
Prediction of Optimal Conditions of Hydrogenation Reaction Using the Likelihood Ranking Approach

 and
and 
Abstract
:1. Introduction
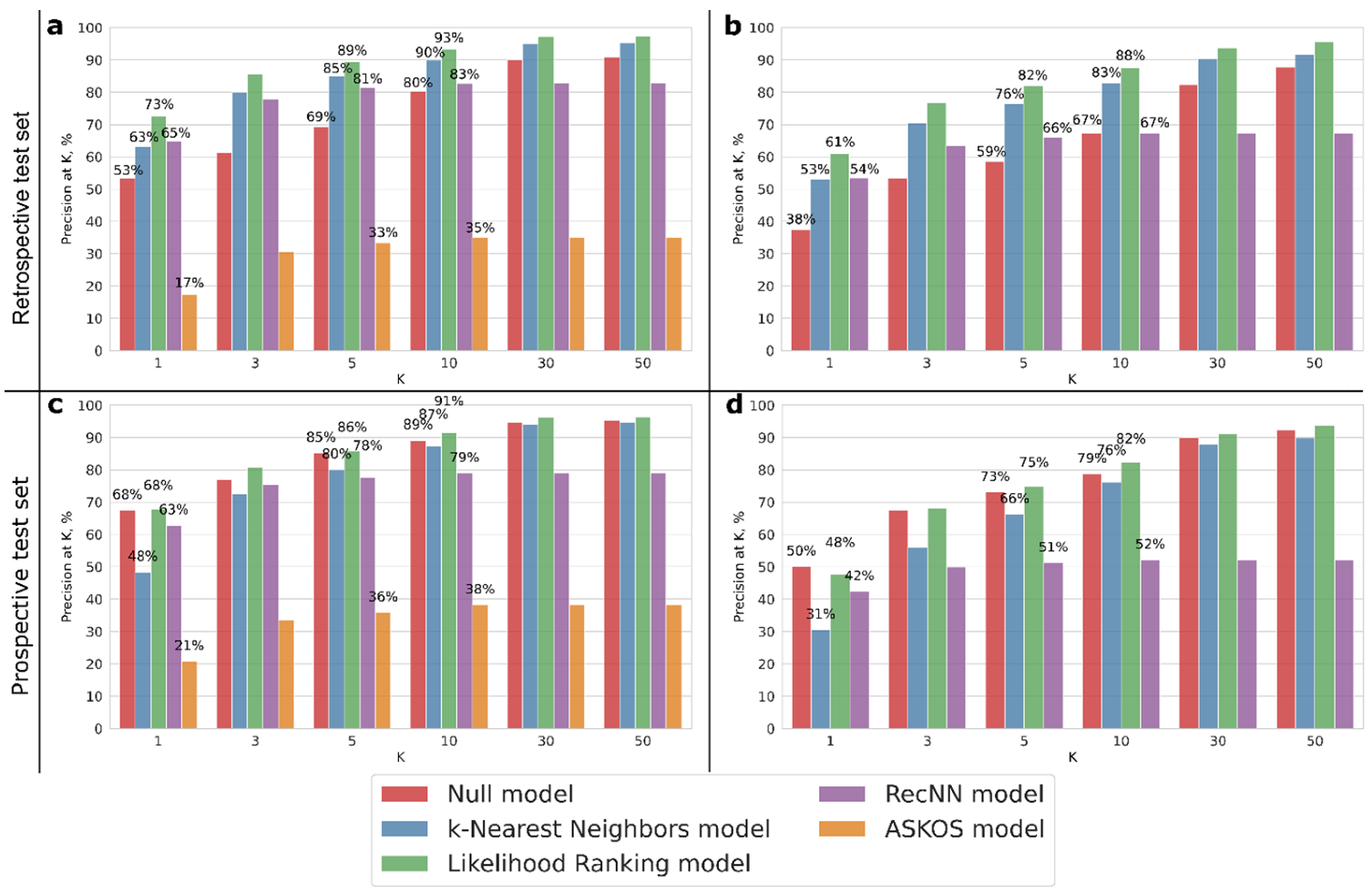
2. Results and Discussion
3. Computational Procedure
3.1. Data Curation
3.2. Training/Test Set Preparation
3.3. Descriptors
3.4. Methods
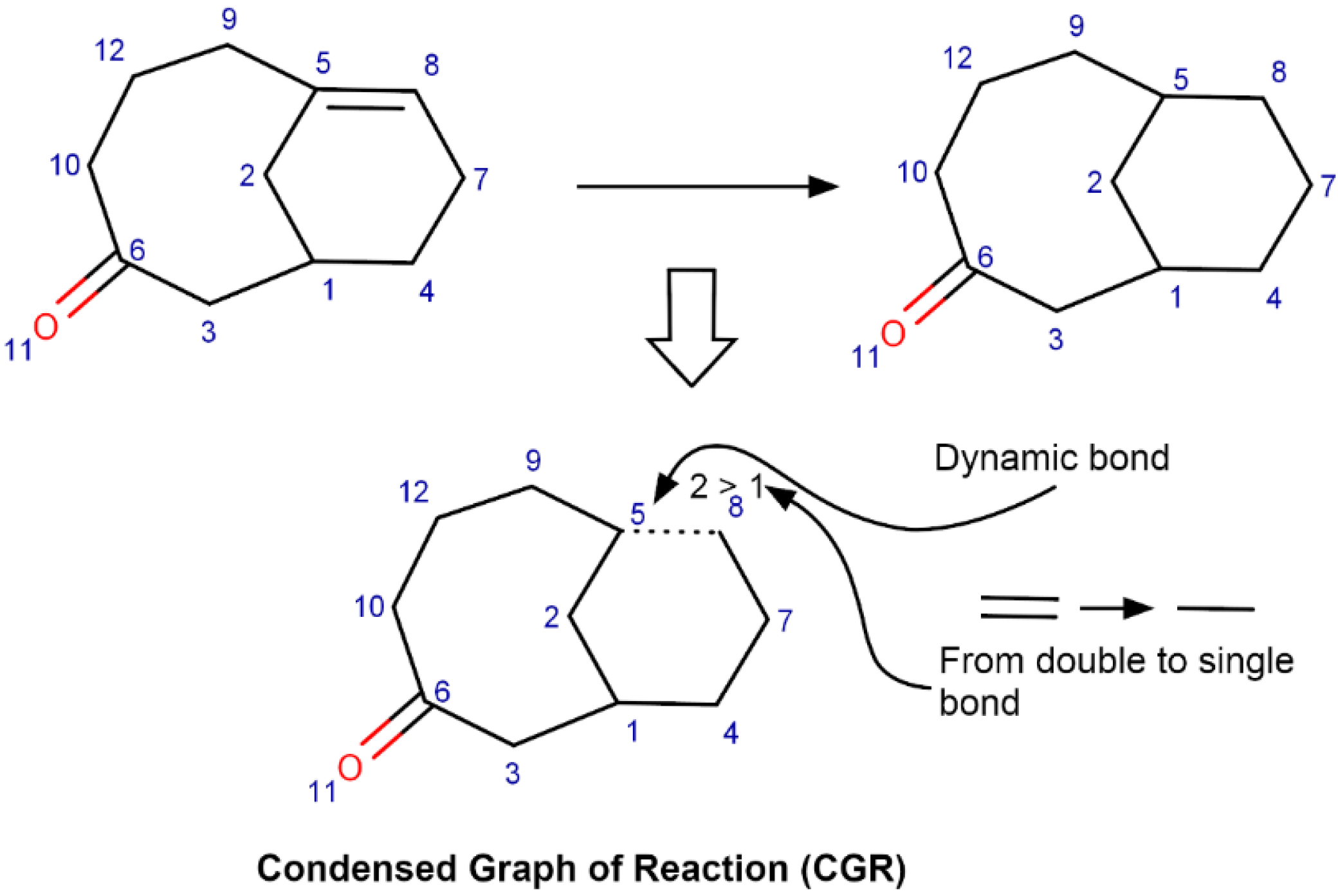
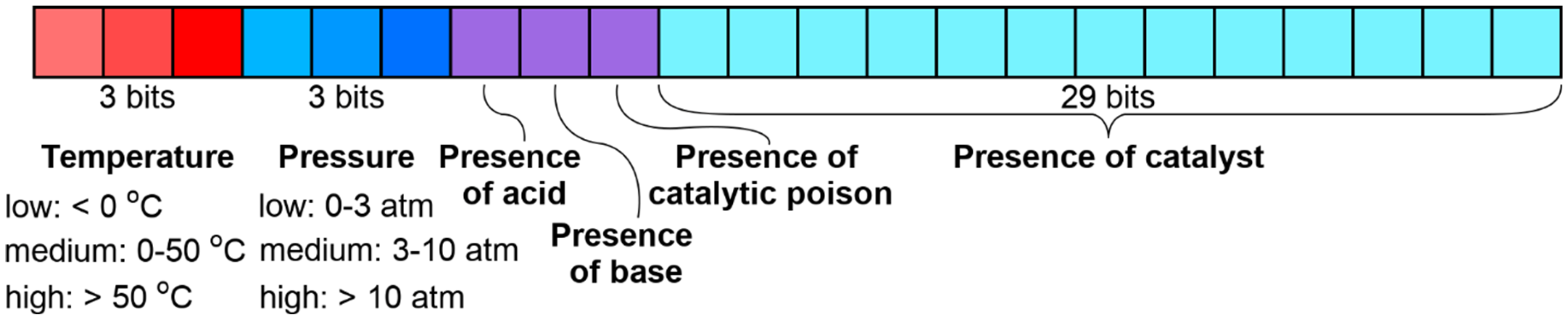
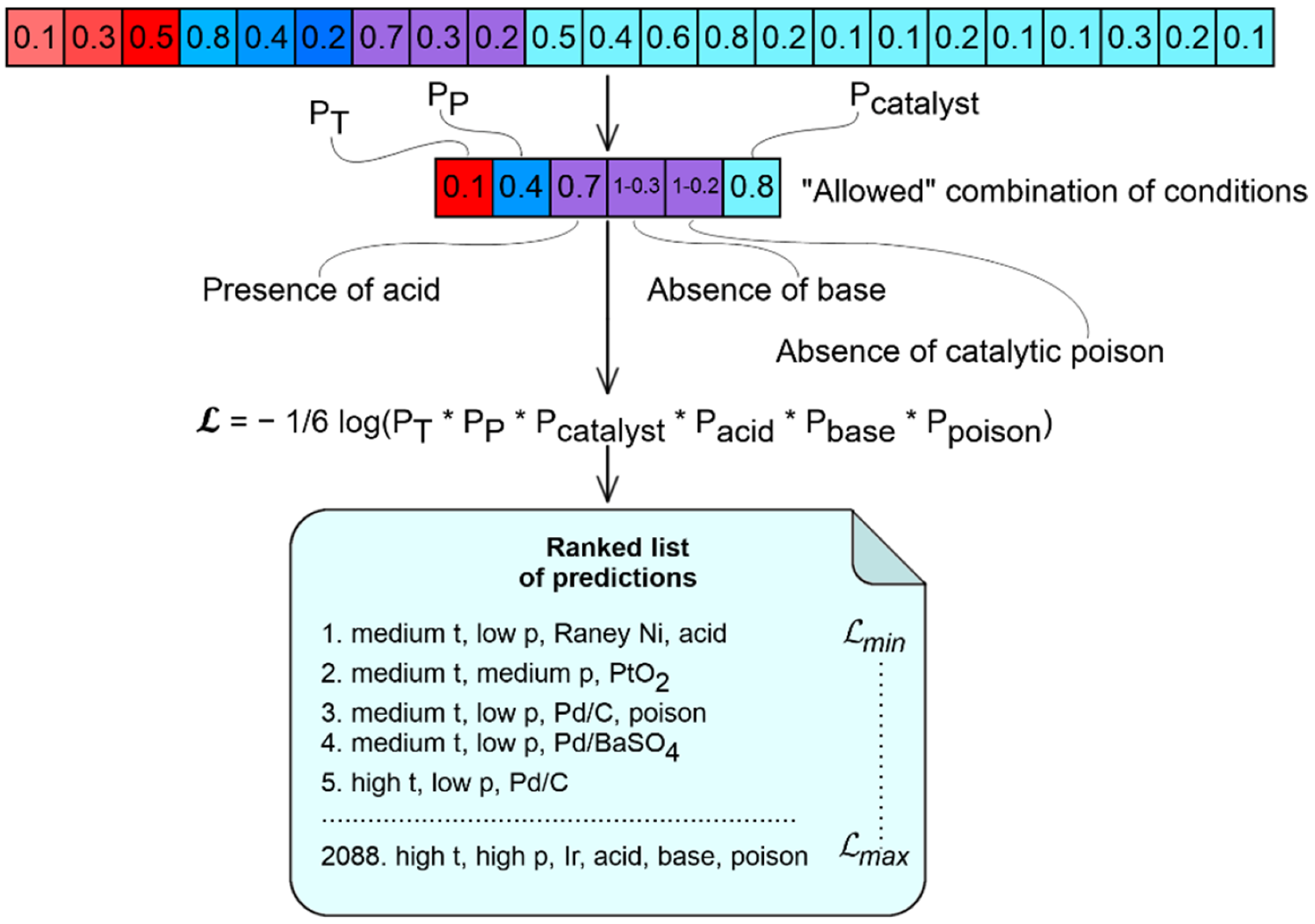
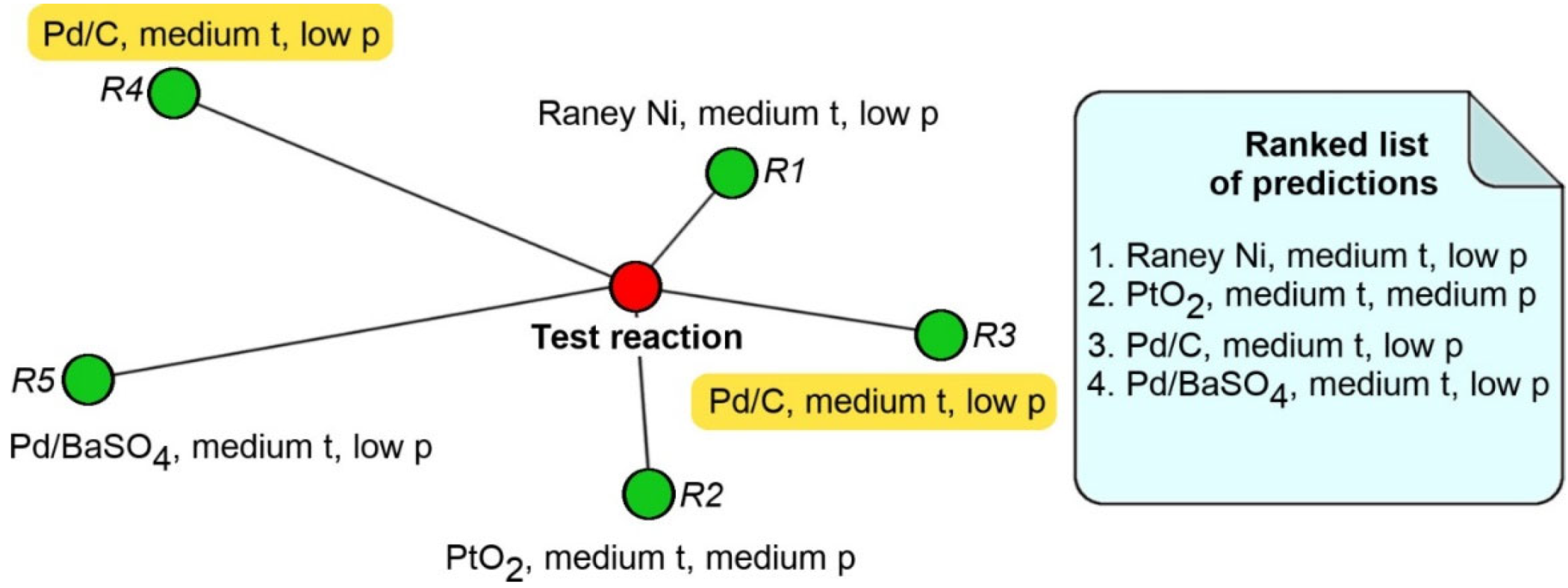
3.4.1. Likelihood Ranking Model
- -
- the number and size of hidden layers;
- -
- the number of layers: 1–3;
- -
- the number of neurons: 10, 20, 40, 80, 100, 200, 400, 800 (for 1–3 layered networks, the upper hidden layer does not have more neurons than the previous one), 1000, 2000, 3000, 4000 (for networks with a single hidden layer);
- -
- the number of learning epochs: 10–500;
- -
- the dropout value: 0–0.5 (for 2–3 layered networks only).
3.4.2. Null Model
3.4.3. k Nearest Neighbors Approach
3.4.4. Model from ASKOS
3.4.5. RecNN
3.5. Model Performance Assessment
3.6. Applicability Domain
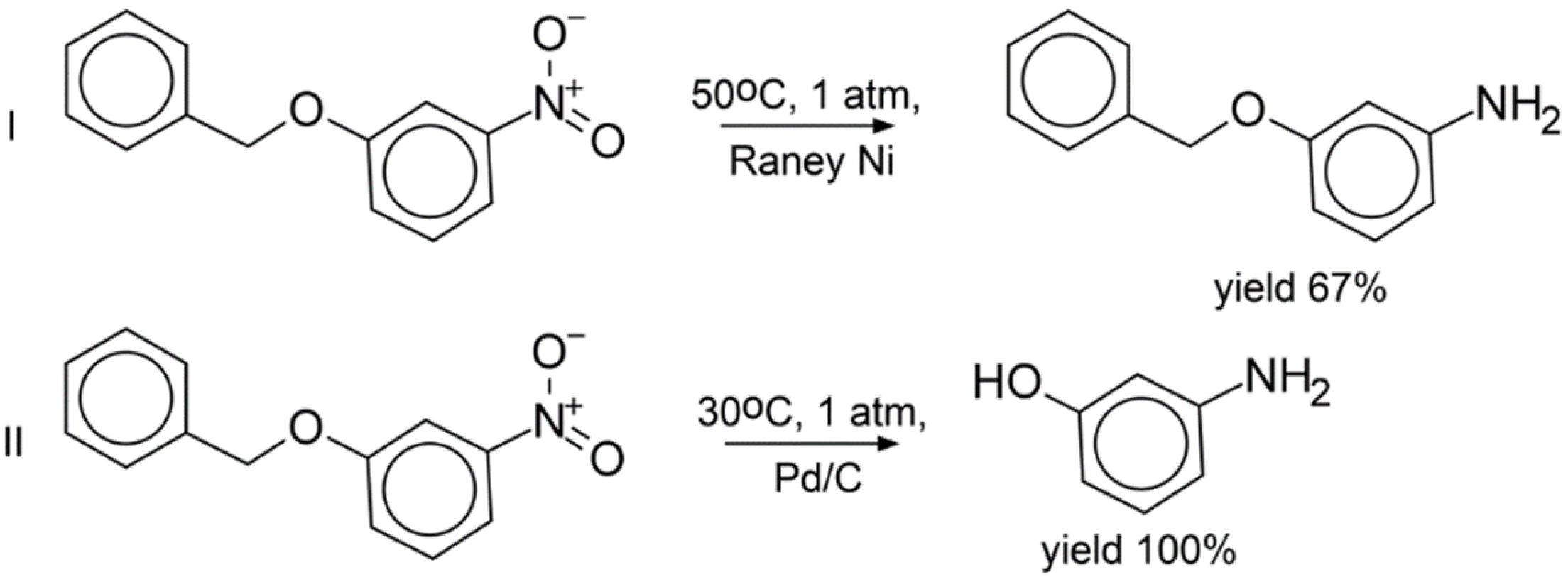
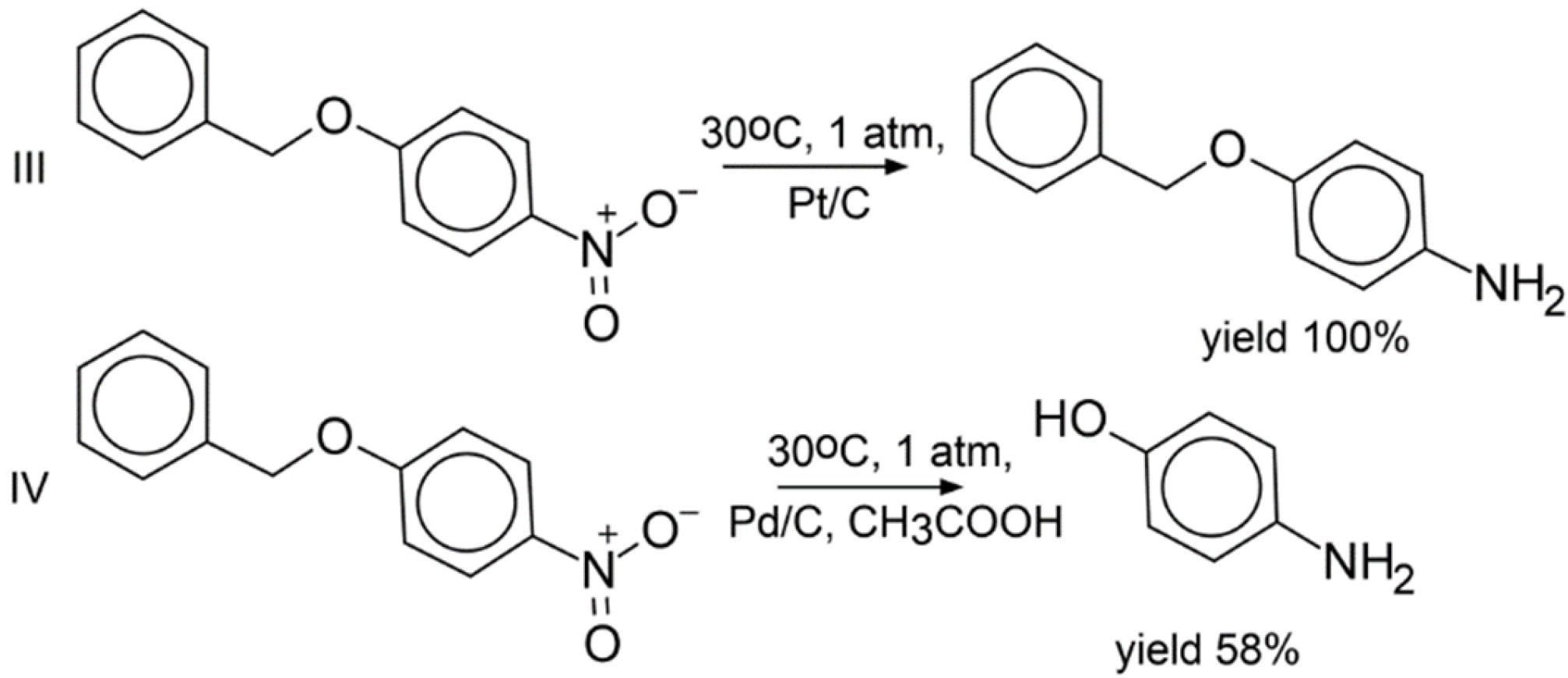
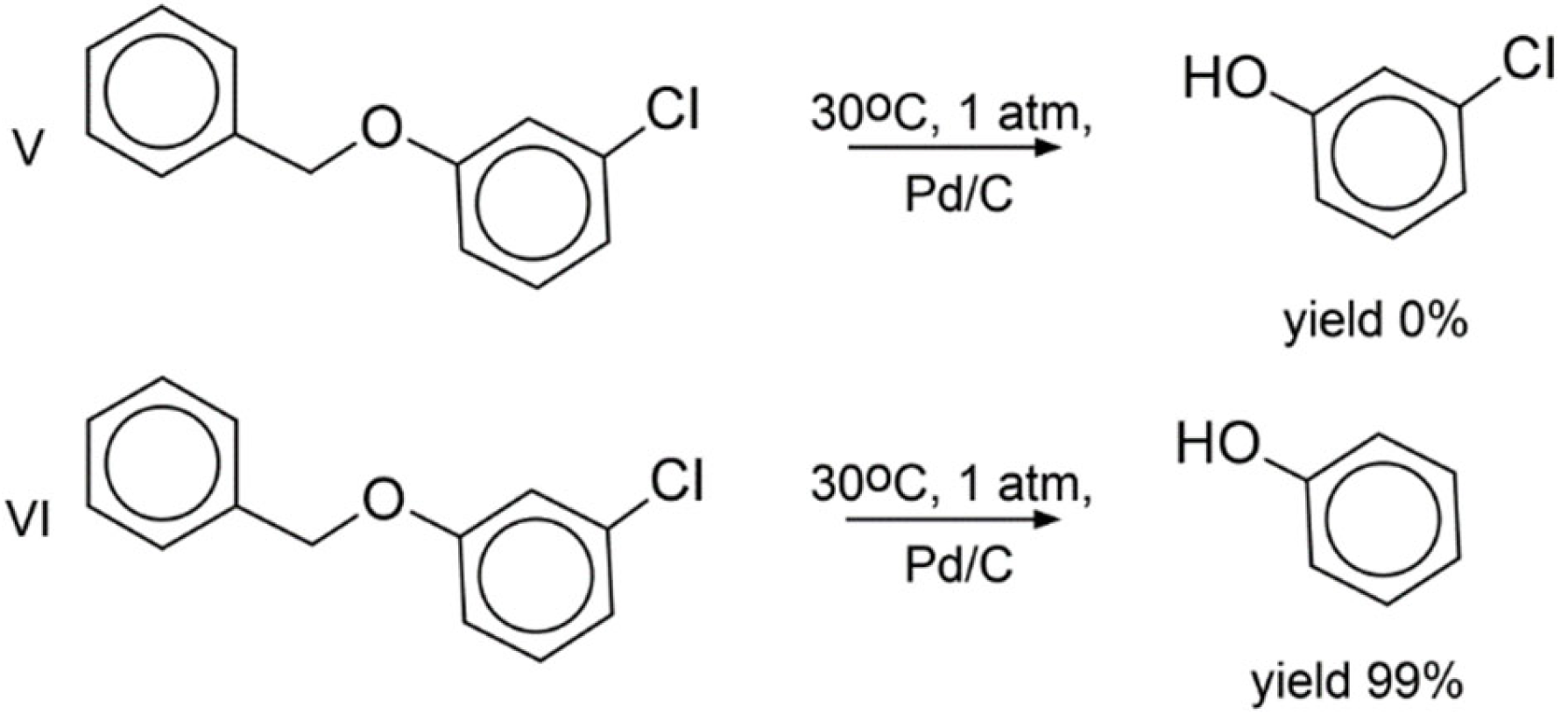
3.7. Experimental Validation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| kNN | k nearest neighbor approach |
| AD | applicability domain |
| QSAR | quantitative structure-activity relationship |
| LRM | Likelihood ranking model |
| NM | null model |
References
- Sanderson, K. Automation: Chemistry Shoots for the Moon. Nature 2019, 568, 577–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coley, C.W.; Thomas, D.A.; Lummiss, J.A.M.; Jaworski, J.N.; Breen, C.P.; Schultz, V.; Hart, T.; Fishman, J.S.; Rogers, L.; Gao, H.; et al. A Robotic Platform for Flow Synthesis of Organic Compounds Informed by AI Planning. Science 2019, 365, eaax1566. [Google Scholar] [CrossRef] [PubMed]
- Burger, B.; Maffettone, P.M.; Gusev, V.V.; Aitchison, C.M.; Bai, Y.; Wang, X.; Li, X.; Alston, B.M.; Li, B.; Clowes, R.; et al. A Mobile Robotic Chemist. Nature 2020, 583, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Steiner, S.; Wolf, J.; Glatzel, S.; Andreou, A.; Granda, J.M.; Keenan, G.; Hinkley, T.; Aragon-Camarasa, G.; Kitson, P.J.; Angelone, D.; et al. Organic Synthesis in a Modular Robotic System Driven by a Chemical Programming Language. Science 2019, 363, eaav2211. [Google Scholar] [CrossRef] [Green Version]
- Mehr, S.H.M.; Craven, M.; Leonov, A.I.; Keenan, G.; Cronin, L. A Universal System for Digitization and Automatic Execution of the Chemical Synthesis Literature. Science 2020, 370, 101–108. [Google Scholar] [CrossRef]
- Struebing, H.; Ganase, Z.; Karamertzanis, P.G.; Siougkrou, E.; Haycock, P.; Piccione, P.M.; Armstrong, A.; Galindo, A.; Adjiman, C.S. Computer-Aided Molecular Design of Solvents for Accelerated Reaction Kinetics. Nat. Chem. 2013, 5, 952–957. [Google Scholar] [CrossRef] [Green Version]
- Gao, H.; Struble, T.J.; Coley, C.W.; Wang, Y.; Green, W.H.; Jensen, K.F. Using Machine Learning to Predict Suitable Conditions for Organic Reactions. ACS Cent. Sci. 2018, 4, 1465–1476. [Google Scholar] [CrossRef] [Green Version]
- Marcou, G.; de Sousa, J.A.; Latino, D.A.R.S.; de Luca, A.; Horvath, D.; Rietsch, V.; Varnek, A. Expert System for Predicting Reaction Conditions: The Michael Reaction Case. J. Chem. Inf. Model. 2015, 55, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.I.; Madzhidov, T.I.; Klimchuk, O.; Nugmanov, R.I.; Antipin, I.S.; Varnek, A. Automatized Assessment of Protective Group Reactivity: A Step Toward Big Reaction Data Analysis. J. Chem. Inf. Model. 2016, 56, 2140–2148. [Google Scholar] [CrossRef]
- Segler, M.H.S.; Waller, M.P. Modelling Chemical Reasoning to Predict and Invent Reactions. Chem.-A Eur. J. 2017, 23, 6118–6128. [Google Scholar] [CrossRef] [Green Version]
- Walker, E.; Kammeraad, J.; Goetz, J.; Robo, M.T.; Tewari, A.; Zimmerman, P.M. Learning To Predict Reaction Conditions: Relationships between Solvent, Molecular Structure, and Catalyst. J. Chem. Inf. Model. 2019, 59, 3645–3654. [Google Scholar] [CrossRef] [PubMed]
- Schneider, N.; Lowe, D.M.; Sayle, R.A.; Tarselli, M.A.; Landrum, G.A. Big Data from Pharmaceutical Patents: A Computational Analysis of Medicinal Chemists’ Bread and Butter. J. Med. Chem. 2016, 59, 4385–4402. [Google Scholar] [CrossRef] [PubMed]
- Gimadiev, T.; Lin, A.; Afonina, V.; Batyrshin, D.; Nugmanov, R.; Akhmetshin, T.; Sidorov, P.; Dyubankova, N.; Verhoeven, J.; Wegner, J.; et al. Reaction Data Curation I: Chemical Structures and Transformations Standardization. Mol. Inform. 2021, 40, 2100119. [Google Scholar] [CrossRef] [PubMed]
- Nugmanov, R.I.; Mukhametgaleev, R.N.; Akhmetshin, T.; Gimadiev, T.R.; Afonina, V.A.; Madzhidov, T.I.; Varnek, A. CGRtools: Python Library for Molecule, Reaction, and Condensed Graph of Reaction Processing. J. Chem. Inf. Model. 2019, 59, 2516–2521. [Google Scholar] [CrossRef]
- Standardizer, J. 19.4.0, ChemAxon Ltd.: Chem. 2019. Available online: www.chemaxon.com (accessed on 22 November 2021).
- Chen, W.L.; Chen, D.Z.; Taylor, K.T. Automatic Reaction Mapping and Reaction Center Detection. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2013, 3, 560–593. [Google Scholar] [CrossRef]
- Dobson, N.A.; Eglinton, G.; Krishnamurti, M.; Raphael, R.A.; Willis, R.G. Selective Catalytic Hydrogenation of Acetylenes. Tetrahedron 1961, 16, 16–24. [Google Scholar] [CrossRef]
- McEwen, A.B.; Guttieri, M.J.; Maier, W.F.; Laine, R.M.; Shvo, Y. Metallic Palladium, the Actual Catalyst in Lindlar and Rosenmund Reductions? J. Org. Chem. 1983, 48, 4436–4438. [Google Scholar] [CrossRef]
- King, A.O.; Shinkai, I. Palladium on Calcium Carbonate (Lead Poisoned). In e-EROS Encyclopedia of Reagents for Organic Synthesis; Crich, D., Ed.; John Wiley & Sons, Ltd.: Chichester, UK, 2001. [Google Scholar] [CrossRef]
- Lindlar, H. Ein Neuer Katalysator Für Selektive Hydrierungen. Helv. Chim. Acta 1952, 35, 446–450. [Google Scholar] [CrossRef]
- Tian, J.; Chen, W.; Wu, P.; Zhu, Z.; Li, X. Cu–Mg–Zr/SiO2 Catalyst for the Selective Hydrogenation of Ethylene Carbonate to Methanol and Ethylene Glycol. Catal. Sci. Technol. 2018, 8, 2624–2635. [Google Scholar] [CrossRef]
- Xie, Y.; Wang, H.; Liu, X.; Xia, Y. Zirconium Tripolyphosphate as an Efficient Catalyst for the Hydrogenation of Ethyl Levulinate to γ-Valerolactone with Isopropanol as Hydrogen Donor. React. Kinet. Mech. Catal. 2018, 125, 71–84. [Google Scholar] [CrossRef]
- Maxted, E.B. The Poisoning of Metallic Catalysts. Adv. Catal. 1951, 129–178. [Google Scholar] [CrossRef]
- Hagen, J. Industrial Catalysis; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2015. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, C.; Liu, H.; Qiu, J.; Bao, X. Ag/SiO2: A Novel Catalyst with High Activity and Selectivity for Hydrogenation of Chloronitrobenzenes. Chem. Commun. 2005, 42, 5298. [Google Scholar] [CrossRef]
- Bullock, R.M. Molybdenum and Tungsten Catalysts for Hydrogenation, Hydrosilylation and Hydrolysis. In Catalysis without Precious Metals; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2010; pp. 51–81. [Google Scholar] [CrossRef]
- Shiekh, B.A.; Kaur, D.; Kumar, S. Bio-Mimetic Self-Assembled Computationally Designed Catalysts of Mo and W for Hydrogenation of CO2/Dehydrogenation of HCOOH Inspired by the Active Site of Formate Dehydrogenase. Phys. Chem. Chem. Phys. 2019, 21, 21370–21380. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Torres, A.; Aguilar-Calderón, J.R.; Encerrado-Manriquez, A.M.; Pink, M.; Metta-Magaña, A.J.; Lee, W.; Fortier, S. Titanium-Mediated Catalytic Hydrogenation of Monocyclic and Polycyclic Arenes. Chem.–A Eur. J. 2020, 26, 2803–2807. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; van Putten, R.; Kulyaev, P.O.; Filonenko, G.A.; Pidko, E.A. Computational Insights into the Catalytic Role of the Base Promoters in Ester Hydrogenation with Homogeneous Non-Pincer-Based Mn-P,N Catalyst. J. Catal. 2018, 363, 136–143. [Google Scholar] [CrossRef]
- Wang, L.; Dong, Y.; Yan, T.; Hu, Z.; Jelle, A.A.; Meira, D.M.; Duchesne, P.N.; Loh, J.Y.Y.; Qiu, C.; Storey, E.E.; et al. Black Indium Oxide a Photothermal CO2 Hydrogenation Catalyst. Nat. Commun. 2020, 11, 2432. [Google Scholar] [CrossRef]
- Varnek, A.; Fourches, D.; Hoonakker, F.; Solovev, V.P. Substructural Fragments: An Universal Language to Encode Reactions, Molecular and Supramolecular Structures. J. Comput. Aided. Mol. Des. 2005, 19, 693–703. [Google Scholar] [CrossRef]
- Marcou, G.; Solovev, V.P.; Horvath, D.; Varnek, A. ISIDA Fragmentor 2017—User Manual. Available online: http://infochim.u-strasbg.fr/downloads/manuals/Fragmentor2017/Fragmentor2017_Manual_nov2017.pdf (accessed on 22 November 2021).
- Rakhimbekova, A.; Madzhidov, T.I.; Nugmanov, R.I.; Gimadiev, T.R.; Baskin, I.I.; Varnek, A. Comprehensive Analysis of Applicability Domains of QSPR Models for Chemical Reactions. Int. J. Mol. Sci. 2020, 21, 5542. [Google Scholar] [CrossRef]
- Ross, D.A.; Lim, J.; Lin, R.-S.; Yang, M.-H. Incremental Learning for Robust Visual Tracking. Int. J. Comput. Vis. 2008, 77, 125–141. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-Learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Chollet, F. Keras. 2015. Available online: https://keras.io (accessed on 22 November 2021).
- Abadi, M.; Barham, P.; Chen, J.; Chen, Z.; Davis, A.; Dean, J.; Devin, M.; Ghemawat, S.; Irving, G.; Isard, M.; et al. TensorFlow: A System for Large-Scale Machine Learning. In Proceedings of the 12th USENIX Symposium on Operating Systems Design and Implementation, OSDI 2016; USENIX Association: Berkeley, CA, USA, 2016; pp. 265–283. [Google Scholar]
- Gao, H.; Struble, T.J.; Coley, C.W.; Wang, Y.; Green, W.H.; Jensen, K.F. Github Page of Paper “Using Machine Learning to Predict Suitable Conditions for Organic Reactions”. Available online: https://github.com/Coughy1991/Reaction_condition_recommendation (accessed on 22 November 2021).
- Liu, T.-Y. Learning to Rank for Information Retrieval. Found. Trends® Inf. Retr. 2007, 3, 225–331. [Google Scholar] [CrossRef]
- Armarego, W.L.F.; Chai, C.L.L. Purification of Organic Chemicals. Purif. Lab. Chem. 2009. [Google Scholar] [CrossRef]
- Zhao, Y.; Liu, Y.; Chen, D.; Wei, Z.; Liu, W.; Gong, P. Synthesis and Biological Evaluation of 1H-Benzimidazol-5-Ols as Potent HBV Inhibitors. Bioorganic Med. Chem. Lett. 2010, 20, 7230–7233. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Song, G. Combined Microwave and Ultrasound Assisted Williamson Ether Synthesis in the Absence of Phase-Transfer Catalysts. Green Chem. 2002, 4, 349–351. [Google Scholar] [CrossRef]
- Chakraborti, A.K.; Chankeshwara, S.V. Counterattack Mode Differential Acetylative Deprotection of Phenylmethyl Ethers: Applications to Solid Phase Organic Reactions. J. Org. Chem. 2009, 74, 1367–1370. [Google Scholar] [CrossRef]
- Johnson, J.; Douze, M.; Jégou, H. Billion-Scale Similarity Search with GPUs. arXiv 2017, arXiv:1702.08734. [Google Scholar] [CrossRef] [Green Version]
- FAISS. 2017. Available online: https://ai.facebook.com/tools/faiss/ (accessed on 22 November 2021).









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mode | TOP 1 | TOP 3 | TOP 5 | TOP 10 | ||||
|---|---|---|---|---|---|---|---|---|
| LRM | NM | LRM | NM | LRM | NM | LRM | NM | |
| catalyst | 82 | 66 | 93 | 75 | 96 | 85 | 98 | 91 |
| pressure | 78 | 59 | 100 | 100 | 100 | 100 | 100 | 100 |
| additives | 91 | 87 | 99 | 97 | 99 | 100 | 99 | 100 |
| temperature | 93 | 85 | 100 | 100 | 100 | 100 | 100 | 100 |
| catalyst, pressure | 67 | 44 | 85 | 66 | 90 | 71 | 95 | 81 |
| additives, pressure | 72 | 52 | 93 | 87 | 97 | 92 | 99 | 98 |
| catalyst, additives | 76 | 58 | 89 | 67 | 92 | 76 | 96 | 84 |
| catalyst, pressure | 67 | 44 | 85 | 66 | 90 | 71 | 95 | 81 |
| catalyst, temperature | 78 | 60 | 90 | 70 | 93 | 76 | 96 | 86 |
| temperature, pressure | 75 | 56 | 93 | 85 | 98 | 97 | 100 | 100 |
| catalyst, pressure, additives | 63 | 39 | 80 | 58 | 86 | 64 | 91 | 73 |
| catalyst, temperature, additives | 73 | 53 | 86 | 61 | 89 | 69 | 93 | 80 |
| catalyst, temperature, pressure | 65 | 42 | 81 | 60 | 86 | 65 | 91 | 75 |
| temperature, pressure, additives | 69 | 50 | 88 | 74 | 93 | 84 | 97 | 94 |
| catalyst, additives, pressure, temperature | 61 | 38 | 77 | 53 | 82 | 59 | 88 | 67 |
| Before Standardization | After Standardization | |
|---|---|---|
| Initial dataset | 345,619/566,896 | |
| Includes temperature | 175,697/295,331 | 140,843/208,671 |
| Includes pressure | 150,100/215,191 | 113,057/151,154 |
| Includes catalysts | 207,070/275,546 | 279,739/419,223 * |
| Includes temperature and pressure | 93,769/143,138 | 68,711/96,755 |
| Includes temperature, pressure and catalysts | 36,684/52,367 | 57,427/72,871 * |
| Modeling dataset (T, P and single catalyst) | 54,345/66,840 | |
| Dataset | Number of Reactions | Number of Transformations | Number of Unique Combinations of Condition Components |
|---|---|---|---|
| Modeling set | 66,840 | 54,345 | 507 |
| Training set | 42,541 | 40,870 | 494 |
| Retrospective test set | 5103 | 4888 | 264 |
| Prospective test set | 5011 | 4992 | 144 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Afonina, V.A.; Mazitov, D.A.; Nurmukhametova, A.; Shevelev, M.D.; Khasanova, D.A.; Nugmanov, R.I.; Burilov, V.A.; Madzhidov, T.I.; Varnek, A. Prediction of Optimal Conditions of Hydrogenation Reaction Using the Likelihood Ranking Approach. Int. J. Mol. Sci. 2022, 23, 248. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23010248
Afonina VA, Mazitov DA, Nurmukhametova A, Shevelev MD, Khasanova DA, Nugmanov RI, Burilov VA, Madzhidov TI, Varnek A. Prediction of Optimal Conditions of Hydrogenation Reaction Using the Likelihood Ranking Approach. International Journal of Molecular Sciences. 2022; 23(1):248. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23010248
Chicago/Turabian StyleAfonina, Valentina A., Daniyar A. Mazitov, Albina Nurmukhametova, Maxim D. Shevelev, Dina A. Khasanova, Ramil I. Nugmanov, Vladimir A. Burilov, Timur I. Madzhidov, and Alexandre Varnek. 2022. "Prediction of Optimal Conditions of Hydrogenation Reaction Using the Likelihood Ranking Approach" International Journal of Molecular Sciences 23, no. 1: 248. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23010248