVID-Track: World and USA SARS-CoV-2 testing and COVID-19 tracking. Biodata Min. 2021, 14, 1–15. [Google Scholar] [CrossRef]
- Grasselli, G.; Zangrillo, A.; Zanella, A.; Antonelli, M.; Cabrini, L.; Castelli, A.; Cereda, D.; Coluccello, A.; Foti, G.; Fumagalli, R.; et al. Baseline Characteristics and Outcomes of 1591 Patients Infected With SARS-CoV-2 Admitted to ICUs of the Lombardy Region, Italy. JAMA 2020, 323, 1574–1581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borba, M.G.S.; Val, F.F.A.; Sampaio, V.S.; Alexandre, M.A.A.; Melo, G.C.; Brito, M.; Mourão, M.P.G.; Brito-Sousa, J.D.; Baía-da-Silva, D.; Guerra, M.V.F.; et al. Effect of High vs Low Doses of Chloroquine Diphosphate as Adjunctive Therapy for Patients Hospitalized with Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Infection: A Randomized Clinical Trial. JAMA Netw. Open 2020, 3, e208857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontana, F.; Alfano, G.; Mori, G.; Amurri, A.; Tei, L.; Ballestri, M.; Leonelli, M.; Facchini, F.; Damiano, F.; Magistroni, R.; et al. COVID-19 pneumonia in a kidney transplant recipient successfully treated with tocilizumab and hydroxychloroquine. Arab. Archaeol. Epigr. 2020, 20, 1902–1906. [Google Scholar] [CrossRef] [Green Version]
- Meo, S.A.; Klonoff, D.C.; Akram, J. Efficacy of chloroquine and hydroxychloroquine in the treat-ment of COVID-19. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 4539–4547. [Google Scholar] [PubMed]
- Sisti, G.; Schiattarella, A.; Sisti, A. Treatment of COVID-19 in Pregnancy with Hydroxychloroquine and Azithromycin: A case report. Acta Bio-Med. Atenei Parmensis 2020, 91, e2020123. [Google Scholar]
- Lesiak, A.; Narbutt, J.; Sysa-Jedrzejowska, A.; Lukamowicz, J.; McCauliffe, D.P.; Wózniacka, A. Effect of chloroquine phosphate treatment on serum MMP-9 and TIMP-1 levels in patients with systemic lupus erythematosus. Lupus 2010, 19, 683–688. [Google Scholar] [CrossRef] [PubMed]
- Tuomela, J.; Sandholm, J.; Kauppila, J.H.; Lehenkari, P.; Harris, K.W.; Selander, K.S. Chloroquine has tumor-inhibitory and tumor-promoting effects in triple-negative breast cancer. Oncol. Lett. 2013, 6, 1665–1672. [Google Scholar] [CrossRef]
- Lu, L.-H.; Chao, C.-H.; Yeh, T.-M. Inhibition of autophagy protects against sepsis by concurrently attenuating the cytokine storm and vascular leakage. J. Infect. 2019, 78, 178–186. [Google Scholar] [CrossRef]
- Chen, H.-R.; Chuang, Y.-C.; Chao, C.-H.; Yeh, T.-M. Macrophage migration inhibitory factor induces vascular leakage via autophagy. Biol. Open 2015, 4, 244–252. [Google Scholar] [CrossRef] [Green Version]
- Maes, H.; Kuchnio, A.; Carmeliet, P.; Agostinis, P. How to teach an old dog new tricks: Autophagy-independent action of chloroquine on the tumor vasculature. Autophagy 2014, 10, 2082–2084. [Google Scholar] [CrossRef] [Green Version]
- Maes, H.; Kuchnio, A.; Carmeliet, P.; Agostinis, P. Chloroquine anticancer activity is mediated by autophagy-independent effects on the tumor vasculature. Mol. Cell. Oncol. 2016, 3, e970097. [Google Scholar] [CrossRef] [Green Version]
- Maes, H.; Kuchnio, A.; Peric, A.; Moens, S.; Nys, K.; De Bock, K.; Quaegebeur, A.; Schoors, S.; Georgiadou, M.; Wouters, J.; et al. Tumor vessel normalization by chloroquine independent of autophagy. Cancer Cell 2014, 26, 190–206. [Google Scholar] [CrossRef] [Green Version]
- Schaaf, M.B.; Houbaert, D.; Meçe, O.; To, S.K.; Ganne, M.; Maes, H.; Agostinis, P. Lysosomal Pathways and Autophagy Distinctively Control Endothelial Cell Behavior to Affect Tumor Vasculature. Front. Oncol. 2019, 9, 171. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yuan, X.; Tang, Y.; Wang, B.; Deng, Z.; Huang, Y.; Liu, F.; Zhao, Z.; Zhang, Y. Hydroxychloroquine is a novel therapeutic approach for rosacea. Int. Immunopharmacol. 2020, 79, 106178. [Google Scholar] [CrossRef]
- Rolain, J.-M.; Colson, P.; Raoult, D. Recycling of chloroquine and its hydroxyl analogue to face bacterial, fungal and viral infections in the 21st century. Int. J. Antimicrob. Agents 2007, 30, 297–308. [Google Scholar] [CrossRef]
- Chen, Z.; Hu, J.; Zhang, Z.; Jiang, S.; Han, S.; Yan, D.; Zhuang, R.; Hu, B.; Zhang, Z. Efficacy of hydroxychloroquine in patients with COVID-19: Results of a randomized clinical trial. medRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Fatima, U.; Rizvi, S.S.A.; Fatima, S.; Hassan, I. Impact of Hydroxychloroquine/Chloroquine in COVID-19 Therapy: Two Sides of the Coin. J. Interf. Cytokine Res. 2020, 40, 469–471. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Deng, X.; Gao, J.; Gao, W.; Xiao, H.; Wang, X.; Zhang, Y. Autophagy mediates the secretion of macrophage migration inhibitory factor from cardiomyocytes upon serum-starvation. Sci. China Life Sci. 2019, 62, 1038–1046. [Google Scholar] [CrossRef]
- Liu, X.; Li, Z.; Liu, S.; Sun, J.; Chen, Z.; Jiang, M.; Zhang, Q.; Wei, Y.; Wang, X.; Huang, Y.-Y.; et al. Potential therapeutic effects of dipyridamole in the severely ill patients with COVID-19. Acta Pharm. Sin. B 2020, 10, 1205–1215. [Google Scholar] [CrossRef]
- Gentile, D.; Fuochi, V.; Rescifina, A.; Furneri, P.M. New Anti-SARS-CoV-2 Targets for Quinoline Derivatives Chloroquine and Hydroxychloroquine. Int. J. Mol. Sci. 2020, 21, 5856. [Google Scholar] [CrossRef]
- Taccone, F.S.; Gorham, J.; Vincent, J.-L. Hydroxychloroquine in the management of critically ill patients with COVID-19: The need for an evidence base. Lancet Respir. Med. 2020, 8, 539–541. [Google Scholar] [CrossRef]
- Docherty, K.F.; Jhund, P.S.; Inzucchi, E.S.; Køber, L.; Kosiborod, M.N.; Martinez, A.F.; Ponikowski, P.; DeMets, D.L.; Sabatine, M.S.; Bengtsson, O.; et al. Effects of dapagliflozin in DAPA-HF according to background heart failure therapy. Eur. Heart J. 2020, 41, 2379–2392. [Google Scholar] [CrossRef] [Green Version]
- Costanzo, M.; De Giglio, M.A.R.; Roviello, G.N. SARS-CoV-2: Recent Reports on Antiviral Therapies Based on Lopinavir/Ritonavir, Darunavir/Umifenovir, Hydroxychloroquine, Remdesivir, Favipiravir and other Drugs for the Treatment of the New Coronavirus. Curr. Med. Chem. 2020, 27, 4536–4541. [Google Scholar] [CrossRef]
- Chiotos, K.; Hayes, M.; Kimberlin, D.W.; Jones, S.B.; James, S.H.; Pinninti, S.G.; Yarbrough, A.; Abzug, M.J.; MacBrayne, C.E.; Soma, V.L.; et al. Multicenter Interim Guidance on Use of Antivirals for Children with Coronavirus Disease 2019/Severe Acute Respiratory Syndrome Coronavirus 2. J. Pediatr. Infect. Dis. Soc. 2021, 10, 34–48. [Google Scholar] [CrossRef]
- Freedberg, D.E.; Conigliaro, J.; Wang, T.C.; Tracey, K.J.; Callahan, M.V.; Abrams, J.A.; Sobieszczyk, M.E.; Markowitz, D.D.; Gupta, A.; O’Donnell, M.R.; et al. Famotidine Use Is Associated with Improved Clinical Outcomes in Hospitalized COVID-19 Patients: A Propensity Score Matched Retrospective Cohort Study. Gastroenterology 2020, 159, 1129–1131.e3. [Google Scholar] [CrossRef]
- Ii, R.B.H.; Cannon, T.; Rappai, M.; Studdard, J.; Paul, D.; Dooley, T.P. Dual-histamine receptor blockade with cetirizine—Famotidine reduces pulmonary symptoms in COVID-19 patients. Pulm. Pharmacol. Ther. 2020, 63, 101942. [Google Scholar] [CrossRef]
- Mather, J.F.; Seip, R.L.; McKay, R.G. Impact of Famotidine Use on Clinical Outcomes of Hospitalized Patients with COVID-19. Am. J. Gastroenterol. 2020, 115, 1617–1623. [Google Scholar] [CrossRef]
- Samimagham, H.R.; Azad, M.H.; Haddad, M.; Arabi, M.; Hooshyar, D.; KazemiJahromi, M. The Efficacy of Famotidine in improvement of outcomes in Hospitalized COVID-19 Patients: A structured summary of a study protocol for a randomised controlled trial. Trials 2020, 21, 1–3. [Google Scholar] [CrossRef]
- Lalu, M.M.; Gao, C.Q.; Schulz, R. Matrix metalloproteinase inhibitors attenuate endotoxemia induced cardiac dysfunction: A potential role for MMP-9. Biochem. Hypertrophy Heart Fail. 2003, 251, 61–66. [Google Scholar] [CrossRef]
- Amin, M.; Pushpakumar, S.; Muradashvili, N.; Kundu, S.; Tyagi, S.C.; Sen, U. Regulation and involvement of matrix metalloproteinases in vascular diseases. Front. Biosci. 2016, 21, 89–118. [Google Scholar]
- Cabral-Pacheco, G.A.; Garza-Veloz, I.; La Rosa, C.C.-D.; Ramirez-Acuña, J.M.; Perez-Romero, B.A.; Guerrero-Rodriguez, J.F.; Martinez-Avila, N.; Martinez-Fierro, M.L. The Roles of Matrix Metalloproteinases and Their Inhibitors in Human Diseases. Int. J. Mol. Sci. 2020, 21, 9739. [Google Scholar] [CrossRef] [PubMed]
- Cohen, L.; Sagi, I.; Bigelman, E.; Solomonov, I.; Aloshin, A.; Ben-Shoshan, J.; Rozenbaum, Z.; Keren, G.; Entin-Meer, M. Cardiac remodeling secondary to chronic volume overload is attenuated by a novel MMP9/2 blocking antibody. PLoS ONE 2020, 15, e0231202. [Google Scholar]
- Tyagi, S.C.; Kumar, S.G.; Haas, S.J.; Reddy, H.K.; Voelker, D.J.; Hayden, M.R.; Demmy, T.L.; Schmaltz, R.A.; Curtis, J.J. Post-transcriptional Regulation of Extracellular Matrix Metalloproteinase in Human Heart End-stage Failure Secondary to Ischemic Cardiomyopathy. J. Mol. Cell. Cardiol. 1996, 28, 1415–1428. [Google Scholar] [CrossRef]
- Hu, J.; Steen, P.V.D.; Sang, Q.-X.A.; Opdenakker, G. Matrix metalloproteinase inhibitors as therapy for inflammatory and vascular diseases. Nat. Rev. Drug Discov. 2007, 6, 480–498. [Google Scholar] [CrossRef] [PubMed]
- Camp, T.M.; Tyagi, S.C.; Aru, G.M.; Hayden, M.R.; Mehta, J.L.; Tyagi, S.C. Doxycycline ameliorates ischemic and Border-Zone remodeling and endothelial dysfunction after myocardial infarction in rats. J. Heart Lung Transpl. 2004, 23, 729–736. [Google Scholar] [CrossRef]
- El-Din, A.N.; Ata, K.A.E.-S.A.; Abdel-Gawad, A.R.; Fahmy, N.F. Impact of High Serum Levels of MMP-7, MMP-9, TGF-β and PDGF Macrophage Activation Markers on Severity of COVID-19 in Obese-Diabetic Patients. Infect. Drug Resist. 2021, 14, 4015–4025. [Google Scholar] [CrossRef]
- D’avila-Mesquita, C.; Couto, A.E.; Campos, L.C.; Vasconcelos, T.F.; Michelon-Barbosa, J.; Corsi, C.A.; Mestriner, F.; Petroski-Moraes, B.C.; Garbellini-Diab, M.J.; Couto, D.M.; et al. MMP-2 and MMP-9 levels in plasma are altered and associated with mortality in COVID-19 patients. Biomed. Pharmacother. 2021, 142, 112067. [Google Scholar] [CrossRef]
- Lartey, N.L.; Valle-Reyes, S.; Vargas-Robles, H.; Jiménez-Camacho, K.E.; Guerrero-Fonseca, I.M.; Castellanos-Martínez, R.; Montoya-García, A.; García-Cordero, J.; Cedillo-Barrón, L.; Nava, P.; et al. ADAM17/MMP inhibition prevents neutrophilia and lung injury in a mouse model of COVID-19. J. Leukocyte Biol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Syed, F.; Li, W.; Relich, R.F.; Russell, P.M.; Zhang, S.; Zimmerman, M.K.; Yu, Q. Excessive Matrix Metalloproteinase-1 and Hyperactivation of Endothelial Cells Occurred in COVID-19 Patients and Were Associated with the Severity of COVID-19. J. Infect. Dis. 2021, 224, 60–69. [Google Scholar] [CrossRef]
- Ueland, T.; Holter, J.C.; Holten, A.R.; Müller, K.E.; Lind, A.; Bekken, G.K.; Dudman, S.; Aukrust, P.; Dyrhol-Riise, A.M.; Heggelund, L. Distinct and early increase in circulating MMP-9 in COVID-19 patients with respiratory failure. J. Infect. 2020, 81, e41–e43. [Google Scholar] [CrossRef]
- Rucklidge, G.J.; Milne, G.; McGaw, B.A.; Milne, E.; Robins, S.P. Turnover rates of different collagen types measured by isotope ratio mass spectrometry. Biochim. Biophys. Acta (BBA) Gen. Subj. 1992, 1156, 57–61. [Google Scholar] [CrossRef]
- Rottlaender, D.; Boengler, K.; Wolny, M.; Michels, G.; Endres-Becker, J.; Motloch, L.J.; Schwaiger, A.; Buechert, A.; Schulz, R.; Heusch, G.; et al. Connexin 43 acts as a cytoprotective mediator of signal transduction by stimulating mitochondrial K(ATP) channels in mouse cardiomyocytes. J. Clin. Investig. 2012, 122, 47–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boengler, K.; Ruiz-Meana, M.; Gent, S.; Ungefug, E.; Soetkamp, D.; Miro-Casas, E.; Cabestrero, A.; Fernandez-Sanz, C.; Semenzato, M.; Di Lisa, F.; et al. Mitochondrial connexin 43 impacts on respiratory complex I activity and mitochondrial oxygen consumption. J. Cell. Mol. Med. 2012, 16, 1649–1655. [Google Scholar] [CrossRef] [PubMed]
- Givvimani, S.; Pushpakumar, S.; Veeranki, S.; Tyagi, S.C. Dysregulation of Mfn2 and Drp-1 proteins in heart failure. Can. J. Physiol. Pharm. Ther. 2014, 92, 583–591. [Google Scholar] [CrossRef] [Green Version]
- Givvimani, S.; Munjal, C.; Tyagi, N.; Sen, U.; Metreveli, N.; Tyagi, S.C. Mitochondrial division/mitophagy inhibitor (Mdivi) Ameliorates Pressure Overload Induced Heart Failure. PLoS ONE 2012, 7, e32388. [Google Scholar] [CrossRef]
- Garcia, R.; Diebold, S. Simple, rapid, and effective method of producing aortocaval shunts in the rat. Cardiovasc. Res. 1990, 24, 430–432. [Google Scholar] [CrossRef] [PubMed]
- Turcani, M.; Jacob, R. Minoxidil accelerates heart failure development in rats with ascending aortic constriction. Can. J. Physiol. Pharmacol. 1998, 76, 613–620. [Google Scholar] [CrossRef]
- Tsoporis, J.; Leenen, F.H. Effects of arterial vasodilators on cardiac hypertrophy and sympathetic activity in rats. Hypertension 1988, 11, 376–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qureshi, S.T.; Skamene, E.; Malo, D. Comparative Genomics and Host Resistance against Infectious Diseases. Emerg. Infect. Dis. 1999, 5, 36–47. [Google Scholar] [CrossRef]
- Miller, A.; Mujumdar, V.; Shek, E.; Guillot, J.; Angelo, M.; Palmer, L.; Tyagi, S.C. Hyperhomocyst(e)inemia induces multiorgan damage. Heart Vessel. 2000, 15, 135–143. [Google Scholar] [CrossRef]
- Singh, M.; Hardin, S.J.; George, A.K.; Eyob, W.; Stanisic, D.; Pushpakumar, S.; Tyagi, S.C. Epigenetics, 1-Carbon Metabolism, and Homocysteine during Dysbiosis. Front. Physiol. 2021, 11, 617953. [Google Scholar] [CrossRef]
- Stanisic, D.; George, A.K.; Smolenkova, I.; Singh, M.; Tyagi, S.C. Hyperhomocysteinemia: An instigating factor for periodontal disease. Can. J. Physiol. Pharmacol. 2021, 99, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, S.C.; Matsubara, L.; Weber, K.T. Direct extraction and estimation of collagenase(s) activity by zymography in microquantities of rat myocardium and uterus. Clin. Biochem. 1993, 26, 191–198. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of Structural Proteins during the Assembly of the Head of Bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Givvimani, S.; Pushpakumar, S.; Metreveli, N.; Veeranki, S.; Kundu, S.; Tyagi, S. Role of mitochondrial fission and fusion in cardiomyocyte contractility. Int. J. Cardiol. 2015, 187, 325–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moshal, K.S.; Kumar, M.; Tyagi, N.; Mishra, P.K.; Metreveli, N.; Rodriguez, W.E.; Tyagi, S.C. Restoration of contractility in hyperhomocysteinemia by cardiac-specific deletion of NMDA-R1. Am. J. Physiol. Circ. Physiol. 2009, 296, H887–H892. [Google Scholar] [CrossRef] [Green Version]
- Moshal, K.S.; Tipparaju, S.M.; Vacek, T.P.; Kumar, M.; Singh, M.; Frank, I.E.; Patibandla, P.K.; Tyagi, N.; Rai, J.; Metreveli, N.; et al. Mitochondrial matrix metalloproteinase activation decreases myocyte contractility in hyperhomocysteinemia. Am. J. Physiol. Circ. Physiol. 2008, 295, H890–H897. [Google Scholar] [CrossRef] [PubMed]
- Vacek, T.P.; Metreveli, N.; Tyagi, N.; Vacek, J.C.; Pagni, S.; Tyagi, S.C. Electrical stimulation of cardiomyocytes activates mitochondrial matrix metalloproteinase causing electrical remodeling. Biochem. Biophys. Res. Commun. 2011, 404, 762–766. [Google Scholar] [CrossRef] [Green Version]
- Tyagi, S.C. Homocyst(E)Ine and Heart Disease: Pathophysiology of Extracellular Matrix. Clin. Exp. Hypertens. 1999, 21, 181–198. [Google Scholar] [CrossRef]
- McMillan, A.; Hazen, S.L. Gut Microbiota Involvement in Ventricular Remodeling Post–Myocardial Infarction. Circulation 2019, 139, 660–662. [Google Scholar] [CrossRef]
- Wang, J.; Morgan, J.P. Endocardial endothelium modulates myofilament Ca2+ responsiveness in aequorin-loaded ferret myocardium. Circ. Res. 1992, 70, 754–760. [Google Scholar] [CrossRef] [Green Version]
- Gattuso, A.; Mazza, R.; Pellegrino, D.; Tota, B. Endocardial endothelium mediates luminal ACh-NO signaling in isolated frog heart. Am. J. Physiol. Content 1999, 276, H633–H641. [Google Scholar] [CrossRef] [PubMed]
- Cox, M.J.; Sood, H.S.; Hunt, M.J.; Chandler, D.; Henegar, J.R.; Aru, G.M.; Tyagi, S.C. Apoptosis in the left ventricle of chronic volume overload causes endocardial endothelial dysfunction in rats. Am. J. Physiol. Circ. Physiol. 2002, 282, H1197–H1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mujumdar, V.S.; Tyagi, S.C. Temporal regulation of extracellular matrix components in transition from compensatory hypertrophy to decompensatory heart failure. J. Hypertens. 1999, 17, 261–270. [Google Scholar] [CrossRef]
- Limberg, J.K.; Casey, D.P.; Trinity, J.D.; Nicholson, W.T.; Wray, D.W.; Tschakovsky, M.E.; Green, D.J.; Hellsten, Y.; Fadel, P.J.; Joyner, M.J.; et al. Assessment of resistance vessel function in human skeletal muscle: Guidelines for experimental design, Doppler ultrasound, and pharmacology. Am. J. Physiol. Circ. Physiol. 2020, 318, H301–H325. [Google Scholar] [CrossRef] [Green Version]
- Givvimani, S.; Sen, U.; Tyagi, N.; Munjal, C.; Tyagi, S.C. X-ray imaging of differential vascular density in MMP-9−/−, PAR-1−/+, hyperhomocysteinemic (CBS−/+) and diabetic (Ins2−/+) mice. Arch. Physiol. Biochem. 2011, 117, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myojin, K.; Taguchi, A.; Umetani, K.; Fukushima, K.; Nishiura, N.; Matsuyama, T.; Kimura, H.; Stern, D.; Imai, Y.; Mori, H. Visualization of Intracerebral Arteries by Synchrotron Radiation Microangiography. Am. J. Neuroradiol. 2007, 28, 953–957. [Google Scholar]
- Muradashvili, N.; Tyagi, R.; Lominadze, D. A Dual-Tracer Method for Differentiating Transendothelial Transport from Paracellular Leakage in Vivo and in Vitro. Front. Physiol. 2012, 3, 166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- George, A.K.; Behera, J.; Kelly, K.E.; Mondal, N.; Richardson, K.P.; Tyagi, N. Exercise Mitigates Alcohol Induced Endoplasmic Reticulum Stress Mediated Cognitive Impairment through ATF6-Herp Signaling. Sci. Rep. 2018, 8, 5158. [Google Scholar] [CrossRef]
- George, A.K.; Homme, R.P.; Majumder, A.; Laha, A.; Metreveli, N.; Sandhu, H.S.; Tyagi, S.C.; Singh, M. Hydrogen sulfide intervention in cystathionine-β-synthase mutant mouse helps restore ocular homeostasis. Int. J. Ophthalmol. 2019, 12, 754–764. [Google Scholar] [CrossRef] [PubMed]
- Kunkel, G.H.; Kunkel, C.J.; Ozuna, H.; Miralda, I.; Tyagi, S.C. TFAM overexpression reduces pathological cardiac remodeling. Mol. Cell. Biochem. 2018, 454, 139–152. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Homme, R.P.; George, A.K.; Singh, M.; Smolenkova, I.; Zheng, Y.; Pushpakumar, S.; Tyagi, S.C. Mechanism of Blood–Heart-Barrier Leakage: Implications for COVID-19 Induced Cardiovascular Injury. Int. J. Mol. Sci. 2021, 22, 13546. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222413546
Homme RP, George AK, Singh M, Smolenkova I, Zheng Y, Pushpakumar S, Tyagi SC. Mechanism of Blood–Heart-Barrier Leakage: Implications for COVID-19 Induced Cardiovascular Injury. International Journal of Molecular Sciences. 2021; 22(24):13546. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222413546
Chicago/Turabian StyleHomme, Rubens P., Akash K. George, Mahavir Singh, Irina Smolenkova, Yuting Zheng, Sathnur Pushpakumar, and Suresh C. Tyagi. 2021. "Mechanism of Blood–Heart-Barrier Leakage: Implications for COVID-19 Induced Cardiovascular Injury" International Journal of Molecular Sciences 22, no. 24: 13546. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222413546