
Neurofibromatosis Type 1 Gene Alterations Define Specific Features of a Subset of Glioblastomas

 ,
,  , and
, and
Abstract
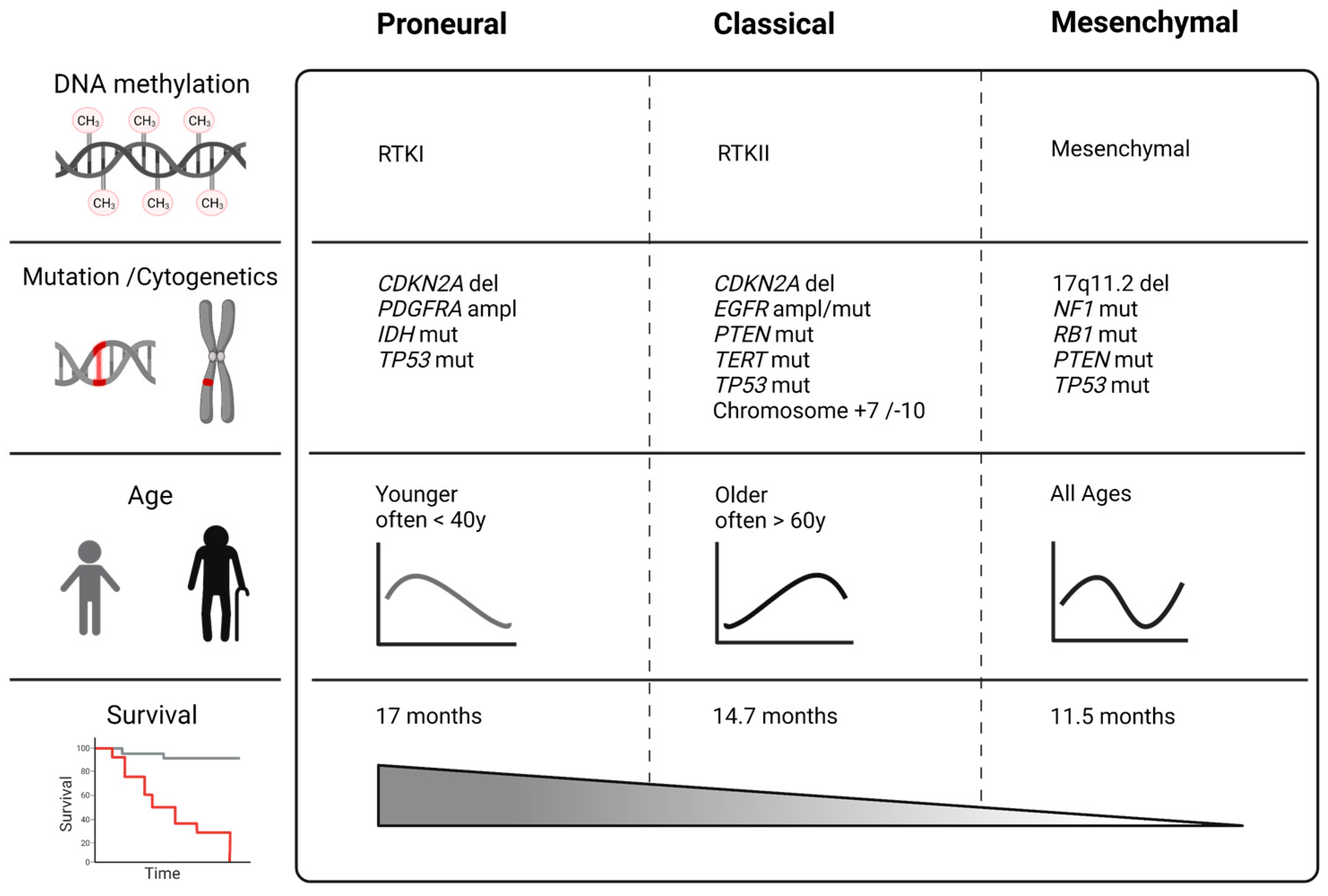
:1. Molecular Subtypes of Glioblastomas
2. The Neurofibromatosis Type 1 (NF1) Gene in Normal Tissue
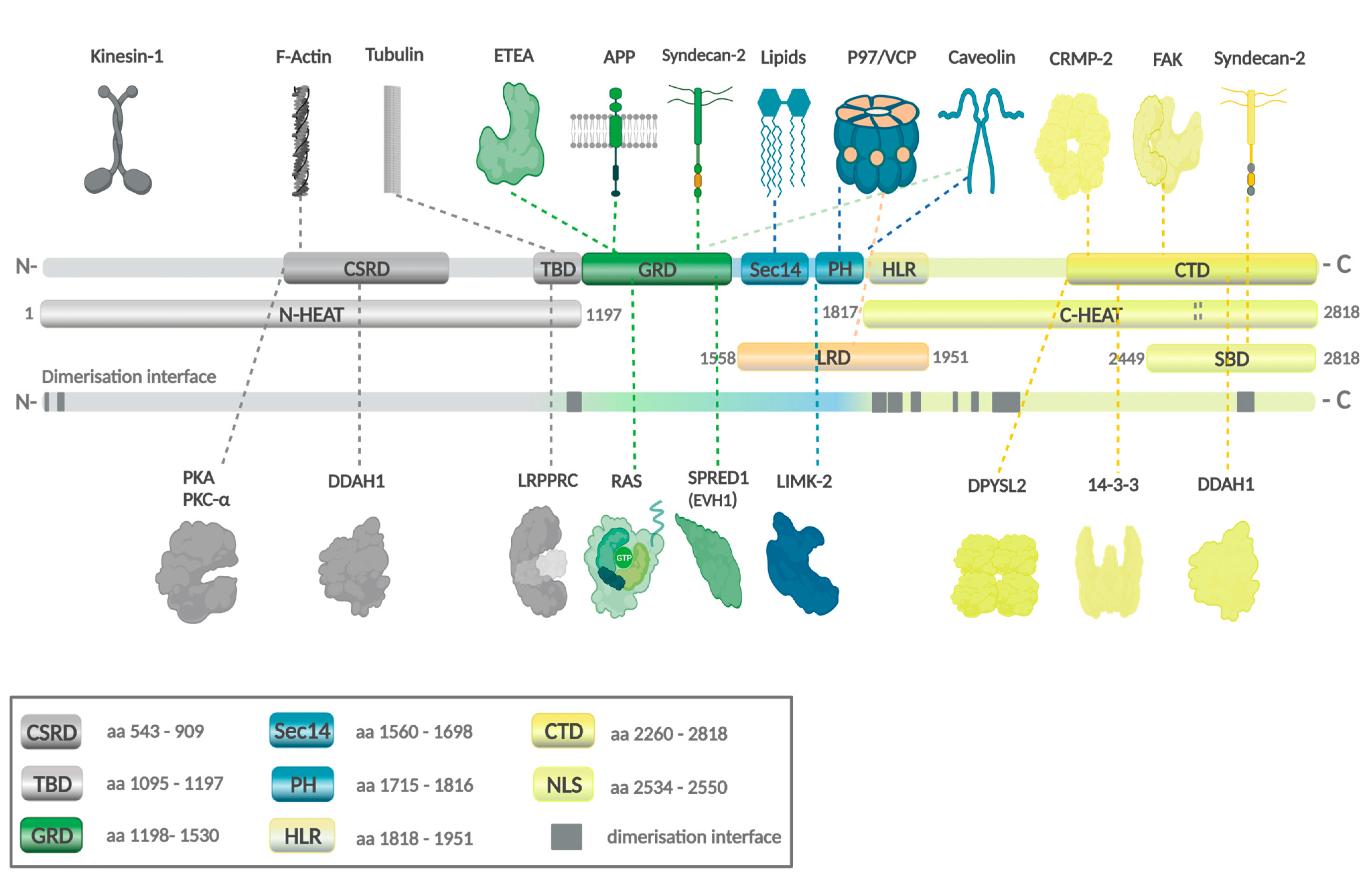
3. The Neurofibromatosis Type 1 (NF1) Gene in Neoplasia
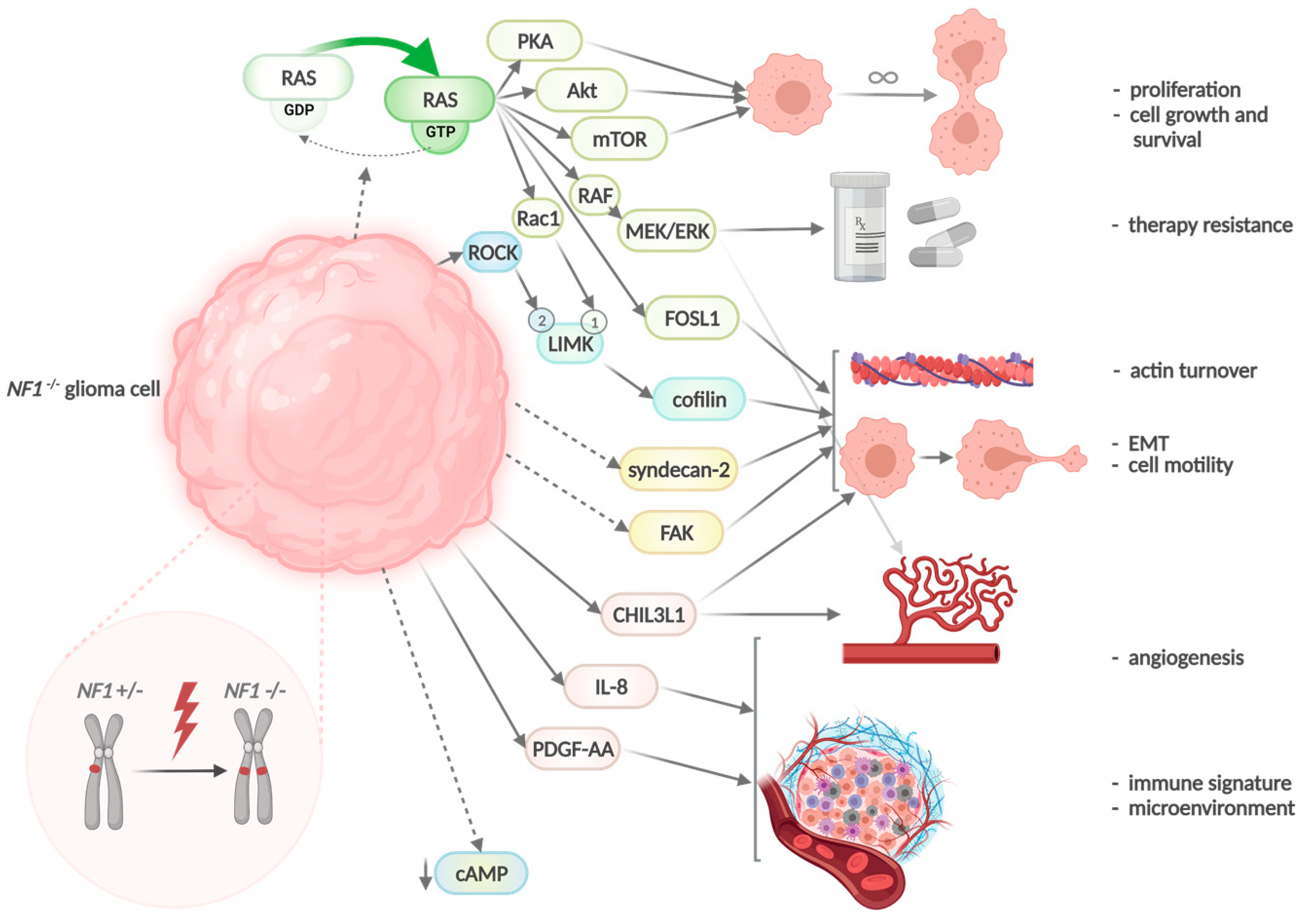
4. The Neurofibromatosis Type 1 (NF1) Gene in GBM
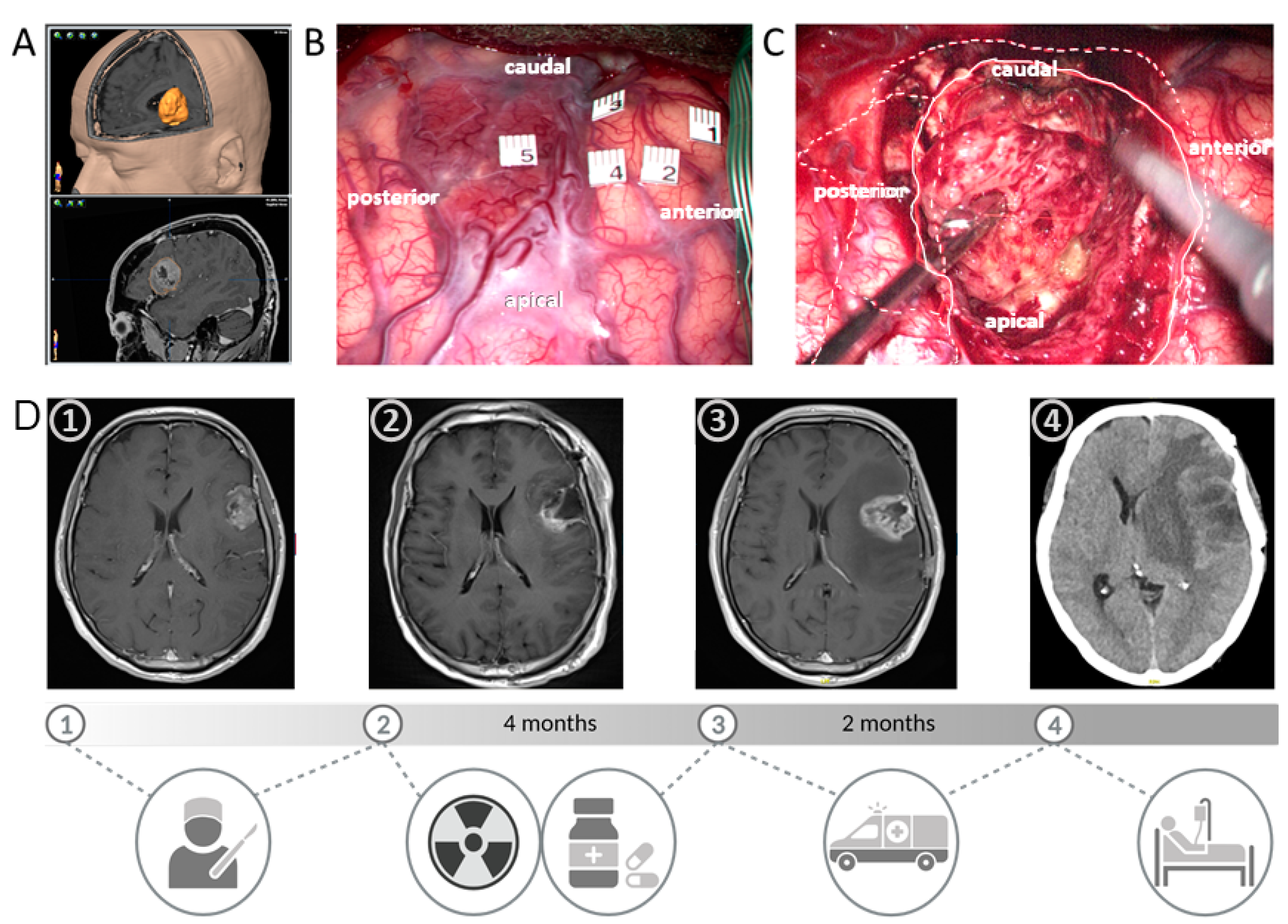
5. Mesenchymal Glioblastomas Accumulate Neurofibromatosis Type 1 (NF1) Gene Alterations
6. NF1 Mutations and Glioma Invasiveness
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Ostrom, Q.T.; Gittleman, H.; Truitt, G.; Boscia, A.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2011–2015. Neuro-Oncollogy 2018, 20, iv1–iv86. [Google Scholar] [CrossRef] [Green Version]
- Wesseling, P.; Capper, D. WHO 2016 Classification of gliomas. Neuropathol. Appl. Neurobiol. 2018, 44, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Delgado-López, P.D.; Corrales-García, E.M. Survival in glioblastoma: A review on the impact of treatment modalities. Clin. Transl. Oncol. 2016, 18, 1062–1071. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma: A Randomized Clinical Trial. JAMA 2017, 318, 2306–2316. [Google Scholar] [CrossRef] [Green Version]
- Wirsching, H.G.; Galanis, E.; Weller, M. Glioblastoma. Handb. Clin. Neurol. 2016, 134, 381–397. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Lukas, R.V.; Wainwright, D.A.; Ladomersky, E.; Sachdev, S.; Sonabend, A.M.; Stupp, R. Newly Diagnosed Glioblastoma: A Review on Clinical Management. Oncology 2019, 33, 91–100. [Google Scholar] [PubMed]
- Brown, T.J.; Brennan, M.C.; Li, M.; Church, E.W.; Brandmeir, N.J.; Rakszawski, K.L.; Patel, A.S.; Rizk, E.B.; Suki, D.; Sawaya, R.; et al. Association of the Extent of Resection With Survival in Glioblastoma: A Systematic Review and Meta-analysis. JAMA Oncol. 2016, 2, 1460–1469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bette, S.; Barz, M.; Wiestler, B.; Huber, T.; Gerhardt, J.; Buchmann, N.; Combs, S.E.; Schmidt-Graf, F.; Delbridge, C.; Zimmer, C.; et al. Prognostic Value of Tumor Volume in Glioblastoma Patients: Size Also Matters for Patients with Incomplete Resection. Ann. Surg. Oncol. 2018, 25, 558–564. [Google Scholar] [CrossRef]
- Herrlinger, U.; Tzaridis, T.; Mack, F.; Steinbach, J.P.; Schlegel, U.; Sabel, M.; Hau, P.; Kortmann, R.D.; Krex, D.; Grauer, O.; et al. Lomustine-temozolomide combination therapy versus standard temozolomide therapy in patients with newly diagnosed glioblastoma with methylated MGMT promoter (CeTeG/NOA-09): A randomised, open-label, phase 3 trial. Lancet 2019, 393, 678–688. [Google Scholar] [CrossRef]
- Perry, A.; Wesseling, P. Histologic classification of gliomas. Handb. Clin. Neurol. 2016, 134, 71–95. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Burger, P.; Ellison, D.W.; Reifenberger, G.; von Deimling, A.; Aldape, K.; Brat, D.; Collins, V.P.; Eberhart, C.; et al. International Society Of Neuropathology-Haarlem consensus guidelines for nervous system tumor classification and grading. Brain Pathol. 2014, 24, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Capper, D.; Jones, D.T.W.; Sill, M.; Hovestadt, V.; Schrimpf, D.; Sturm, D.; Koelsche, C.; Sahm, F.; Chavez, L.; Reuss, D.E.; et al. DNA methylation-based classification of central nervous system tumours. Nature 2018, 555, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Ceccarelli, M.; Barthel, F.P.; Malta, T.M.; Sabedot, T.S.; Salama, S.R.; Murray, B.A.; Morozova, O.; Newton, Y.; Radenbaugh, A.; Pagnotta, S.M.; et al. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell 2016, 164, 550–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Angelo, F.; Ceccarelli, M.; Tala; Garofano, L.; Zhang, J.; Frattini, V.; Caruso, F.P.; Lewis, G.; Alfaro, K.D.; Bauchet, L.; et al. The molecular landscape of glioma in patients with Neurofibromatosis 1. Nat. Med. 2019, 25, 176–187. [Google Scholar] [CrossRef]
- Brat, D.J.; Aldape, K.; Colman, H.; Holland, E.C.; Louis, D.N.; Jenkins, R.B.; Kleinschmidt-DeMasters, B.K.; Perry, A.; Reifenberger, G.; Stupp, R.; et al. cIMPACT-NOW update 3: Recommended diagnostic criteria for “Diffuse astrocytic glioma, IDH-wildtype, with molecular features of glioblastoma, WHO grade IV”. Acta Neuropathol. 2018, 136, 805–810. [Google Scholar] [CrossRef] [Green Version]
- Louis, D.N.; Wesseling, P.; Aldape, K.; Brat, D.J.; Capper, D.; Cree, I.A.; Eberhart, C.; Figarella-Branger, D.; Fouladi, M.; Fuller, G.N.; et al. cIMPACT-NOW update 6: New entity and diagnostic principle recommendations of the cIMPACT-Utrecht meeting on future CNS tumor classification and grading. Brain Pathol. 2020, 30, 844–856. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; deCarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56.e46. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Smith-Cohn, M.; Cohen, A.L.; Colman, H. Glioma Subclassifications and Their Clinical Significance. Neurotherapeutics 2017, 14, 284–297. [Google Scholar] [CrossRef] [Green Version]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abedalthagafi, M.; Barakeh, D.; Foshay, K.M. Immunogenetics of glioblastoma: The future of personalized patient management. NPJ Precis. Oncol. 2018, 2, 27. [Google Scholar] [CrossRef] [PubMed]
- Sturm, D.; Witt, H.; Hovestadt, V.; Khuong-Quang, D.A.; Jones, D.T.; Konermann, C.; Pfaff, E.; Tonjes, M.; Sill, M.; Bender, S.; et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 2012, 22, 425–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Fletcher, M.; Gu, Z.; Wang, Q.; Costa, B.; Bertoni, A.; Man, K.H.; Schlotter, M.; Felsberg, J.; Mangei, J.; et al. Glioblastoma epigenome profiling identifies SOX10 as a master regulator of molecular tumour subtype. Nat. Commun. 2020, 11, 6434. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, T.; Riester, M.; Cheng, Y.K.; Huse, J.T.; Squatrito, M.; Helmy, K.; Charles, N.; Michor, F.; Holland, E.C. Most human non-GCIMP glioblastoma subtypes evolve from a common proneural-like precursor glioma. Cancer Cell 2014, 26, 288–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fedele, M.; Cerchia, L.; Pegoraro, S.; Sgarra, R.; Manfioletti, G. Proneural-Mesenchymal Transition: Phenotypic Plasticity to Acquire Multitherapy Resistance in Glioblastoma. Int. J. Mol. Sci. 2019, 20, 2746. [Google Scholar] [CrossRef] [Green Version]
- Segerman, A.; Niklasson, M.; Haglund, C.; Bergström, T.; Jarvius, M.; Xie, Y.; Westermark, A.; Sönmez, D.; Hermansson, A.; Kastemar, M.; et al. Clonal Variation in Drug and Radiation Response among Glioma-Initiating Cells Is Linked to Proneural-Mesenchymal Transition. Cell Rep. 2016, 17, 2994–3009. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [Green Version]
- Behnan, J.; Finocchiaro, G.; Hanna, G. The landscape of the mesenchymal signature in brain tumours. Brain 2019, 142, 847–866. [Google Scholar] [CrossRef] [Green Version]
- Bhat, K.P.L.; Balasubramaniyan, V.; Vaillant, B.; Ezhilarasan, R.; Hummelink, K.; Hollingsworth, F.; Wani, K.; Heathcock, L.; James, J.D.; Goodman, L.D.; et al. Mesenchymal differentiation mediated by NF-kappaB promotes radiation resistance in glioblastoma. Cancer Cell 2013, 24, 331–346. [Google Scholar] [CrossRef] [Green Version]
- Rutledge, W.C.; Kong, J.; Gao, J.; Gutman, D.A.; Cooper, L.A.; Appin, C.; Park, Y.; Scarpace, L.; Mikkelsen, T.; Cohen, M.L.; et al. Tumor-infiltrating lymphocytes in glioblastoma are associated with specific genomic alterations and related to transcriptional class. Clin. Cancer Res. 2013, 19, 4951–4960. [Google Scholar] [CrossRef] [Green Version]
- Barker, D.; Wright, E.; Nguyen, K.; Cannon, L.; Fain, P.; Goldgar, D.; Bishop, D.T.; Carey, J.; Baty, B.; Kivlin, J.; et al. Gene for von Recklinghausen neurofibromatosis is in the pericentromeric region of chromosome 17. Science 1987, 236, 1100–1102. [Google Scholar] [CrossRef] [Green Version]
- Wallace, M.R.; Andersen, L.B.; Saulino, A.M.; Gregory, P.E.; Glover, T.W.; Collins, F.S. A de novo Alu insertion results in neurofibromatosis type 1. Nature 1991, 353, 864–866. [Google Scholar] [CrossRef] [PubMed]
- Viskochil, D.; Buchberg, A.M.; Xu, G.; Cawthon, R.M.; Stevens, J.; Wolff, R.K.; Culver, M.; Carey, J.C.; Copeland, N.G.; Jenkins, N.A.; et al. Deletions and a translocation interrupt a cloned gene at the neurofibromatosis type 1 locus. Cell 1990, 62, 187–192. [Google Scholar] [CrossRef]
- Luijten, M.; Wang, Y.; Smith, B.T.; Westerveld, A.; Smink, L.J.; Dunham, I.; Roe, B.A.; Hulsebos, T.J. Mechanism of spreading of the highly related neurofibromatosis type 1 (NF1) pseudogenes on chromosomes 2, 14 and 22. Eur. J. Hum. Genet. 2000, 8, 209–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martín, Y.; Dopazo, A.; Hernández-Chico, C. Progress and challenges in developing a molecular diagnostic test for neurofibromatosis type 1. Expert Rev. Mol. Diagn. 2011, 11, 671–673. [Google Scholar] [CrossRef]
- Nishi, T.; Lee, P.S.; Oka, K.; Levin, V.A.; Tanase, S.; Morino, Y.; Saya, H. Differential expression of two types of the neurofibromatosis type 1 (NF1) gene transcripts related to neuronal differentiation. Oncogene 1991, 6, 1555–1559. [Google Scholar] [PubMed]
- Gutman, D.H.; Andersen, L.B.; Cole, J.L.; Swaroop, M.; Collins, F.S. An alternatively-spliced mRNA in the carboxy terminus of the neurofibromatosis type 1 (NF1) gene is expressed in muscle. Hum. Mol. Genet. 1993, 2, 989–992. [Google Scholar] [CrossRef] [PubMed]
- Danglot, G.; Regnier, V.; Fauvet, D.; Vassal, G.; Kujas, M.; Bernheim, A. Neurofibromatosis 1 (NF1) mRNAs expressed in the central nervous system are differentially spliced in the 5’ part of the gene. Hum. Mol. Genet. 1995, 4, 915–920. [Google Scholar] [CrossRef]
- Kaufmann, D.; Muller, R.; Kenner, O.; Leistner, W.; Hein, C.; Vogel, W.; Bartelt, B. The N-terminal splice product NF1-10a-2 of the NF1 gene codes for a transmembrane segment. Biochem. Biophys. Res. Commun. 2002, 294, 496–503. [Google Scholar] [CrossRef]
- Fagerberg, L.; Hallström, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteom. MCP 2014, 13, 397–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, L.B.; Ballester, R.; Marchuk, D.A.; Chang, E.; Gutmann, D.H.; Saulino, A.M.; Camonis, J.; Wigler, M.; Collins, F.S. A conserved alternative splice in the von Recklinghausen neurofibromatosis (NF1) gene produces two neurofibromin isoforms, both of which have GTPase-activating protein activity. Mol. Cell. Biol. 1993, 13, 487–495. [Google Scholar] [PubMed] [Green Version]
- Anastasaki, C.; Le, L.Q.; Kesterson, R.A.; Gutmann, D.H. Updated nomenclature for human and mouse neurofibromatosis type 1 genes. Neurol. Genet. 2017, 3, e169. [Google Scholar] [CrossRef] [Green Version]
- Messiaen, L.M. Molecular Diagnosis for NF1. In Multidisciplinary Approach to Neurofibromatosis Type 1; Tadini, G., Legius, E., Brems, H., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 15–34. [Google Scholar]
- Li, S.; Janosch, P.; Tanji, M.; Rosenfeld, G.C.; Waymire, J.C.; Mischak, H.; Kolch, W.; Sedivy, J.M. Regulation of Raf-1 kinase activity by the 14-3-3 family of proteins. EMBO J. 1995, 14, 685–696. [Google Scholar] [CrossRef] [PubMed]
- Hinman, M.N.; Sharma, A.; Luo, G.; Lou, H. Neurofibromatosis type 1 alternative splicing is a key regulator of Ras signaling in neurons. Mol. Cell. Biol. 2014, 34, 2188–2197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danglot, G.; Teinturier, C.; Duverger, A.; Bernheim, A. Tissue-specific alternative splicing of neurofibromatosis 1 (NF1) mRNA. Biomed. Pharmacother. 1994, 48, 365–372. [Google Scholar] [CrossRef]
- Vandenbroucke, I.; Vandesompele, J.; De Paepe, A.; Messiaen, L. Quantification of NF1 transcripts reveals novel highly expressed splice variants. FEBS Lett. 2002, 522, 71–76. [Google Scholar] [CrossRef] [Green Version]
- Gutmann, D.H.; Geist, R.T.; Rose, K.; Wright, D.E. Expression of two new protein isoforms of the neurofibromatosis type 1 gene product, neurofibromin, in muscle tissues. Dev. Dyn. 1995, 202, 302–311. [Google Scholar] [CrossRef]
- Bergoug, M.; Doudeau, M.; Godin, F.; Mosrin, C.; Vallée, B.; Bénédetti, H. Neurofibromin Structure, Functions and Regulation. Cells 2020, 9, 2365. [Google Scholar] [CrossRef]
- Gutmann, D.H.; Zhang, Y.; Hirbe, A. Developmental regulation of a neuron-specific neurofibromatosis 1 isoform. Ann. Neurol. 1999, 46, 777–782. [Google Scholar] [CrossRef]
- Geist, R.T.; Gutmann, D.H. Expression of a developmentally-regulated neuron-specific isoform of the neurofibromatosis 1 (NF1) gene. Neurosci. Lett. 1996, 211, 85–88. [Google Scholar] [CrossRef]
- Gutmann, D.H.; Cole, J.L.; Collins, F.S. Modulation of neurofibromatosis type 1 gene expression during in vitro myoblast differentiation. J. Neurosci. Res. 1994, 37, 398–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skuse, G.R.; Cappione, A.J. RNA processing and clinical variability in neurofibromatosis type I (NF1). Hum. Mol. Genet. 1997, 6, 1707–1712. [Google Scholar] [CrossRef] [Green Version]
- Park, V.M.; Kenwright, K.A.; Sturtevant, D.B.; Pivnick, E.K. Alternative splicing of exons 29 and 30 in the neurofibromatosis type 1 gene. Hum. Genet. 1998, 103, 382–385. [Google Scholar] [CrossRef] [PubMed]
- Scheffzek, K.; Ahmadian, M.R.; Kabsch, W.; Wiesmuller, L.; Lautwein, A.; Schmitz, F.; Wittinghofer, A. The Ras-RasGAP complex: Structural basis for GTPase activation and its loss in oncogenic Ras mutants. Science 1997, 277, 333–338. [Google Scholar] [CrossRef] [Green Version]
- Sherekar, M.; Han, S.W.; Ghirlando, R.; Messing, S.; Drew, M.; Rabara, D.; Waybright, T.; Juneja, P.; O’Neill, H.; Stanley, C.B.; et al. Biochemical and structural analyses reveal that the tumor suppressor neurofibromin (NF1) forms a high-affinity dimer. J. Biol. Chem. 2020, 295, 1105–1119. [Google Scholar] [CrossRef]
- Lupton, C.J.; Bayly-Jones, C.; D’Andrea, L.; Huang, C.; Schittenhelm, R.B.; Venugopal, H.; Whisstock, J.C.; Halls, M.L.; Ellisdon, A.M. The cryo-EM structure of the neurofibromin dimer reveals the molecular basis for von Recklinghausen disease. Biorxiv 2021. [Google Scholar] [CrossRef]
- Tokuo, H.; Yunoue, S.; Feng, L.; Kimoto, M.; Tsuji, H.; Ono, T.; Saya, H.; Araki, N. Phosphorylation of neurofibromin by cAMP-dependent protein kinase is regulated via a cellular association of N G, N G-dimethylarginine dimethylaminohydrolase. FEBS Lett. 2001, 494, 48–53. [Google Scholar] [CrossRef] [Green Version]
- Mangoura, D.; Sun, Y.; Li, C.; Singh, D.; Gutmann, D.H.; Flores, A.; Ahmed, M.; Vallianatos, G. Phosphorylation of neurofibromin by PKC is a possible molecular switch in EGF receptor signaling in neural cells. Oncogene 2006, 25, 735–745. [Google Scholar] [CrossRef] [Green Version]
- Leondaritis, G.; Petrikkos, L.; Mangoura, D. Regulation of the Ras-GTPase activating protein neurofibromin by C-tail phosphorylation: Implications for protein kinase C/Ras/extracellular signal-regulated kinase 1/2 pathway signaling and neuronal differentiation. J. Neurochem. 2009, 109, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Koliou, X.; Fedonidis, C.; Kalpachidou, T.; Mangoura, D. Nuclear import mechanism of neurofibromin for localization on the spindle and function in chromosome congression. J. Neurochem. 2016, 136, 78–91. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Yunoue, S.; Tokuo, H.; Ozawa, T.; Zhang, D.; Patrakitkomjorn, S.; Ichimura, T.; Saya, H.; Araki, N. PKA phosphorylation and 14-3-3 interaction regulate the function of neurofibromatosis type I tumor suppressor, neurofibromin. FEBS Lett. 2004, 557, 275–282. [Google Scholar] [CrossRef] [Green Version]
- Gregory, P.; Gutmann, D.; Mitchell, A.; Park, S.; Boguski, M.; Jacks, T.; Wood, D.; Jove, R.; Collins, F. Neurofibromatosis type 1 gene product (neurofibromin) associates with microtubules. Somat. Cell Mol. Genet. 1993, 19, 265–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Angelo, I.; Welti, S.; Bonneau, F.; Scheffzek, K. A novel bipartite phospholipid-binding module in the neurofibromatosis type 1 protein. EMBO Rep. 2006, 7, 174–179. [Google Scholar] [CrossRef] [Green Version]
- Boyanapalli, M.; Lahoud, O.B.; Messiaen, L.; Kim, B.; Anderle de Sylor, M.S.; Duckett, S.J.; Somara, S.; Mikol, D.D. Neurofibromin binds to caveolin-1 and regulates ras, FAK, and Akt. Biochem. Biophys. Res. Commun. 2006, 340, 1200–1208. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Du, J.; Shi, D.; Ji, F.; Ji, Y.; Pan, J.; Lv, F.; Zhang, Y.; Zhang, J. Knockdown of MSI2 inhibits metastasis by interacting with caveolin-1 and inhibiting its ubiquitylation in human NF1-MPNST cells. Cell Death Dis. 2020, 11, 489. [Google Scholar] [CrossRef] [PubMed]
- Scheffzek, K.; Welti, S. Pleckstrin homology (PH) like domains—Versatile modules in protein-protein interaction platforms. FEBS Lett. 2012, 586, 2662–2673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratner, N.; Miller, S.J. A RASopathy gene commonly mutated in cancer: The neurofibromatosis type 1 tumour suppressor. Nat. Rev. Cancer 2015, 15, 290–301. [Google Scholar] [CrossRef]
- Gouzi, J.Y.; Moressis, A.; Walker, J.A.; Apostolopoulou, A.A.; Palmer, R.H.; Bernards, A.; Skoulakis, E.M. The receptor tyrosine kinase Alk controls neurofibromin functions in Drosophila growth and learning. PLoS Genet. 2011, 7, e1002281. [Google Scholar] [CrossRef]
- Li, X.; Gao, M.; Choi, J.M.; Kim, B.J.; Zhou, M.T.; Chen, Z.; Jain, A.N.; Jung, S.Y.; Yuan, J.; Wang, W.; et al. Clustered, Regularly Interspaced Short Palindromic Repeats (CRISPR)/Cas9-coupled Affinity Purification/Mass Spectrometry Analysis Revealed a Novel Role of Neurofibromin in mTOR Signaling. Mol. Cell. Proteomics 2017, 16, 594–607. [Google Scholar] [CrossRef] [Green Version]
- Nada, S.; Mori, S.; Takahashi, Y.; Okada, M. p18/LAMTOR1: A late endosome/lysosome-specific anchor protein for the mTORC1/MAPK signaling pathway. Methods Enzymol. 2014, 535, 249–263. [Google Scholar] [CrossRef] [PubMed]
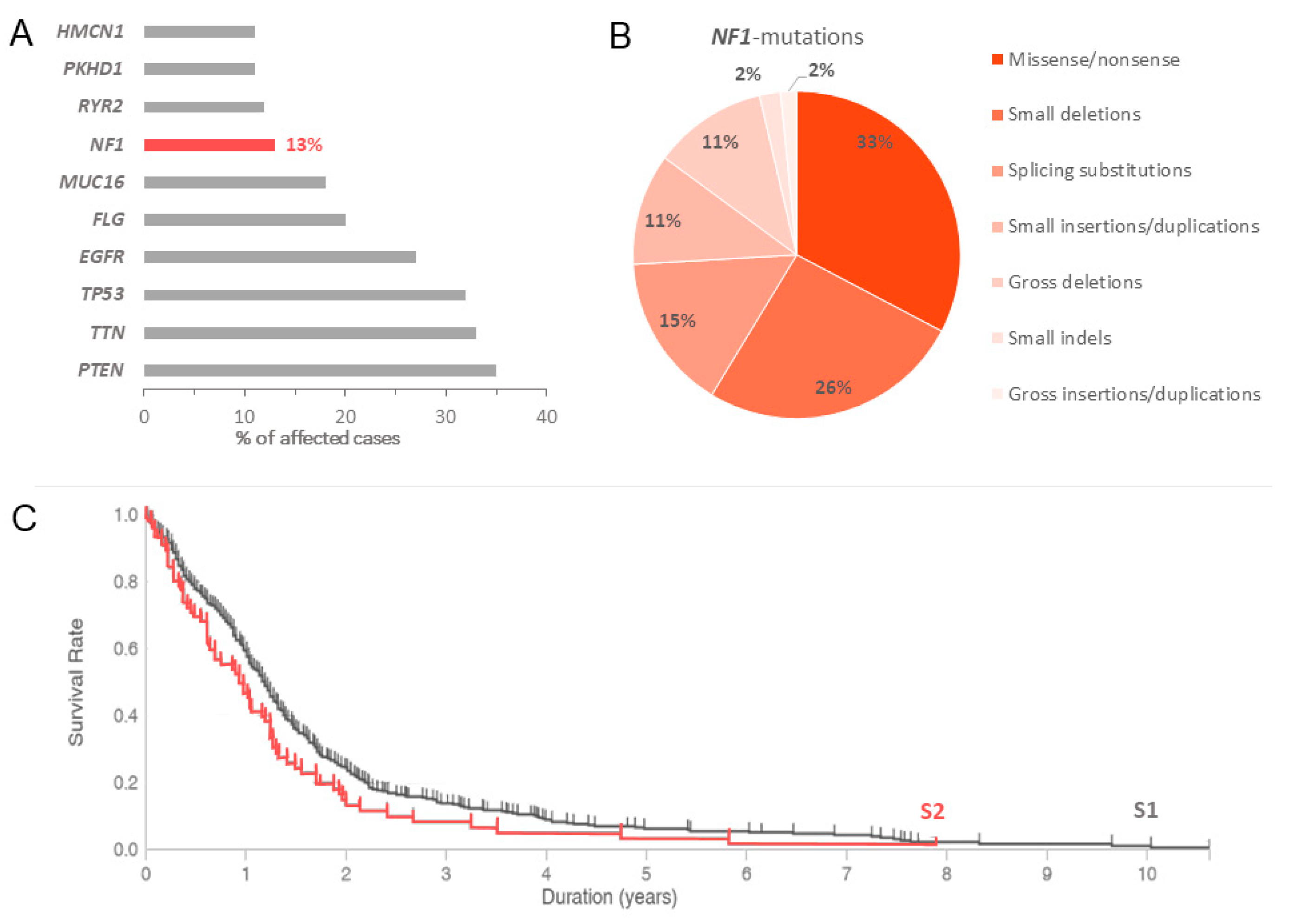
- Philpott, C.; Tovell, H.; Frayling, I.M.; Cooper, D.N.; Upadhyaya, M. The NF1 somatic mutational landscape in sporadic human cancers. Hum. Genom. 2017, 11, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Messiaen, L.; Callens, T.; Mortier, G.; Beysen, D.; Vandenbroucke, I.; Van Roy, N.; Speleman, F.; Paepe, A.D. Exhaustive mutation analysis of the NF1 gene allows identification of 95% of mutations and reveals a high frequency of unusual sloicng defects. Hum. Mutat. 2000, 15, 541–555. [Google Scholar] [CrossRef]
- Tamura, R. Current Understanding of Neurofibromatosis Type 1, 2, and Schwannomatosis. Int. J. Mol. Sci. 2021, 22, 5850. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.J.; Rangwala, F.; Williams, J.; Ackerman, P.; Kong, S.; Jegga, A.G.; Kaiser, S.; Aronow, B.J.; Frahm, S.; Kluwe, L.; et al. Large-scale molecular comparison of human schwann cells to malignant peripheral nerve sheath tumor cell lines and tissues. Cancer Res. 2006, 66, 2584–2591. [Google Scholar] [CrossRef] [Green Version]
- Vallee, B.; Doudeau, M.; Godin, F.; Gombault, A.; Tchalikian, A.; de Tauzia, M.L.; Benedetti, H. Nf1 RasGAP inhibition of LIMK2 mediates a new cross-talk between Ras and Rho pathways. PLoS ONE 2012, 7, e47283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whittaker, S.R.; Theurillat, J.P.; Van Allen, E.; Wagle, N.; Hsiao, J.; Cowley, G.S.; Schadendorf, D.; Root, D.E.; Garraway, L.A. A genome-scale RNA interference screen implicates NF1 loss in resistance to RAF inhibition. Cancer Discov. 2013, 3, 350–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nissan, M.H.; Pratilas, C.A.; Jones, A.M.; Ramirez, R.; Won, H.; Liu, C.; Tiwari, S.; Kong, L.; Hanrahan, A.J.; Yao, Z.; et al. Loss of NF1 in cutaneous melanoma is associated with RAS activation and MEK dependence. Cancer Res. 2014, 74, 2340–2350. [Google Scholar] [CrossRef] [Green Version]
- Maertens, O.; Johnson, B.; Hollstein, P.; Frederick, D.T.; Cooper, Z.A.; Messiaen, L.; Bronson, R.T.; McMahon, M.; Granter, S.; Flaherty, K.; et al. Elucidating distinct roles for NF1 in melanomagenesis. Cancer Discov. 2013, 3, 338–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holzel, M.; Huang, S.; Koster, J.; Ora, I.; Lakeman, A.; Caron, H.; Nijkamp, W.; Xie, J.; Callens, T.; Asgharzadeh, S.; et al. NF1 is a tumor suppressor in neuroblastoma that determines retinoic acid response and disease outcome. Cell 2010, 142, 218–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Bruin, E.C.; Cowell, C.; Warne, P.H.; Jiang, M.; Saunders, R.E.; Melnick, M.A.; Gettinger, S.; Walther, Z.; Wurtz, A.; Heynen, G.J.; et al. Reduced NF1 expression confers resistance to EGFR inhibition in lung cancer. Cancer Discov. 2014, 4, 606–619. [Google Scholar] [CrossRef] [Green Version]
- Harder, A. MEK inhibitors—Novel targeted therapies of neurofibromatosis associated benign and malignant lesions. Biomark. Res. 2021, 9, 26. [Google Scholar] [CrossRef]
- Ozawa, T.; Araki, N.; Yunoue, S.; Tokuo, H.; Feng, L.; Patrakitkomjorn, S.; Hara, T.; Ichikawa, Y.; Matsumoto, K.; Fujii, K.; et al. The neurofibromatosis type 1 gene product neurofibromin enhances cell motility by regulating actin filament dynamics via the Rho-ROCK-LIMK2-cofilin pathway. J. Biol. Chem. 2005, 280, 39524–39533. [Google Scholar] [CrossRef] [Green Version]
- Buchstaller, J.; McKeever, P.E.; Morrison, S.J. Tumorigenic cells are common in mouse MPNSTs but their frequency depends upon tumor genotype and assay conditions. Cancer Cell 2012, 21, 240–252. [Google Scholar] [CrossRef] [Green Version]
- Wilson, B.N.; John, A.M.; Handler, M.Z.; Schwartz, R.A. Neurofibromatosis type 1: New developments in genetics and treatment. J. Am. Acad. Dermatol. 2021, 84, 1667–1676. [Google Scholar] [CrossRef] [PubMed]
- Mazuelas, H.; Carrio, M.; Serra, E. Modeling tumors of the peripheral nervous system associated with Neurofibromatosis type 1: Reprogramming plexiform neurofibroma cells. Stem Cell Res. 2020, 49, 102068. [Google Scholar] [CrossRef] [PubMed]
- Hirbe, A.C.; Gutmann, D.H. Neurofibromatosis type 1: A multidisciplinary approach to care. Lancet Neurol. 2014, 13, 834–843. [Google Scholar] [CrossRef] [Green Version]
- Gutmann, D.H.; Ferner, R.E.; Listernick, R.H.; Korf, B.R.; Wolters, P.L.; Johnson, K.J. Neurofibromatosis type 1. Nat. Rev. Dis. Primers 2017, 3, 17004. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.L.; Gutmann, D.H. Neurofibromatosis type 1. Handb. Clin. Neurol. 2015, 132, 75–86. [Google Scholar] [CrossRef]
- Karaconji, T.; Whist, E.; Jamieson, R.V.; Flaherty, M.P.; Grigg, J.R.B. Neurofibromatosis Type 1: Review and Update on Emerging Therapies. Asia Pac. J. Ophthalmol. 2019, 8, 62–72. [Google Scholar] [CrossRef]
- Melloni, G.; Eoli, M.; Cesaretti, C.; Bianchessi, D.; Ibba, M.C.; Esposito, S.; Scuvera, G.; Morcaldi, G.; Micheli, R.; Piozzi, E.; et al. Risk of Optic Pathway Glioma in Neurofibromatosis Type 1: No Evidence of Genotype-Phenotype Correlations in A Large Independent Cohort. Cancers 2019, 11, 1838. [Google Scholar] [CrossRef] [Green Version]
- Legius, E.; Messiaen, L.; Wolkenstein, P.; Pancza, P.; Avery, R.A.; Berman, Y.; Blakeley, J.; Babovic-Vuksanovic, D.; Cunha, K.S.; Ferner, R.; et al. Revised diagnostic criteria for neurofibromatosis type 1 and Legius syndrome: An international consensus recommendation. Genet. Med. 2021, 23, 1506–1513. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herting, C.J.; Chen, Z.; Pitter, K.L.; Szulzewsky, F.; Kaffes, I.; Kaluzova, M.; Park, J.C.; Cimino, P.J.; Brennan, C.; Wang, B.; et al. Genetic driver mutations define the expression signature and microenvironmental composition of high-grade gliomas. Glia 2017, 65, 1914–1926. [Google Scholar] [CrossRef]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849.e821. [Google Scholar] [CrossRef]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e814. [Google Scholar] [CrossRef] [Green Version]
- Wei, C.J.; Gu, S.C.; Ren, J.Y.; Gu, Y.H.; Xu, X.W.; Chou, X.; Lian, X.; Huang, X.; Li, H.Z.; Gao, Y.S.; et al. The impact of host immune cells on the development of neurofibromatosis type 1: The abnormal immune system provides an immune microenvironment for tumorigenesis. Neurooncol. Adv. 2019, 1, vdz037. [Google Scholar] [CrossRef]
- Wood, M.D.; Mukherjee, J.; Pieper, R.O. Neurofibromin knockdown in glioma cell lines is associated with changes in cytokine and chemokine secretion in vitro. Sci. Rep. 2018, 8, 5805. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.; Smithson, L.J.; Ma, Y.; Hambardzumyan, D.; Gutmann, D.H. Ccl5 establishes an autocrine high-grade glioma growth regulatory circuit critical for mesenchymal glioblastoma survival. Oncotarget 2017, 8, 32977–32989. [Google Scholar] [CrossRef] [Green Version]
- Fletcher, J.S.; Pundavela, J.; Ratner, N. After Nf1 loss in Schwann cells, inflammation drives neurofibroma formation. Neurooncol. Adv. 2020, 2, i23–i32. [Google Scholar] [CrossRef]
- Guo, X.; Pan, Y.; Xiong, M.; Sanapala, S.; Anastasaki, C.; Cobb, O.; Dahiya, S.; Gutmann, D.H. Midkine activation of CD8(+) T cells establishes a neuron-immune-cancer axis responsible for low-grade glioma growth. Nat. Commun. 2020, 11, 2177. [Google Scholar] [CrossRef]
- Xu, S.; Tang, L.; Li, X.; Fan, F.; Liu, Z. Immunotherapy for glioma: Current management and future application. Cancer Lett. 2020, 476, 1–12. [Google Scholar] [CrossRef]
- Zhu, X.; Fujita, M.; Snyder, L.A.; Okada, H. Systemic delivery of neutralizing antibody targeting CCL2 for glioma therapy. J. Neurooncol. 2011, 104, 83–92. [Google Scholar] [CrossRef] [Green Version]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef] [Green Version]
- Butowski, N.; Colman, H.; De Groot, J.F.; Omuro, A.M.; Nayak, L.; Wen, P.Y.; Cloughesy, T.F.; Marimuthu, A.; Haidar, S.; Perry, A.; et al. Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: An Ivy Foundation Early Phase Clinical Trials Consortium phase II study. Neuro-Oncolgy 2016, 18, 557–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omuro, A.; Vlahovic, G.; Lim, M.; Sahebjam, S.; Baehring, J.; Cloughesy, T.; Voloschin, A.; Ramkissoon, S.H.; Ligon, K.L.; Latek, R.; et al. Nivolumab with or without ipilimumab in patients with recurrent glioblastoma: Results from exploratory phase I cohorts of CheckMate 143. Neuro-Oncolgy 2018, 20, 674–686. [Google Scholar] [CrossRef]
- Schalper, K.A.; Rodriguez-Ruiz, M.E.; Diez-Valle, R.; Lopez-Janeiro, A.; Porciuncula, A.; Idoate, M.A.; Inoges, S.; de Andrea, C.; Lopez-Diaz de Cerio, A.; Tejada, S.; et al. Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nat. Med. 2019, 25, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.R.; Hugo, W.; Lee, A.H.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M.; et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat. Med. 2019, 25, 477–486. [Google Scholar] [CrossRef]
- Colombo, F.; Barzon, L.; Franchin, E.; Pacenti, M.; Pinna, V.; Danieli, D.; Zanusso, M.; Palu, G. Combined HSV-TK/IL-2 gene therapy in patients with recurrent glioblastoma multiforme: Biological and clinical results. Cancer Gene. Ther. 2005, 12, 835–848. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, T.; Kayama, T.; Nishikawa, R.; Takahashi, H.; Hashimoto, N.; Takahashi, J.; Aoki, T.; Sugiyama, K.; Ogura, M.; Natsume, A.; et al. A multicenter phase I trial of combination therapy with interferon-beta and temozolomide for high-grade gliomas (INTEGRA study): The final report. J. Neuro-Oncol. 2011, 104, 573–577. [Google Scholar] [CrossRef]
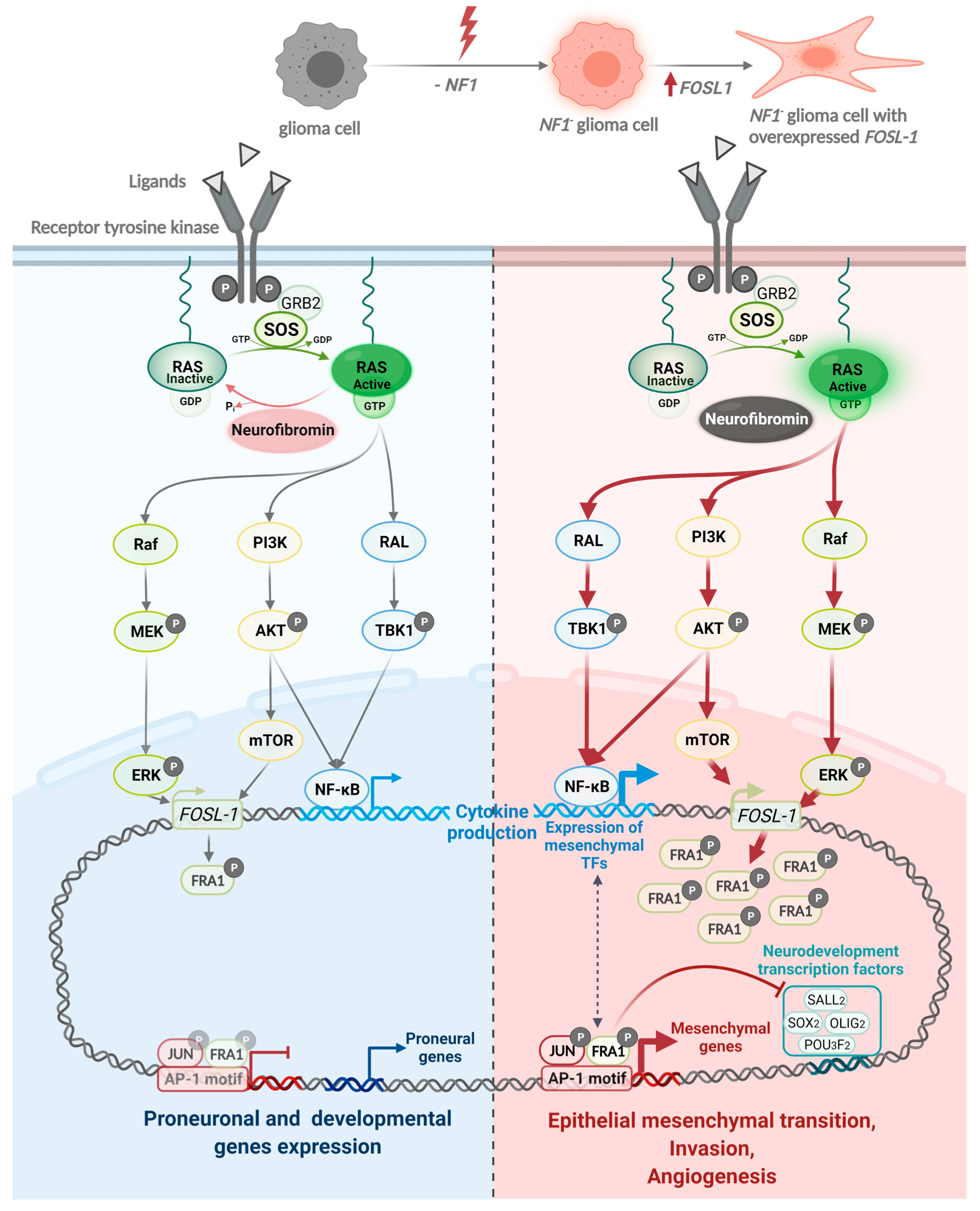
- Marques, C.; Unterkircher, T.; Kroon, P.; Oldrini, B.; Izzo, A.; Dramaretska, Y.; Ferrarese, R.; Kling, E.; Schnell, O.; Nelander, S.; et al. NF1 regulates mesenchymal glioblastoma plasticity and aggressiveness through the AP-1 transcription factor FOSL1. Elife 2021, 10, e64846. [Google Scholar] [CrossRef]
- Starinsky-Elbaz, S.; Faigenbloom, L.; Friedman, E.; Stein, R.; Kloog, Y. The pre-GAP-related domain of neurofibromin regulates cell migration through the LIM kinase/cofilin pathway. Mol. Cell. Neurosci. 2009, 42, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Larribere, L.; Cakrapradipta Wibowo, Y.; Patil, N.; Abba, M.; Tundidor, I.; Aguinon Olivares, R.G.; Allgayer, H.; Utikal, J. NF1-RAC1 axis regulates migration of the melanocytic lineage. Transl. Oncol. 2020, 13, 100858. [Google Scholar] [CrossRef] [PubMed]
- Manetti, F. LIM kinases are attractive targets with many macromolecular partners and only a few small molecule regulators. Med. Res. Rev. 2012, 32, 968–998. [Google Scholar] [CrossRef]
- Lin, Y.L.; Lei, Y.T.; Hong, C.J.; Hsueh, Y.P. Syndecan-2 induces filopodia and dendritic spine formation via the neurofibromin-PKA-Ena/VASP pathway. J. Cell Biol. 2007, 177, 829–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kweh, F.; Zheng, M.; Kurenova, E.; Wallace, M.; Golubovskaya, V.; Cance, W.G. Neurofibromin physically interacts with the N-terminal domain of focal adhesion kinase. Mol. Carcinog. 2009, 48, 1005–1017. [Google Scholar] [CrossRef] [Green Version]
- Tsai, P.I.; Wang, M.; Kao, H.H.; Cheng, Y.J.; Walker, J.A.; Chen, R.H.; Chien, C.T. Neurofibromin mediates FAK signaling in confining synapse growth at Drosophila neuromuscular junctions. J. Neurosci. 2012, 32, 16971–16981. [Google Scholar] [CrossRef]
- Errico, A.; Stocco, A.; Riccardi, V.M.; Gambalunga, A.; Bassetto, F.; Grigatti, M.; Ferlosio, A.; Tadini, G.; Garozzo, D.; Ferraresi, S.; et al. Neurofibromin Deficiency and Extracellular Matrix Cooperate to Increase Transforming Potential through FAK-Dependent Signaling. Cancers 2021, 13, 2329. [Google Scholar] [CrossRef] [PubMed]
- Arima, Y.; Hayashi, H.; Kamata, K.; Goto, T.M.; Sasaki, M.; Kuramochi, A.; Saya, H. Decreased expression of neurofibromin contributes to epithelial-mesenchymal transition in neurofibromatosis type 1. Exp. Dermatol. 2010, 19, e136–e141. [Google Scholar] [CrossRef]
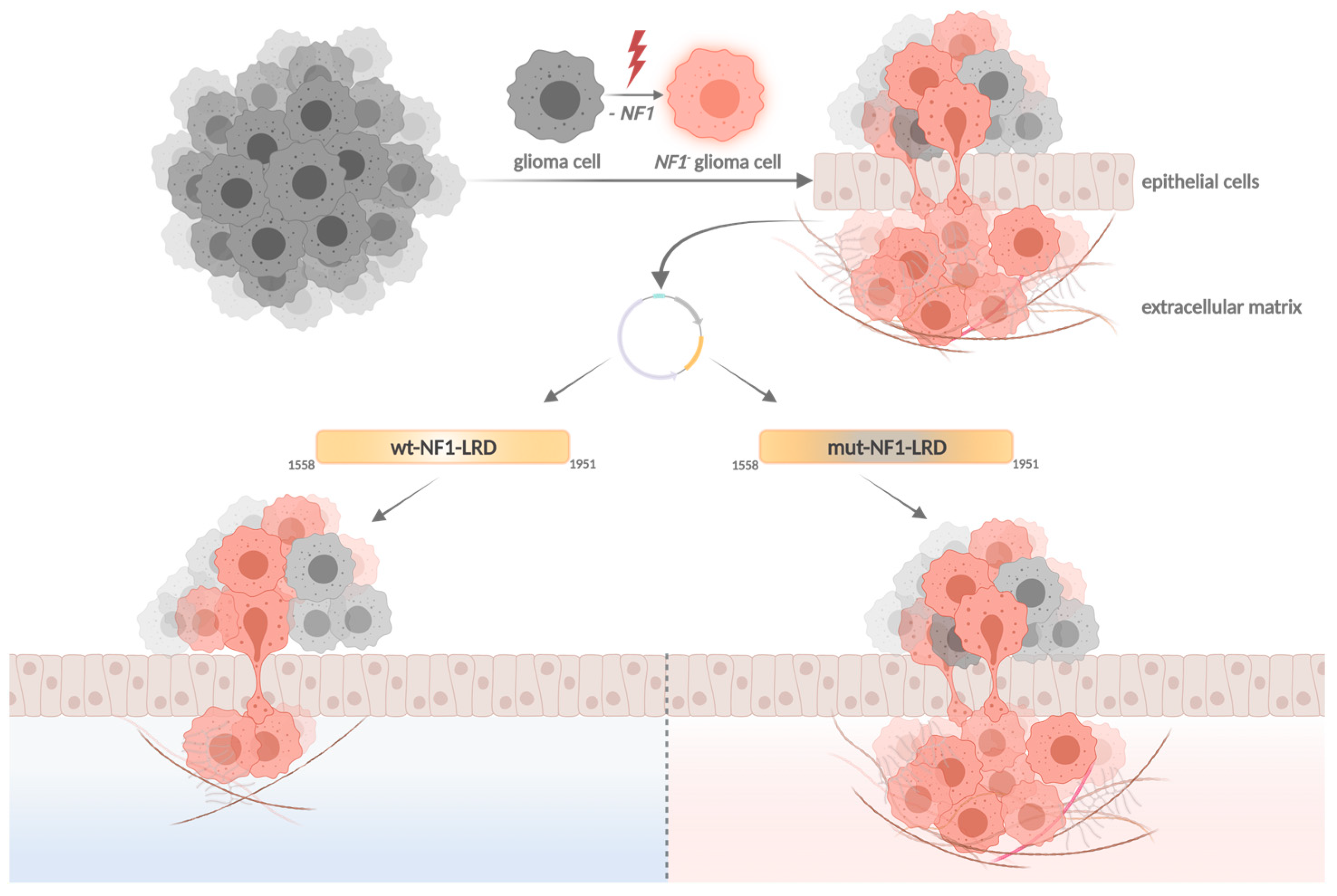
- Fadhlullah, S.F.B.; Halim, N.B.A.; Yeo, J.Y.T.; Ho, R.L.Y.; Um, P.; Ang, B.T.; Tang, C.; Ng, W.H.; Virshup, D.M.; Ho, I.A.W. Pathogenic mutations in neurofibromin identifies a leucine-rich domain regulating glioma cell invasiveness. Oncogene 2019, 38, 5367–5380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Zhou, R.; Qu, Y.; Shu, M.; Guo, S.; Bai, Z. Lipoamide Inhibits NF1 Deficiency-induced Epithelial-Mesenchymal Transition in Murine Schwann Cells. Arch. Med. Res. 2017, 48, 498–505. [Google Scholar] [CrossRef] [PubMed]
- Welti, S.; Fraterman, S.; D’Angelo, I.; Wilm, M.; Scheffzek, K. The sec14 homology module of neurofibromin binds cellular glycerophospholipids: Mass spectrometry and structure of a lipid complex. J. Mol. Biol. 2007, 366, 551–562. [Google Scholar] [CrossRef] [PubMed]
- Lacy, S.E.; Barrans, S.L.; Beer, P.A.; Painter, D.; Smith, A.G.; Roman, E.; Cooke, S.L.; Ruiz, C.; Glover, P.; Van Hoppe, S.J.L.; et al. Targeted sequencing in DLBCL, molecular subtypes, and outcomes: A Haematological Malignancy Research Network report. Blood 2020, 135, 1759–1771. [Google Scholar] [CrossRef]
- Griffiths, S.; Thompson, P.; Frayling, I.; Upadhyaya, M. Molecular diagnosis of neurofibromatosis type 1: 2 years experience. Fam. Cancer 2007, 6, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Shaw, K.; Phillips, A.; Cooper, D.N. The Human Gene Mutation Database: Building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum. Genet. 2014, 133, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Mao, B.; Chen, S.; Chen, X.; Yu, X.; Zhai, X.; Yang, T.; Li, L.; Wang, Z.; Zhao, X.; Zhang, X. Clinical characteristics and spectrum of NF1 mutations in 12 unrelated Chinese families with neurofibromatosis type 1. BMC Med. Genet. 2018, 19, 1–9. [Google Scholar] [CrossRef]
- Xu, M.; Xiong, H.; Han, Y.; Li, C.; Mai, S.; Huang, Z.; Ai, X.; Guo, Z.; Zeng, F.; Guo, Q. Identification of Mutation Regions on NF1 Responsible for High- and Low-Risk Development of Optic Pathway Glioma in Neurofibromatosis Type I. Front. Genet. 2018, 9, 270. [Google Scholar] [CrossRef]
- Anastasaki, C.; Gao, F.; Gutmann, D.H. Commentary: Identification of Mutation Regions on NF1 Responsible for High- and Low-Risk Development of Optic Pathway Glioma in Neurofibromatosis Type I. Front. Genet. 2019, 10, 115. [Google Scholar] [CrossRef] [Green Version]
- Schmitt, M.J.; Company, C.; Dramaretska, Y.; Barozzi, I.; Gohrig, A.; Kertalli, S.; Grossmann, M.; Naumann, H.; Sanchez-Bailon, M.P.; Hulsman, D.; et al. Phenotypic Mapping of Pathologic Cross-Talk between Glioblastoma and Innate Immune Cells by Synthetic Genetic Tracing. Cancer Discov. 2021, 11, 754–777. [Google Scholar] [CrossRef]
- Sa, J.K.; Chang, N.; Lee, H.W.; Cho, H.J.; Ceccarelli, M.; Cerulo, L.; Yin, J.; Kim, S.S.; Caruso, F.P.; Lee, M.; et al. Transcriptional regulatory networks of tumor-associated macrophages that drive malignancy in mesenchymal glioblastoma. Genome Biol. 2020, 21, 216. [Google Scholar] [CrossRef]
- Carro, M.S.; Lim, W.K.; Alvarez, M.J.; Bollo, R.J.; Zhao, X.; Snyder, E.Y.; Sulman, E.P.; Anne, S.L.; Doetsch, F.; Colman, H.; et al. The transcriptional network for mesenchymal transformation of brain tumours. Nature 2010, 463, 318–325. [Google Scholar] [CrossRef]
- Cooper, L.A.; Gutman, D.A.; Chisolm, C.; Appin, C.; Kong, J.; Rong, Y.; Kurc, T.; Van Meir, E.G.; Saltz, J.H.; Moreno, C.S.; et al. The tumor microenvironment strongly impacts master transcriptional regulators and gene expression class of glioblastoma. Am. J. Pathol. 2012, 180, 2108–2119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, A.; Wei, J.; Kong, L.Y.; Wang, Y.; Priebe, W.; Qiao, W.; Sawaya, R.; Heimberger, A.B. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro. Oncol. 2010, 12, 1113–1125. [Google Scholar] [CrossRef] [PubMed]
- Gabrusiewicz, K.; Li, X.; Wei, J.; Hashimoto, Y.; Marisetty, A.L.; Ott, M.; Wang, F.; Hawke, D.; Yu, J.; Healy, L.M.; et al. Glioblastoma stem cell-derived exosomes induce M2 macrophages and PD-L1 expression on human monocytes. Oncoimmunology 2018, 7, e1412909. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tumour Entity | Frequency of Somatic Mutation | Frequency of CNV Loss Events |
|---|---|---|
| Uterine Corpus Endometrial Carcinoma | 19.62% | 5.69% |
| Melanoma | 16.63% | 3.63% |
| Glioblastoma multiforme | 12.98% | 3.04% |
| Lung Squamous Cell Carcinoma | 12.73% | 5.78% |
| Lung Adenocarcinoma | 12.52% | 3.31% |
| Angiosarcoma | 11.11% | 1.76% |
| Cervical Squamous Cell Carcinoma and Endocervical Adenocarcinoma | 10.03% | 2.78% |
| Adrenocortical Carcinoma | 9.78% | 3.33% |
| Stomach Adenocarcinoma | 9.55% | 1.16% |
| Paragangliomas and Glomus Tumours | 9.50% | 10.83% |
| Bladder Urothelial Carcinoma | 8.98% | 2.94% |
| Ovarian Serous Cystadenocarcinoma | 7.57% | 14.36% |
| Sarcoma | 7.17% | 17.31% |
| Breast Invasive Carcinoma | 5.58% | 6.06% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scheer, M.; Leisz, S.; Sorge, E.; Storozhuk, O.; Prell, J.; Ho, I.; Harder, A. Neurofibromatosis Type 1 Gene Alterations Define Specific Features of a Subset of Glioblastomas. Int. J. Mol. Sci. 2022, 23, 352. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23010352
Scheer M, Leisz S, Sorge E, Storozhuk O, Prell J, Ho I, Harder A. Neurofibromatosis Type 1 Gene Alterations Define Specific Features of a Subset of Glioblastomas. International Journal of Molecular Sciences. 2022; 23(1):352. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23010352
Chicago/Turabian StyleScheer, Maximilian, Sandra Leisz, Eberhard Sorge, Olha Storozhuk, Julian Prell, Ivy Ho, and Anja Harder. 2022. "Neurofibromatosis Type 1 Gene Alterations Define Specific Features of a Subset of Glioblastomas" International Journal of Molecular Sciences 23, no. 1: 352. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23010352