
Gangliosides as Biomarkers of Human Brain Diseases: Trends in Discovery and Characterization by High-Performance Mass Spectrometry

Abstract
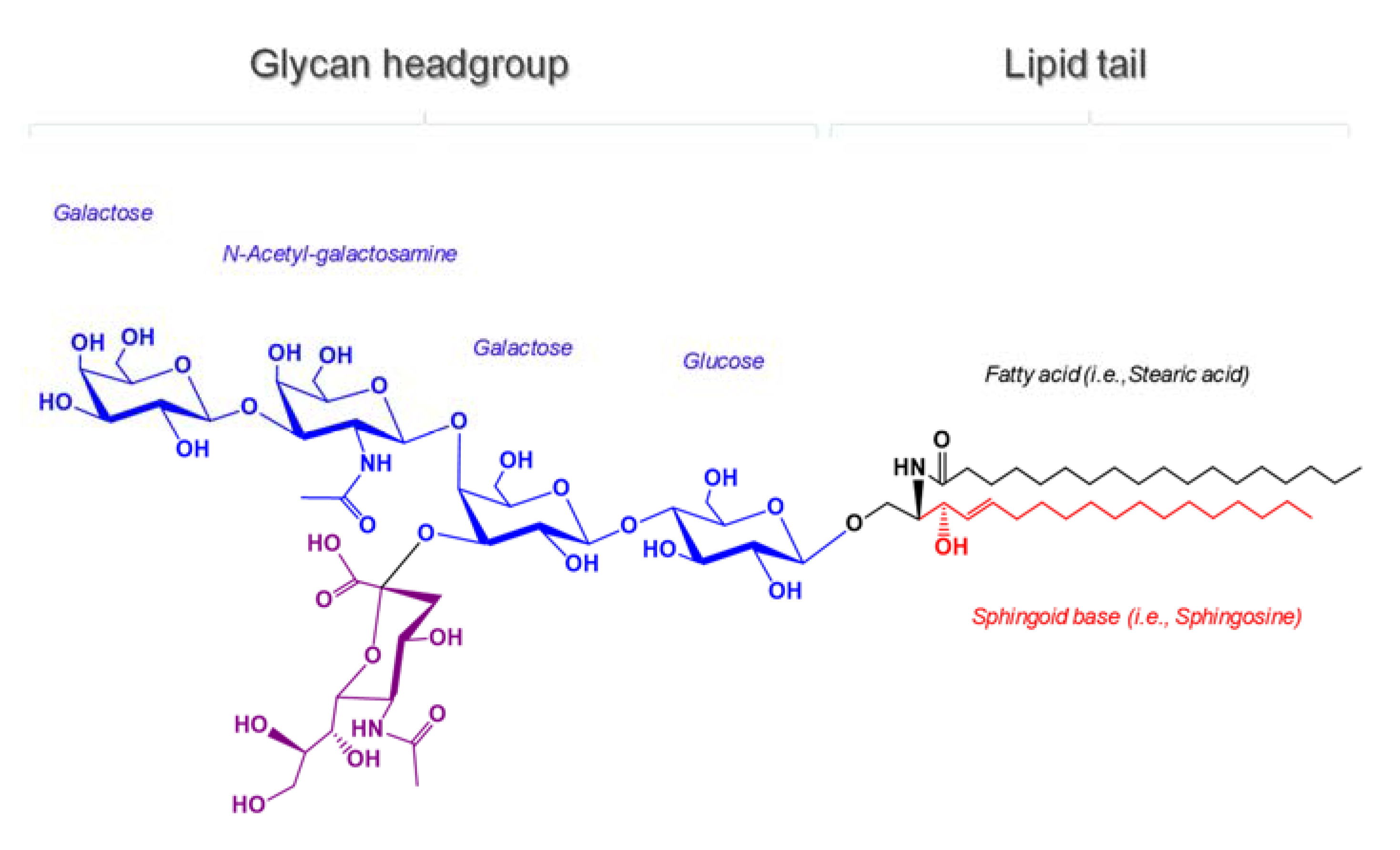
:1. Introduction
2. Neurodegenerative Diseases
2.1. Alzheimer’s Disease
2.2. Parkinson’s Disease
3. Cephalic Disorders
4. Primary Brain Tumors
4.1. Meningioma
4.2. Hemangioma
4.3. Astrocytoma
4.4. Glioblastoma Multiforme
4.5. Gliosarcoma
5. Secondary Brain Tumors
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Vukelić, Ž.; Metelmann, W.; Müthing, J.; Kos, M.; Peter-Katalinić, J. Anencephaly: Structural characterization of gangliosides in defined brain regions. Biol. Chem. 2001, 382, 259–274. [Google Scholar] [CrossRef]
- Schnaar, R.L.; Gerardy-Schahn, R.; Hildebrandt, H. Sialic acids in the brain: Gangliosides and polysialic acid in nervous system development, stability, disease, and regeneration. Physiol. Rev. 2014, 94, 461–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Posse de Chaves, E.; Sipione, S. Sphingolipids and gangliosides of the nervous system in membrane function and dysfunction. FEBS Lett. 2010, 584, 1748–1759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonnino, S.; Chigorno, V. Ganglioside molecular species containing C18-, and C20-sphingosine in mammalian nervous tissues, and neuronal cell cultures. Biochim. Biophys. Acta 2000, 1469, 63–77. [Google Scholar] [CrossRef]
- Yu, R.K.; Tsai, Y.T.; Ariga, T.; Yanagisawa, M. Structures, biosynthesis, and functions of gangliosides—An overview. J. Oleo. Sci. 2011, 60, 537–544. [Google Scholar] [CrossRef] [Green Version]
- Schnaar, R.L. Glycolipid-mediated cell-cell recognition in inflammation, and nerve regeneration. Arch. Biochem. Biophys. 2004, 426, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Schnaar, R.L.; Kinoshita, T. Glycosphingolipids. In Essentials of Glycobiology, 3rd ed.; Varki, A., Cummings, R.D., Eds.; Cold Spring Harbor: New York, NY, USA, 2017. [Google Scholar]
- Wiegandt, H. Structure and Function of Gangliosides; Springer Science & Business Media: New York, NY, USA, 2012. [Google Scholar]
- Zhang, X.; Kiechle, F.L. Glycosphingolipids in health and disease. Ann. Clin. Lab. Sci. 2004, 34, 3–13. [Google Scholar] [PubMed]
- Varki, A.; Schnaar, R.L.; Schauer, R. Sialic acids, and other nonulosonic acids. In Essentials of Glycobiology; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Eds.; Cold Spring Harbor: New York, NY, USA, 2015; pp. 179–195. [Google Scholar]
- Sipione, S.; Monyror, J.; Galleguillos, D.; Steinberg, N.; Kadam, V. Gangliosides in the brain: Physiology, pathophysiology and therapeutic applications. Front Neurosci. 2020, 6, 572965. [Google Scholar] [CrossRef] [PubMed]
- Ledeen, R.; Wu, G. Gangliosides of the nervous system. Methods Mol. Biol. 2018, 1804, 19–55. [Google Scholar] [PubMed]
- Ngamukote, S.; Yanagisawa, M.; Ariga, T.; Ando, S.; Yu, R.K. Developmental changes of glycosphingolipids and expression of glycogenes in mouse brains. J. Neurochem. 2007, 103, 2327–2341. [Google Scholar] [CrossRef] [PubMed]
- Kolter, T. Ganglioside biochemistry. ISRN Biochem. 2012, 2012, 506160. [Google Scholar] [CrossRef] [Green Version]
- Groux-Degroote, S.; Rodríguez-Walker, M.; Dewald, J.H.; Daniotti, J.L.; Delannoy, P. Gangliosides in cancer cell signaling. Prog. Mol. Biol. Transl. Sci. 2018, 156, 197–227. [Google Scholar]
- Liu, J.; Zheng, X.; Pang, X.; Li, L.; Wang, J.; Yang, C.; Du, G. Ganglioside GD3 synthase (GD3S), a novel cancer drug target. Acta Pharm. Sin. 2018, 8, 713–720. [Google Scholar] [CrossRef]
- Trinchera, M.; Parini, R.; Indellicato, R.; Domenighini, R.; Dall’Olio, F. Diseases of ganglioside biosynthesis: An expanding group of congenital disorders of glycosylation. Mol. Genet. Metab. 2018, 124, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Di Pardo, A.; Amico, E.; Maglione, V. Impaired levels of gangliosides in the corpus callosum of huntington disease animal models. Front. Neurosci. 2016, 10, 457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alpaugh, M.; Galleguillos, D.; Forero, J.; Morales, L.C.; Lackey, S.W.; Kar, P.; Di Pardo, A.; Holt, A.; Kerr, B.J.; Todd, K.G.; et al. Disease-modifying effects of ganglioside GM1 in Huntington’s disease models. EMBO Mol. Med. 2017, 9, 1537–1557. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, N.; Matsubara, T.; Fukuda, R.; Yasumori, H.; Hatsuta, H.; Murayama, S.; Sato, T.; Suzuki, A.; Yanagisawa, K. Imbalance in fatty-acid-chain length of gangliosides triggers Alzheimer amyloid deposition in the precuneus. PLoS ONE 2015, 10, e0121356. [Google Scholar] [CrossRef] [Green Version]
- Dukhinova, M.; Veremeyko, T.; Yung, A.W.Y.; Kuznetsova, I.S.; Lau, T.Y.B.; Kopeikina, E.; Chan, A.M.L.; Ponomarev, E.D. Fresh evidence for major brain gangliosides as a target for the treatment of Alzheimer’s disease. Neurobiol. Aging 2019, 77, 128–143. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, K.; Kato, K.; Yanagisawa, K. Ganglioside-mediated assembly of amyloid β-protein: Roles in Alzheimer’s Disease. Prog. Mol. Biol. Transl. Sci. 2018, 156, 413–434. [Google Scholar]
- Ledeen, R.W.; Wu, G. Gangliosides, α-synuclein, and Parkinson’s disease. Prog. Mol. Biol. Transl. Sci. 2018, 156, 435–454. [Google Scholar]
- Belarbi, K.; Cuvelier, E.; Bonte, M.A.; Desplanque, M.; Gressier, B.; Devos, D.; Chartier-Harlin, M.C. Glycosphingolipids and neuroinflammation in Parkinson’s disease. Mol. Neurodegener. 2020, 15, 59. [Google Scholar] [CrossRef] [PubMed]
- Forsayeth, J.; Hadaczek, P. Ganglioside metabolism and Parkinson’s disease. Front. Neurosci. 2018, 12, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yahi, N.; Di Scala, C.; Chahinian, H.; Fantini, J. Innovative treatment targeting gangliosides aimed at blocking the formation of neurotoxic α-synuclein oligomers in Parkinson’s disease. Glycoconj. J. 2021, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bouscary, A.; Quessada, C.; René, F.; Spedding, M.; Turner, B.J.; Henriques, A.; Ngo, S.T.; Loeffler, J.P. Sphingolipids metabolism alteration in the central nervous system: Amyotrophic lateral sclerosis (ALS) and other neurodegenerative diseases. Semin. Cell Dev. Biol. 2021, 112, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Krafft, P.R.; Wang, T.; Zhang, J.H.; Li, L.; Tang, J. Pathophysiology of ganglioside GM1 in ischemic stroke: Ganglioside GM1: A critical review. Cell Transplant. 2019, 28, 657–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malinick, A.S.; Lambert, A.S.; Stuart, D.D.; Li, B.; Puente, E.; Cheng, Q. Detection of multiple sclerosis biomarkers in serum by ganglioside microarrays and surface plasmon resonance imaging. ACS Sens. 2020, 5, 3617–3626. [Google Scholar] [CrossRef]
- Kolyovska, V.; Ivanova, S. Neurodegenerative changes and demyelination in serum igg antibodies to GM1, GD and GM3 gangliosides in patients with secondary progressive multiple sclerosis–Preliminary results. Comptes Rendus L’académie Bulg. Sci. 2019, 72, 115–122. [Google Scholar]
- Simpson, M.A.; Cross, H.; Proukakis, C.; Priestman, D.A.; Neville, D.C.; Reinkensmeier, G.; Wang, H.; Wiznitzer, M.; Gurtz, K.; Verganelaki, A.; et al. Infantile-onset symptomatic epilepsy syndrome caused by a homozygous loss-of-function mutation of GM3 synthase. Nat. Genet. 2004, 36, 1225–1229. [Google Scholar] [CrossRef] [Green Version]
- Groux-Degroote, S.; Guerardel, Y.; Delannoy, P. Gangliosides: Structures, biosynthesis, analysis, and roles in cancer. Chembiochem 2017, 18, 1146–1154. [Google Scholar] [CrossRef] [Green Version]
- Inokuchi, J. GM3 and diabetes. Glycoconj. J. 2014, 31, 193–197. [Google Scholar] [CrossRef]
- Rueda, R. The role of dietary gangliosides on immunity and the prevention of infection. Br. J. Nutr. 2007, 98, S68–S73. [Google Scholar] [CrossRef] [Green Version]
- Magistretti, P.J.; Geisler, F.H.; Schneider, J.S.; Li, P.A.; Fiumelli, H.; Sipione, S. Gangliosides: Treatment avenues in neurodegenerative disease. Front. Neurol. 2019, 10, 859. [Google Scholar] [CrossRef] [Green Version]
- Habchi, J.; Chia, S.; Galvagnion, C.; Michaels, T.C.T.; Bellaiche, M.M.J.; Ruggeri, F.S.; Sanguanini, M.; Idini, I.; Kumita, J.R.; Sparr, E.; et al. Cholesterol catalyses Aβ42 aggregation through a heterogeneous nucleation pathway in the presence of lipid membranes. Nat. Chem. 2018, 10, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.X.; Papadopoulos, V. Function of beta-amyloid in cholesterol transport: A lead to neurotoxicity. FASEB J. 2002, 16, 1677–1679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, S.R.; Wu, Y.; Sui, S.F. Cholesterol is an important factor affecting the membrane insertion of beta-amyloid peptide (A beta 1-40), which may potentially inhibit the fibril formation. J. Biol. Chem. 2002, 277, 6273–6279. [Google Scholar] [CrossRef] [Green Version]
- Williams, T.L.; Serpell, L.C. Membrane and surface interactions of Alzheimer’s Aβ peptide-insights into the mechanism of cytotoxicity. FEBS J. 2011, 278, 3905–3917. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, N.; Matsubara, E.; Maeda, S.; Minagawa, H.; Takashima, A.; Maruyama, W.; Michikawa, M.; Yanagisawa, K. A ganglioside-induced toxic soluble Abeta assembly. Its enhanced formation from Abeta bearing the Arctic mutation. J. Biol. Chem. 2007, 282, 2646–2655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuzaki, K.; Kato, K.; Yanagisawa, K. Abeta polymerization through interaction with membrane gangliosides. Biochim. Biophys. Acta 2010, 1801, 868–877. [Google Scholar] [CrossRef]
- Kakio, A.; Nishimoto, S.; Yanagisawa, K.; Kozutsumi, Y.; Matsuzaki, K. Interactions of amyloid beta-protein with various gangliosides in raft-like membranes: Importance of GM1 ganglioside-bound form as an endogenous seed for Alzheimer amyloid. Biochemistry 2002, 41, 7385–7390. [Google Scholar] [CrossRef]
- Svennerholm, L. Gangliosides–a new therapeutic agent against stroke and Alzheimer’s disease. Life Sci. 1994, 55, 2125–2134. [Google Scholar] [CrossRef]
- Yanagisawa, K.; Odaka, A.; Suzuki, N.; Ihara, Y. GM1 ganglioside-bound amyloid beta-protein (A beta): A possible form of preamyloid in Alzheimer’s disease. Nat. Med. 1995, 1, 1062–1066. [Google Scholar] [CrossRef]
- Yanagisawa, K. GM1 ganglioside and the seeding of amyloid in Alzheimer’s disease: Endogenous seed for Alzheimer amyloid. Neuroscientist 2005, 11, 250–260. [Google Scholar] [CrossRef]
- Yanagisawa, K. Role of gangliosides in Alzheimer’s disease. Biochim. Biophys. Acta. 2007, 1768, 1943–1951. [Google Scholar] [CrossRef] [Green Version]
- Yanagisawa, K. GM1 ganglioside and Alzheimer’s disease. Glycoconj. J. 2015, 32, 87–91. [Google Scholar] [CrossRef]
- Yamamoto, N.; Fukata, Y.; Fukata, M.; Yanagisawa, K. GM1-ganglioside-induced Abeta assembly on synaptic membranes of cultured neurons. Biochim. Biophys. Acta 2007, 1768, 1128–1137. [Google Scholar] [CrossRef] [Green Version]
- Augustinsson, L.E.; Blennow, K.; Blomstrand, C.; Bråne, G.; Ekman, R.; Fredman, P.; Karlsson, I.; Kihlgren, M.; Lehmann, W.; Lekman, A.; et al. Intracerebroventricular administration of GM1 ganglioside to presenile Alzheimer patients. Dement. Geriatr. Cogn. Disord. 1997, 8, 26–33. [Google Scholar] [CrossRef]
- Fernández-Pérez, E.J.; Sepúlveda, F.J.; Peoples, R.; Aguayo, L.G. Role of membrane GM1 on early neuronal membrane actions of Aβ during onset of Alzheimer’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 3105–3116. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, K. Aβ–ganglioside interactions in the pathogenesis of Alzheimer’s disease. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183233. [Google Scholar] [CrossRef] [PubMed]
- Svennerholm, L.; Gottfries, C.G. Membrane lipids, selectively diminished in Alzheimer brains, suggest synapse loss as a primary event in early-onset form (type I) and demyelination in late-onset form (type II). J. Neurochem. 1994, 62, 1039–1047. [Google Scholar] [CrossRef] [PubMed]
- Gylys, K.H.; Fein, J.A.; Yang, F.; Miller, C.A.; Cole, G.M. Increased cholesterol in Abeta-positive nerve terminals from Alzheimer’s disease cortex. Neurobiol. Aging 2007, 28, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhang, K.; Tan, L.; Chen, Y.-H.; Cao, Y.-P. Alterations in cholesterol and ganglioside GM1 content of lipid rafts in platelets from patients with Alzheimer disease. Alzheimer Dis. Assoc. Disord. 2015, 29, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Kalanj, S.; Kracun, I.; Rosner, H.; Cosovic, C. Regional distribution of brain gangliosides in Alzheimer’s disease. Neurol Croat. 1991, 40, 269–281. [Google Scholar]
- Kracun, I.; Kalanj, S.; Talan-Hranilovic, J.; Cosovic, C. Cortical distribution of gangliosides in Alzheimer’s disease. Neurochem. Int. 1992, 20, 433–438. [Google Scholar] [CrossRef]
- Brooksbank, B.W.L.; McGovern, J. Gangliosides in the brain in adult Down’s syndrome and Alzheimer’s disease. Mol. Chem. Neuropathol. 1989, 11, 143–156. [Google Scholar] [CrossRef]
- Crino, P.B.; Ullman, M.D.; Vogt, B.A.; Bird, E.D.; Volicer, L. Brain gangliosides in dementia of the Alzheimer type. Arch. Neurol. 1989, 46, 398–401. [Google Scholar] [CrossRef]
- Ando, S.; Tanaka, Y.; Waki, H.; Kon, K.; Iwamoto, M.; Fukui, F. Gangliosides and sialylcholesterol as modulators of synaptic functions. Ann. N. Y. Acad. Sci. 1998, 845, 232–239. [Google Scholar] [CrossRef]
- Foley, P.; Bradford, H.F.; Docherty, M.; Fillit, H.; Luine, V.N.; McEwen, B.; Bucht, G.; Winblad, B.; Hardy, J. Evidence for the presence of antibodies to cholinergic neurons in the serum of patients with Alzheimer’s disease. J. Neurol. 1988, 235, 466–471. [Google Scholar] [CrossRef]
- Chapman, J.; Bachar, O.; Korczyn, A.D.; Wertman, E.; Michaelson, D.M. Antibodies to cholinergic neurons in Alzheimer’s disease. J. Neurochem. 1988, 51, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Ariga, T.; Yanagisawa, M.; Wakade, C.; Ando, S.; Buccafusco, J.J.; McDonald, M.P.; Yu, R.K. Ganglioside metabolism in a transgenic mouse model of Alzheimer’s disease: Expression of Chol-1α antigens in the brain. ASN Neuro. 2010, 2, e00044. [Google Scholar] [CrossRef] [PubMed]
- Ariga, T.; Kobayashi, K.; Hasegawa, A.; Kiso, M.; Ishida, H.; Miyatake, T. Characterization of high-affinity binding between gangliosides and amyloid β-protein. Arch. Biochem. Biophys. 2001, 388, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, N.; Suzuki, Y.; Makino, Y.; Haga, C.; Kosaka, K.; Iizuka, R. Cell membrane changes in brains manifesting senile plaques: An immunohistochemical study of GM membranous ganglioside. Brain Res. 1990, 522, 152–156. [Google Scholar] [CrossRef]
- Takahashi, H.; Hirokawa, K.; Ando, S.; Obata, K. Immunohistological study on brains of Alzheimer’s disease using antibodies to fetal antigens, C-series gangliosides and microtubule-associated protein 5. Acta Neuropathol. 1991, 81, 626–631. [Google Scholar] [CrossRef] [PubMed]
- Nishinaka, T.; Iwata, D.; Shimada, S.; Kosaka, K.; Suzuki, Y. Anti-ganglioside GD1a monoclonal antibody recognizes senile plaques in the brains of patients with Alzheimer-type dementia. Neurosci. Res. 1993, 17, 171–176. [Google Scholar] [CrossRef]
- Yu, R.K.; Usuki, S.; Ariga, T. Ganglioside molecular mimicry and its pathological roles in Guillain-Barre’ syndrome and related diseases. Infect. Immun. 2006, 74, 6517–6527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuki, N.; Ariga, T. Antibodies to fucogangliosides in neurological diseases. J. Neurol. Sci. 1997, 150, 81–84. [Google Scholar] [CrossRef]
- Miura, Y.; Miyaji, K.; Chai, Y.L.; Chen, C.L.H.; Lai, M.K.P. Autoantibodies to GM1 and GQ1bα are not biological markers of Alzheimer’s disease. J. Alzheimer’s Dis. 2014, 42, 1165–1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caughlin, S.; Maheshwari, S.; Agca, Y.; Agca, C.; Harris, A.J.; Jurcic, K.; Yeung, K.K.C.; Cechetto, D.F.; Whitehead, S.N. Membrane-lipid homeostasis in a prodromal rat model of Alzheimer’s disease: Characteristic profiles in ganglioside distributions during aging detected using MALDI imaging mass spectrometry. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 1327–1338. [Google Scholar] [CrossRef] [PubMed]
- Hirano-Sakamaki, W.; Sugiyama, E.; Hayasaka, T.; Ravid, R.; Setou, M.; Taki, T. Alzheimer’s disease is associated with disordered localization of ganglioside GM1 molecular species in the human dentate gyrus. FEBS Lett. 2015, 589, 3611–3616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaya, I.; Jennische, E.; Dunevall, J.; Lange, S.; Ewing, A.G.; Malmberg, P.; Baykal, A.T.; Fletcher, J.S. Spatial lipidomics reveals region and long chain base specific accumulations of monosialogangliosides in amyloid plaques in familial Alzheimer’s disease mice (5xFAD) brain. ACS Chem. Neurosci. 2020, 11, 14–24. [Google Scholar] [CrossRef]
- Taki, T. An approach to glycobiology from glycolipidomics: Ganglioside molecular scanning in the brains of patients with Alzheimer’s disease by TLC-blot/matrix assisted laser desorption/ionization-time of flight MS. Biol. Pharm. Bull. 2012, 35, 1642–1647. [Google Scholar] [CrossRef] [Green Version]
- Caughlin, S.; Hepburn, J.D.; Park, D.H.; Jurcic, K.; Yeung, K.K.C.; Cechetto, D.F.; Whitehead, S.N. Increased expression of simple ganglioside species gm2 and gm3 detected by maldi imaging mass spectrometry in a combined rat model of Aβ toxicity and stroke. PLoS ONE 2015, 10, e0130364. [Google Scholar] [CrossRef]
- Valdes-Gonzalez, T.; Goto-Inoue, N.; Hirano, W.; Ishiyama, H.; Hayasaka, T.; Setou, M.; Taki, T. New approach for glyco- and lipidomics—molecular scanning of human brain gangliosides by TLC-Blot and MALDI-QIT-TOF MS. J. Neurochem. 2011, 116, 678–683. [Google Scholar] [CrossRef]
- Michno, W.; Wehrli, P.M.; Zetterberg, H.; Blennow, K.; Hanrieder, J. GM1 locates to mature amyloid structures implicating a prominent role for glycolipid-protein interactions in Alzheimer pathology. Biochim. Biophys. Acta Proteins Proteom. 2019, 1867, 458–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Touboul, D.; Gaudin, M. Lipidomics of Alzheimer’s disease. Bioanalysis 2014, 6, 541–561. [Google Scholar] [CrossRef] [PubMed]
- Powers, R.; Lei, S.; Anandhan, A.; Marshall, D.D.; Worley, B.; Cerny, R.L.; Dodds, E.D.; Huang, Y.; Panayiotidis, M.I.; Pappa, A.; et al. Metabolic investigations of the molecular mechanisms associated with Parkinson’s disease. Metabolites 2017, 7, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burbulla, L.F.; Song, P.; Mazzulli, J.R.; Zampese, E.; Wong, Y.C.; Jeon, S.; Santos, D.P.; Blanz, J.; Obermaier, C.D.; Strojny, C.; et al. Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science 2017, 357, 1255–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef]
- Chinta, S.J.; Woods, G.; Demaria, M.; Rane, A.; Zou, Y.; McQuade, A.; Rajagopalan, S.; Limbad, C.; Madden, D.T.; Campisi, J.; et al. Cellular senescence is induced by the environmental neurotoxin paraquat and contributes to neuropathology linked to Parkinson’s disease. Cell Rep. 2018, 22, 930–940. [Google Scholar] [CrossRef] [Green Version]
- El-Battari, A.; Rodriguez, L.; Chahinian, H.; Delézay, O.; Fantini, J.; Yahi, N.; Di Scala, C. Gene therapy strategy for Alzheimer’s and Parkinson’s diseases aimed at preventing the formation of neurotoxic oligomers in SH-SY5Y cells. Int. J. Mol. Sci. 2021, 22, 11550. [Google Scholar] [CrossRef]
- Schneider, J.S.; Aras, R.; Williams, C.K.; Koprich, J.B.; Brotchie, J.M.; Singh, V. GM1 ganglioside modifies α-synuclein toxicity and is neuroprotective in a rat α-synuclein model of Parkinson’s disease. Sci Rep. 2019, 9, 8362. [Google Scholar] [CrossRef] [Green Version]
- Martinez, Z.; Zhu, M.; Han, S.; Fink, A.L. GM1 specifically interacts with a-synuclein and inhibits fibrillation. Biochemistry 2007, 46, 1868–1877. [Google Scholar] [CrossRef]
- Hatzifilippou, E.; Arnaoutoglou, M.; Koutsouraki, E.; Banaki, T.; Costa, V.G.; Baloyannis, S.J. High levels of anti-ganglioside antibodies in patients with Parkinson’s disease associated with cognitive decline. Int. J. Neurorehabil. 2015, 2, 2. [Google Scholar]
- Chiavegatto, S.; Sun, J.; Nelson, R.J.; Schnaar, R.L. A functional role for complex gangliosides: Motor deficits in GM2/GD2 synthase knockout mice. Exp. Neurol. 2000, 166, 227–234. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Lu, Z.H.; Kulkarni, N.; Ledeen, R.W. Deficiency of ganglioside GM1 correlates with Parkinson’s disease in mice, and humans. J. Neurosci. Res. 2012, 90, 1997–2008. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Lu, Z.H.; Seo, J.H.; Alselehdar, S.K.; DeFrees, S.; Ledeen, R.W. Mice deficient in GM1 manifest both motor and non-motor symptoms of Parkinson’s disease; successful treatment with synthetic GM1 ganglioside. Exp. Neurol. 2020, 329, 113284. [Google Scholar] [CrossRef]
- Wu, G.; Lu, Z.H.; Kulkarni, N.; Amin, R.; Ledeen, R.W. Mice lacking major brain gangliosides develop parkinsonism. Neurochem. Res. 2011, 36, 1706–1714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.L.; Duan, W.J.; Lu, D.H.; Ma, X.H.; Li, X.X.; Li, Z.; Bi, W.; Kurihara, H.; Liu, H.Z.; Li, Y.F.; et al. Autophagy-dependent removal of α-synuclein: A novel mechanism of GM1 ganglioside neuroprotection against Parkinson’s disease. Acta Pharmacol. Sin. 2021, 42, 518–528. [Google Scholar] [CrossRef] [PubMed]
- Seyfried, T.N.; Choi, H.; Chevalier, A.; Hogan, D.; Akgoc, Z.; Schneider, J.S. Sex-related abnormalities in substantia nigra lipids in Parkinson’s disease. ASN Neuro. 2018, 10, 1759091418781889. [Google Scholar] [CrossRef] [Green Version]
- Huebecker, M.; Moloney, E.B.; Van der Spoel, A.C.; Priestman, D.A.; Isacson, O.; Hallett, P.J.; Platt, F.M. Reduced sphingolipid hydrolase activities, substrate accumulation and ganglioside decline in Parkinson’s disease. Mol. Neurodegener. 2019, 14, 40. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Zhang, X.; Wang, L.; Yang, C. High performance liquid chromatography-mass spectrometry (LC-MS) based quantitative lipidomics study of ganglioside-NANA-3 plasma to establish its association with Parkinson’s disease patients. Med. Sci. Monit. 2017, 23, 5345–5353. [Google Scholar] [CrossRef]
- Chan, R.B.; Perotte, A.J.; Zhou, B.; Liong, C.; Shorr, E.J.; Marder, K.S.; Kang, U.J.; Waters, C.H.; Levy, O.A.; Xu, Y.; et al. Elevated GM3 plasma concentration in idiopathic Parkinson’s disease: A lipidomic analysis. PLoS ONE 2017, 12, e0172348. [Google Scholar] [CrossRef] [PubMed]
- Boutin, M.; Sun, Y.; Shacka, J.J.; Auray-Blais, C. Tandem mass spectrometry multiplex analysis of glucosylceramide and galactosylceramide isoforms in brain tissues at different stages of Parkinson disease. Anal. Chem. 2016, 88, 1856–1863. [Google Scholar] [CrossRef]
- Chiricozzi, E.; Mauri, L.; Lunghi, G.; Di Biase, E.; Fazzari, M.; Maggioni, M.; Valsecchi, M.; Prioni, S.; Loberto, N.; Pomè, D.Y.; et al. Parkinson’s disease recovery by GM1 oligosaccharide treatment in the B4galnt1+/− mouse model. Sci. Rep. 2019, 9, 19330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cephalic Disorders Fact Sheet. Available online: https://www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Cephalic-Disorders-Fact-Sheet (accessed on 9 December 2021).
- Cephalic Disorders. Available online: https://www.uhhospitals.org/health-information/health-and-wellness-library/article/adult-diseases-and-conditions-v1/cephalic-disorders (accessed on 9 December 2021).
- Anencephaly. Available online: https://rarediseases.info.nih.gov/diseases/5808/anencephaly (accessed on 15 October 2021).
- Colpocephaly. Available online: https://rarediseases.info.nih.gov/diseases/10008/colpocephaly#ref_143 (accessed on 15 October 2021).
- Holoprosencephaly. Available online: https://rarediseases.info.nih.gov/diseases/6665/holoprosencephaly (accessed on 15 October 2021).
- Das, G.; Gayen, S.; Bandyopadhyay, S.; Das, D. Ethmocephaly with amniotic band syndrome. Middle East Afr. J. Ophthalmol. 2012, 19, 429–431. [Google Scholar] [PubMed]
- Hydranencephaly. Available online: https://rarediseases.info.nih.gov/diseases/6681/hydranencephaly (accessed on 17 October 2021).
- Lissencephaly. Available online: https://rarediseases.info.nih.gov/diseases/12291/lissencephaly (accessed on 17 October 2021).
- Microcephaly. Available online: https://www.malacards.org/card/microcephaly?search=Microcephaly (accessed on 17 October 2021).
- Teunissen, M.W.A.; Kamsteeg, E.J.; Sallevelt, S.C.E.H.; Pennings, M.; Bauer, N.; Vermeulen, R.J.; Nicolai, J. Biallelic variants in the COLGALT1 gene causes severe congenital porencephaly: A case report. Neurol. Genet. 2021, 7, e564. [Google Scholar] [CrossRef]
- Porencephaly. Available online: https://www.malacards.org/card/porencephaly?search=Porencephaly (accessed on 17 October 2021).
- Porencephaly Information Page. Available online: https://www.ninds.nih.gov/Disorders/All-Disorders/Porencephaly-Information-Page (accessed on 20 October 2021).
- Schizencephaly Information Page. Available online: https://www.ninds.nih.gov/Disorders/All-Disorders/Schizencephaly-Information-Page (accessed on 20 October 2021).
- Acephaly. Available online: https://maayanlab.cloud/Harmonizome/gene_set/acephaly/MPO+Gene-Phenotype+Associations. (accessed on 20 October 2021).
- Van Groeninghen, J.C.; Franssen, A.M.; Willemsen, W.N.; Nijhuis, J.G.; Puts, J.J. An acardiac acephalic monster. Eur. J. Obstet. Gynecol. Reprod. Biol. 1985, 19, 317–325. [Google Scholar] [CrossRef]
- Exencephaly. Available online: https://radiopaedia.org/articles/13935 (accessed on 11 October 2021).
- Danzer, E.; Rintoul, N.E.; Adzick, N.S. 171—Pathophysiology of neural tube defects. In Fetal and Neonatal Physiology, 5th ed; Polin, R.A., Abman, S.H., Rowitch, D.H., Benitz, W.E., Fox, W.W., Eds.; Elsevier: Amsterdam, The Netherlands, 2017; Volume 2, pp. 1712–1723.e4. [Google Scholar]
- Faye-Petersen, O.; David, E.; Rangwala, N.; Seaman, J.P.; Hua, Z.; Heller, D.S. Otocephaly: Report of five new cases and a literature review. Fetal Pediatr. Pathol. 2006, 25, 277–296. [Google Scholar] [CrossRef]
- Ko, J.M. Genetic syndromes associated with craniosynostosis. J. Korean Neurosurg. Soc. 2016, 59, 187–191. [Google Scholar] [CrossRef]
- Craniosynostosis. Available online: https://www.malacards.org/card/craniosynostosis?search=Craniosynostosis (accessed on 13 October 2021).
- Craniosynostosis Information Page. Available online: https://www.ninds.nih.gov/Disorders/All-Disorders/Craniosynostosis-Information-Page (accessed on 13 October 2021).
- Isolated Brachycephaly. Available online: https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=35099 (accessed on 13 October 2021).
- Oxycephaly. Available online: https://maayanlab.cloud/Harmonizome/gene_set/oxycephaly/HPO+Gene-Disease+Associations (accessed on 17 October 2021).
- Bodian, M. Oxycephaly. J. Am. Med. Assoc. 1950, 143, 15–18. [Google Scholar] [CrossRef]
- Plagiocephaly. Available online: https://www.malacards.org/card/plagiocephaly?search=plagiocephaly (accessed on 17 October 2021).
- Jung, B.K.; Yun, I.S. Diagnosis and treatment of positional plagiocephaly. Arch Craniofac. Surg. 2020, 21, 80–86. [Google Scholar] [CrossRef]
- Mawji, A.; Vollman, A.R.; Hatfield, J.; McNeil, D.A.; Sauve, R. The incidence of positional plagiocephaly: A cohort study. Pediatrics 2013, 132, 298–304. [Google Scholar] [CrossRef] [Green Version]
- Pediatric Sagittal Synostosis. Available online: https://www.ohsu.edu/doernbecher/pediatric-sagittal-synostosis (accessed on 17 October 2021).
- Lajeunie, E.; Merrer, M.L.; Marchac, D.; Renier, D. Syndromal and nonsyndromal primary trigonocephaly: Analysis of a series of 237 patients. Am. J. Med. Genet. 1998, 75, 211–215. [Google Scholar] [CrossRef]
- Alderman, B.W.; Fernbach, S.K.; Greene, C.; Mangione, E.J.; Ferguson, S.W. Diagnostic practice and the estimated prevalence of craniosynostosis in Colorado. Arch. Pediatr. Adolesc. Med. 1997, 151, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.K.; Tsai, Y.-T.; Ariga, T. Functional roles of gangliosides in neurodevelopment: An overview of recent advances. Neurochem. Res. 2012, 37, 1230–1244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, R.K.; Macala, L.J.; Taki, T.; Weinfeld, H.M.; Yu, F.S. Developmental-changes in ganglioside composition and synthesis in embryonic rat-brain. J. Neurochem. 1988, 50, 1825–1829. [Google Scholar] [CrossRef] [PubMed]
- Kracun, I.; Rosner, H.; Drnovsek, V.; Heffer-Lauc, M.; Cosovic, C.; Lauc, G. Human brain gangliosides in development, aging and disease. Int. J. Devel. Biol. 1991, 35, 289–295. [Google Scholar]
- Sarbu, M.; Dehelean, L.; Munteanu, C.V.A.; Vukelic, Z.; Zamfir, A.D. Assessment of ganglioside age-related and topographic specificity in human brain by Orbitrap mass spectrometry. Anal. Biochem. 2017, 521, 40–54. [Google Scholar] [CrossRef]
- Rahmann, H. Brain gangliosides and memory formation. Behav. Brain Res. 1995, 66, 105–116. [Google Scholar] [CrossRef]
- Yu, R.K.; Nakatani, Y.; Yanagisawa, M. The role of glycosphingolipid metabolism in the developing brain. J. Lipid. Res. 2009, 50, S440–S445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fragaki, K.; Ait-El-Mkadem, S.; Chaussenot, A.; Gire, C.; Mengual, R.; Bonesso, L.; Bénéteau, M.; Ricci, J.E.; Desquiret-Dumas, V.; Procaccio, V.; et al. Refractory epilepsy, and mitochondrial dysfunction due to GM3 synthase deficiency. Eur. J. Hum. Genet. 2013, 21, 528–534. [Google Scholar] [CrossRef] [Green Version]
- Boukhris, A.; Schule, R.; Loureiro, J.L.; Lourenco, C.M.; Mundwiller, E.; Gonzalez, M.A.; Charles, P.; Gauthier, J.; Rekik, I.; Acosta Lebrigio, R.F.; et al. Alteration of ganglioside biosynthesis responsible for complex hereditary spastic paraplegia. Am. J. Hum. Genet. 2013, 93, 118–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boccuto, L.; Aoki, K.; Flanagan-Steet, H.; Chen, C.F.; Fan, X.; Bartel, F.; Petukh, M.; Pittman, A.; Saul, R.; Chaubey, A.; et al. A mutation in a ganglioside biosynthetic enzyme, ST3GAL5, results in salt, and pepper syndrome, a neurocutaneous disorder with altered glycolipid, and glycoprotein glycosylation. Hum. Mol. Genet 2014, 23, 418–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nico, D.; Conde, L.; Rivera-Correa, J.L.; Vasconcelos-Dos-Santos, A.; Mesentier-Louro, L.; Freire-de-Lima, L.; Arruda, M.B.; Freire-de-Lima, C.G.; Ferreira, O.D.C., Jr.; Lopes Moreira, M.E.; et al. Prevalence of IgG autoantibodies against GD3 ganglioside in acute Zika virus infection. Front Med. 2018, 9, 5–25. [Google Scholar] [CrossRef]
- Hurtado-Villa, P.; Puerto, A.K.; Victoria, S.; Gracia, G.; Guasmayán, L.; Arce, P.; Álvarez, G.; Blandón, E.; Rengifo, N.; Holguín, J.A.; et al. Raised frequency of microcephaly related to Zika virus infection in two birth defects surveillance systems in Bogota and Cali, Colombia. Pediatr. Infect. Dis. J. 2017, 36, 1017–1019. [Google Scholar] [CrossRef] [PubMed]
- Sarbu, M.; Ica, R.; Petrut, A.; Vukelić, Ž.; Munteanu, C.V.A.; Petrescu, A.J.; Zamfir, A.D. Gangliosidome of human anencephaly: A high resolution multistage mass spectrometry study. Biochimie 2019, 163, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Almeida, R.; Mosoarca, C.; Chirita, M.; Udrescu, V.; Dinca, N.; Vukelić, Z.; Allen, M.; Zamfir, A.D. Coupling of fully automated chip-based electrospray ionization to high-capacity ion trap mass spectrometer for ganglioside analysis. Anal. Biochem. 2008, 378, 43–52. [Google Scholar] [CrossRef]
- Mosoarca, C.; Vukelić, Z.; Zamfir, A.D. Mapping and sequencing of gangliosides from anencephaly by electrospray ionization high capacity ion trap mass spectrometry. In Applications of Mass Spectrometry in Life Safety; Popescu, C., Zamfir, A.D., Dinca, N., Eds.; Springer: Dordrecht, The Netherlands, 2008; pp. 71–82. [Google Scholar]
- Perkins, A.; Liu, G. Primary brain tumors in adults: Diagnosis and treatment. Am. Fam. Physician. 2016, 93, 211–217. [Google Scholar]
- Chandana, S.R.; Movva, S.; Arora, M.; Singh, T. Primary brain tumors in adults. Am. Fam Physician. 2008, 77, 1423–1430. [Google Scholar] [PubMed]
- Dorsey, J.F.; Salinas, R.D.; Dang, M.; Alonso-Basanta, M.; Judy, K.D.; Maity, A.; Lustig, R.A.; Lee, J.Y.K.; Philips, P.C.; Pruitt, A. Cancer of the central nervous system. In Abeloff’s Clinical Oncology, 6th ed.; Niederhuber, J.E., Armitage, J.O., Doroshow, J.H., Kastan, M.B., Tepper, J.E., Eds.; Elsevier: Philadelphia, PA, USA, 2020. [Google Scholar]
- Flangea, C.; Serb, A.; Sisu, E.; Zamfir, A.D. Chip-based nanoelectrospray mass spectrometry of brain gangliosides. Biochim. Biophys. Acta 2011, 1811, 513–535. [Google Scholar] [CrossRef]
- Krafft, C.; Neudert, L.; Simat, T.; Salzer, R. Near infrared Raman spectra of human brain lipids. Spectrochim. Acta. A Mol. Biomol. Spectrosc. 2005, 61, 1529–1535. [Google Scholar] [CrossRef]
- Steiner, G.; Shaw, A.; Choo-Smith, L.P.; Abuid, M.H.; Schackert, G.; Sobottka, S.; Steller, W.; Salzer, R.; Mantsch, H.H. Distinguishing and grading human gliomas by IR spectroscopy. Biopolymers 2003, 72, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Marta, G.N.; Correa, S.F.; Teixeira, M.J. Meningioma: Review of the literature with emphasis on the approach to radiotherapy. Expert. Rev. Anticancer Ther. 2011, 11, 1749–1758. [Google Scholar] [CrossRef]
- Galhom, A.E.; Madawi, A.A.; Ellabban, M.M. Surgical outcomes and predictors of complication in elderly patients with meningiomas. Egypt J. Neurol. Psychiatry Neurosurg. 2018, 54, 3. [Google Scholar] [CrossRef]
- Kaul, D.; Budach, V.; Graaf, L.; Gollrad, J.; Badakhshi, H. Outcome of elderly patients with meningioma after image-guided stereotactic radiotherapy: A study of 100 cases. Biomed. Res. Int. 2015, 2015, 868401. [Google Scholar] [CrossRef]
- Poon, M.T.C.; Fung, L.H.K.; Pu, J.K.S.; Leung, G.K.K. Outcome comparison between younger and older patients undergoing intracranial meningioma resections. J. Neuro-Oncol. 2013, 114, 219–227. [Google Scholar] [CrossRef]
- Adamson, D.C.; Rasheed, B.A.; McLendon, R.E.; Bigner, D.D. Central nervous system. Cancer Biomark. 2010, 9, 193–210. [Google Scholar] [CrossRef]
- Schiopu, C.; Vukelić, Z.; Capitan, F.; Kalanj-Bognar, S.; Sisu, E.; Zamfir, A.D. Chip-nanoelectrospray quadrupole time-of-flight tandem mass spectrometry of meningioma gangliosides: A preliminary study. Electrophoresis 2012, 33, 1778–1786. [Google Scholar] [CrossRef]
- Kostic, D.; Bucheit, F. Gangliosides in human brain tumors. Life Sci. 1970, 9, 589–596. [Google Scholar] [CrossRef]
- Sunder-Plassmann, M.; Bernheimer, H. Ganglioside in meningiomen und hirnhiiuten. Acta Neuropathol. 1974, 27, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Aruna, R.M.; Balasubramanian, K.A.; Mathai, K.V.; Basu, D. Isolation and characterization of glycolipids and glycosaminoglycans from meningiomas. Indian J. Med. Res. 1973, 61, 1688–1693. [Google Scholar] [PubMed]
- Berra, B.; Riboni, L.; De Gasperi, R.; Gaini, S.M.; Ragnotti, G. Modifications of ganglioside patterns in human meningiomas. J. Neurochem. 1983, 40, 777–782. [Google Scholar] [CrossRef]
- Davidsson, P.; Fredman, P.; Collins, V.P.; von Holst, H.; Mänsson, J.E.; Svennerholm, L. Ganglioside composition in human meningiomas. J. Neurochem. 1989, 53, 705–709. [Google Scholar] [CrossRef]
- Fredman, P.; Dumanski, J.; Davidsson, P.; Svennerholm, L.; Collins, V.P. Expression of the ganglioside GD3 in human meningiomas is associated with monosomy of chromosome 22. J. Neurochem. 1990, 55, 1838–1840. [Google Scholar] [CrossRef] [PubMed]
- Radić, B.; Vukelić, Z.; Bognar, S.K. Serum gangliosides in patients with brain tumors. Coll. Antropol. 2008, 32, 171–175. [Google Scholar]
- Zamfir, A.D.; Serb, A.; Vukelic´, Z.; Flangea, C.; Schiopu, C.; Fabris, D.; Kalanj-Bognar, S.; Capitan, F.; Sisu, E. Assessment of the molecular expression and structure of gangliosides in brain metastasis of lung adenocarcinoma by an advanced approach based on fully automated chip-nanoelectrospray mass spectrometry. J. Am. Soc. Mass Spectrom. 2011, 22, 2145–2159. [Google Scholar] [CrossRef] [Green Version]
- Vukelić, Z.; Kalanj-Bognar, S.; Froesch, M.; Bîndila, L.; Radić, B.; Allen, M.; Peter-Katalinić, J.; Zamfir, A.D. Human gliosarcoma-associated ganglioside composition is complex and distinctive as evidenced by high-performance mass spectrometric determination and structural characterization. Glycobiology 2007, 17, 504–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fredman, P.; von Holst, H.; Collins, V.P.; Dellheden, B.; Svennerholm, L. Expression of gangliosides GD3 and 3’-isoLM1 in autopsy brains from patients with malignant tumors. J. Neurochem. 1993, 60, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Schiopu, C.; Flangea, C.; Capitan, F.; Serb, A.; Vukelić, Z.; Kalanj-Bognar, S.; Sisu, E.; Przybylski, M.; Zamfir, A.D. Determination of ganglioside composition and structure in human brain hemangioma by chip-based nanoelectrospray ionization tandem mass spectrometry. Anal. Bioanal. Chem. 2009, 395, 2465–2477. [Google Scholar]
- Kawaguchi, A.; Kunimoto, K.; Inaba, Y.; Mikita, N.; Kaminaka, C.; Kanazawa, N.; Yamamoto, Y.; Kakimoto, N.; Suenaga, T.; Takeuchi, T.; et al. Distribution analysis of infantile hemangioma or capillary malformation on the head and face in Japanese patients. J. Dermatol. 2019, 46, 849–852. [Google Scholar] [CrossRef]
- Lewis, D.; Vaidya, R. Hepatic Hemangioma; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Lescura, C.M.; de Andrade, B.A.B.; Bezerra, K.T.; Agostini, M.; Ankha, M.V.A.; de Castro, F.; Carvalho, Y.R.; Romanach, M.J.; Anbinder, A.L. Oral intramuscular hemangioma: Report of three cases. J. Cutan. Pathol. 2019, 46, 603–608. [Google Scholar] [CrossRef]
- Mukherjee, B.; Moosa, S.; Rajagopal, R. Cavernous hemangioma of the conjunctiva. Indian J. Ophthalmol. 2019, 67, 2061. [Google Scholar] [CrossRef]
- Dauge, C.; Fenouil, T.; Petit, T.; Jeanne-Pasquier, C.; Collardeau–Frachon, S. Pulmonary infantile hemangioma mimicking a congenital cystic adenomatoid malformation. Pediatr. Dev. Pathol. 2019, 22, 480–485. [Google Scholar] [CrossRef]
- Mosnier, J.F.; Brunon, J.; Nuti, C. Histopathology of central nervous system cavernomas. Neurochirurgie 2007, 53, 131–135. [Google Scholar] [CrossRef]
- Bokhari, M.R.; Al-Dhahir, M.A. Brain Cavernous Angiomas; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Ica, R.; Simulescu, A.; Sarbu, M.; Munteanu, C.V.A.; Vukelić, Ž.; Zamfir, A.D. High resolution mass spectrometry provides novel insights into the ganglioside pattern of brain cavernous hemangioma. Anal. Biochem. 2020, 609, 113976. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, M.; Gupta, V. Astrocytom; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Hirtz, A.; Rech, F.; Dubois-Pot-Schneider, H.; Dumond, H. Astrocytoma: A hormone-sensitive tumor? Int. J. Mol. Sci. 2020, 21, 9114. [Google Scholar] [CrossRef] [PubMed]
- Wessels, P.H.; Weber, W.E.; Raven, G.; Ramaekers, F.C.; Hopman, A.H.; Twijnstra, A. Supratentorial grade II astrocytoma: Biological features and clinical course. Lancet. Neurol. 2003, 2, 395–403. [Google Scholar] [CrossRef]
- Zamfir, A.D.; Fabris, D.; Capitan, F.; Munteanu, C.V.A.; Vukelić, Z.; Flangea, C. Profiling and sequence analysis of gangliosides in human astrocytoma by high-resolution mass spectrometry. Anal. Bioanal. Chem. 2013, 405, 7321–7335. [Google Scholar] [CrossRef]
- Bartik, P.; Maglott, A.; Entlicher, G.; Vestweber, D.; Takeda, K.; Martin, S.; Dontenwill, M. Detection of a hypersialylated β1 integrin endogenously expressed in the human astrocytoma cell line A172. Int. J. Oncol. 2008, 32, 1021–1031. [Google Scholar] [CrossRef] [Green Version]
- Kanderi, T.; Gupta, V. Glioblastoma Multiforme; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Iwasawa, T.; Zhang, P.; Ohkawa, Y.; Momota, H.; Wakabayashi, T.; Ohmi, Y.; Bhuiyan, R.H.; Furukawa, K.; Furukawa, K. Enhancement of malignant properties of human glioma cells by ganglioside GD3/GD2. Int. J. Oncol. 2018, 52, 1255–1266. [Google Scholar] [CrossRef] [Green Version]
- Birks, S.M.; Danquah, J.O.; King, L.; Vlasak, R.; Gorecki, D.C.; Pilkington, G.J. Targeting the GD3 acetylation pathway selectively induces apoptosis in glioblastoma. Neuro. Oncol. 2011, 13, 950–960. [Google Scholar] [CrossRef] [Green Version]
- Buckner, J.C.; Brown, P.D.; O’Neill, P.; Meyer, F.B.; Wetmore, C.J.; Uhm, J.H. Central Nervous System Tumours. Mayo Clin. Proc. 2007, 82, 1271–1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, G.; Manjila, S.; Sakla, N.; True, A.; Wardeh Amr, H.; Beig, N.; Vaysberg, A.; Matthews, J.; Prasanna, P.; Spektor, V. Radiomics and radiogenomics in gliomas: A contemporary update. Br. J. Cancer 2021, 125, 641–657. [Google Scholar] [CrossRef]
- Urbańska, K.; Sokołowska, J.; Szmidt, M.; Sysa, P. Glioblastoma multiforme—An overview. Contemp. Oncol. 2014, 18, 307–312. [Google Scholar]
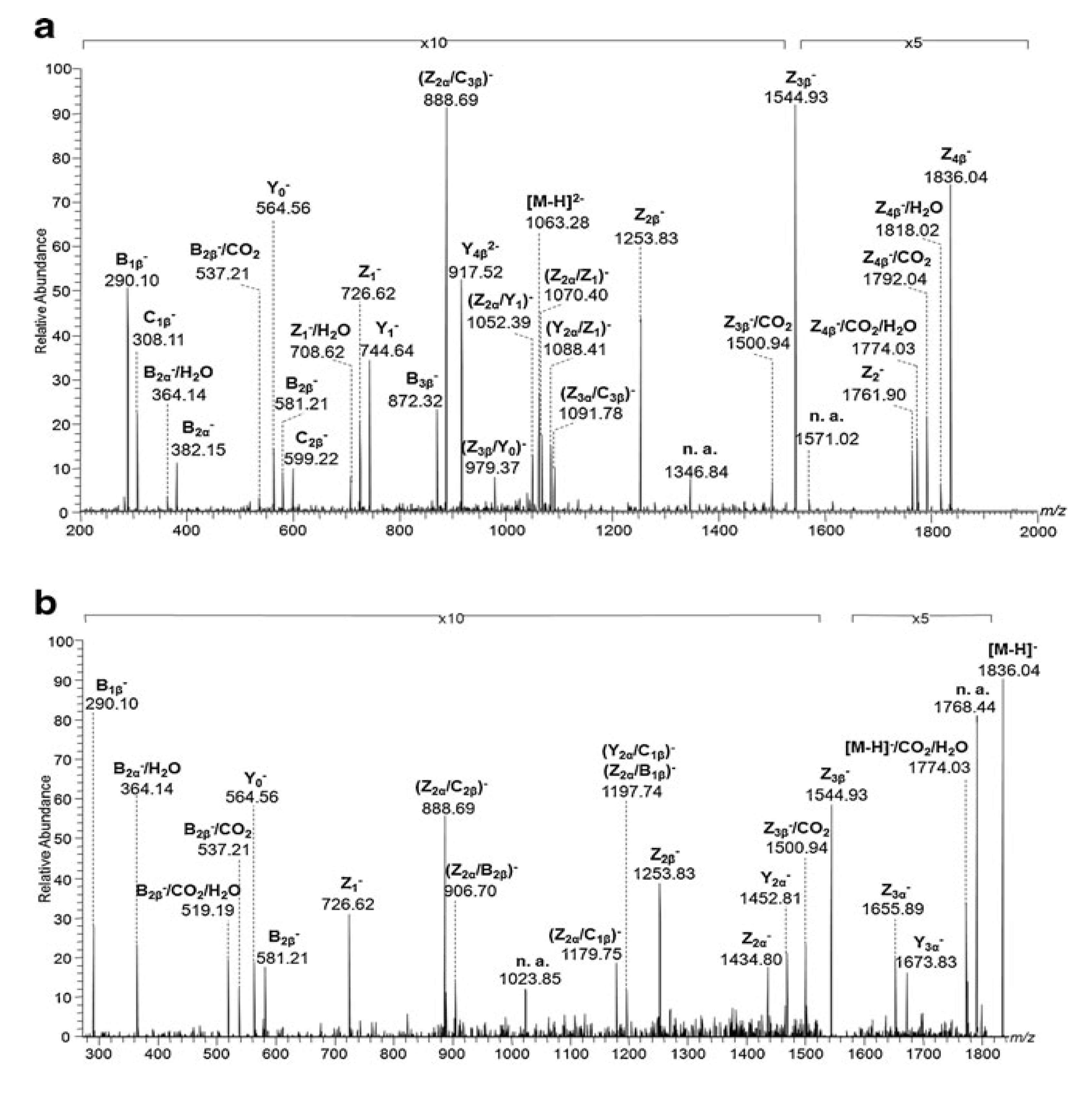
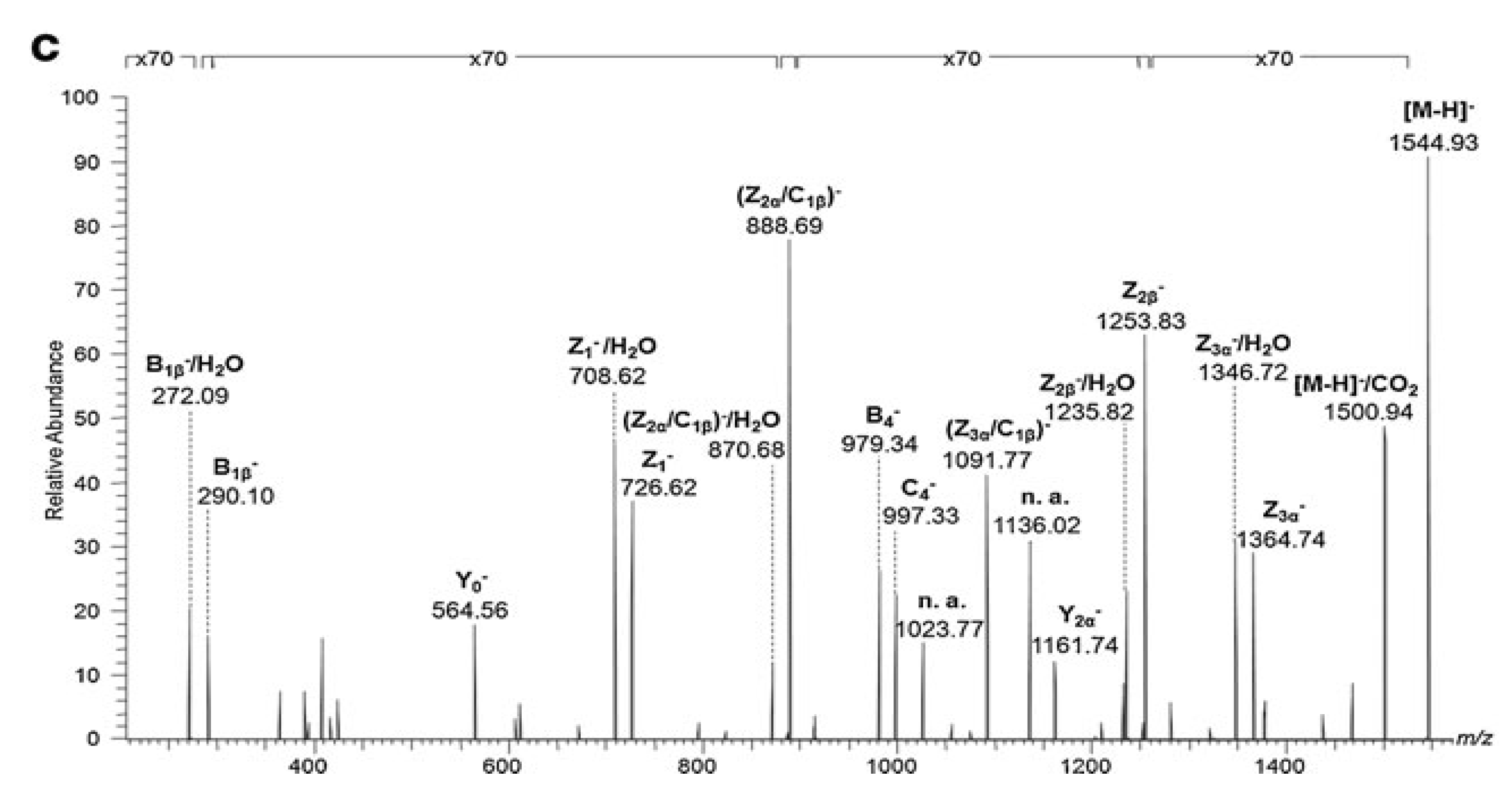
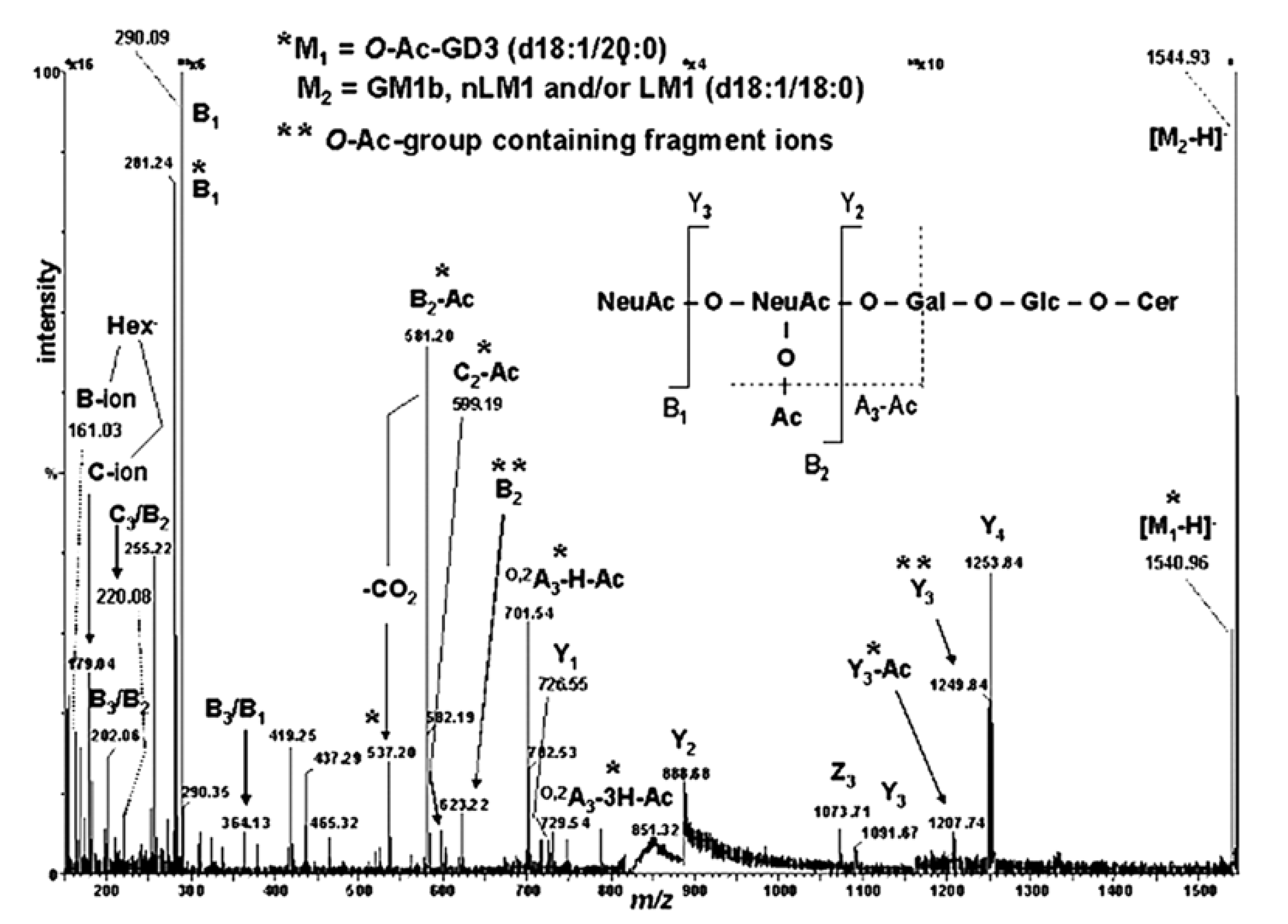
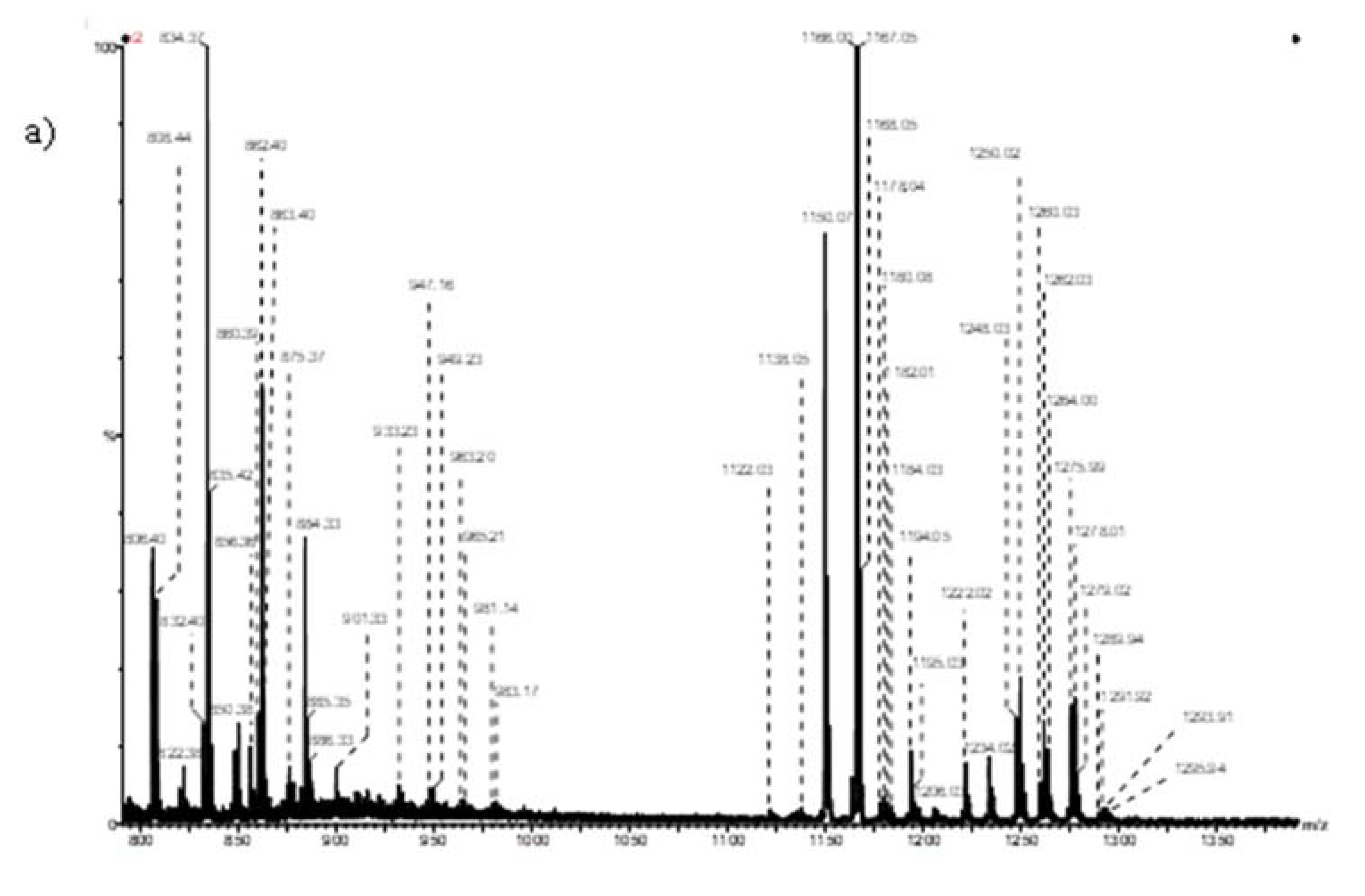
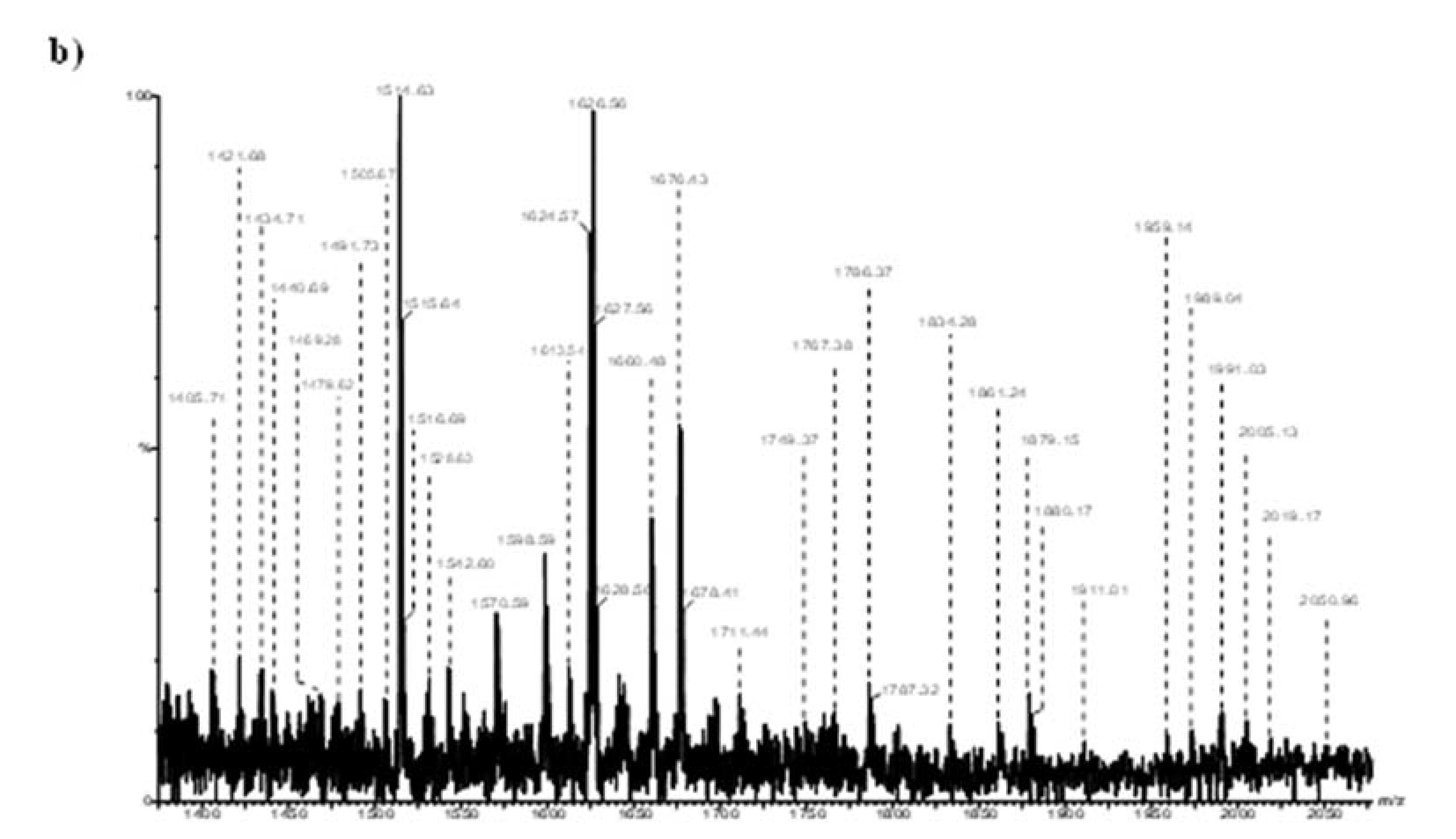
- Sarbu, M.; Petrica, L.; Clemmer, D.E.; Vukelić, Ž.; Zamfir, A.D. Gangliosides of human glioblastoma multiforme: A comprehensive mapping and structural analysis by ion mobility tandem mass spectrometry. J. Am. Soc. Mass Spectrom. 2021, 32, 1249–1257. [Google Scholar] [CrossRef]
- Mangiola, A.; de Bonis, P.; Maira, G.; Balducci, M.; Sica, G.; Lama, G.; Lauriola, L.; Anile, C. Invasive tumor cells and prognosis in a selected population of patients with glioblastoma multiforme. Cancer 2008, 113, 841–846. [Google Scholar] [CrossRef]
- Yeh, S.C.; Wang, P.Y.; Lou, Y.W.; Khoo, K.H.; Hsiao, M.; Hsu, T.L.; Wong, C.H. Glycolipid GD3 and GD3 synthase are key drivers for glioblastoma stem cells and tumorigenicity. Proc. Natl. Acad. Sci. USA 2016, 113, 5592–5597. [Google Scholar] [CrossRef] [Green Version]
- Fredman, P.; von Hoist, H.; Collins, V.P.; Granholm, L.; Svennerholm, L. Sialyllactotetraosylceramide, a ganglioside marker for human malignant gliomas. J. Neurochem. 1988, 50, 912–919. [Google Scholar] [CrossRef] [PubMed]
- Oblinger, J.L.; Pearl, D.K.; Boardman, C.L.; Saqr, H.; Prior, T.W.; Scheithauer, B.W.; Jenkins, R.B.; Burger, P.C.; Yates, A.J. Diagnostic and prognostic value of glycosyltransferase mRNA in glioblastoma multiforme patients. Neuropathol. Appl. Neurobiol. 2006, 32, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Fabris, D.; Rožman, M.; Sajko, T.; Vukelić, Ž. Aberrant ganglioside composition in glioblastoma multiforme and peritumoral tissue: A mass spectrometry characterization. Biochimie 2017, 137, 56–68. [Google Scholar] [CrossRef]
- Von Neubeck, C.; Seidlitz, A.; Kitzler, H.H.; Beuthien-Baumann, B.; Krause, M. Glioblastoma multiforme: Emerging treatments and stratification markers beyond new drugs. Br. J. Radiol. 2015, 88, 20150354. [Google Scholar] [CrossRef] [Green Version]
- Sarbu, M.; Clemmer, D.E.; Zamfir, A.D. Ion mobility mass spectrometry of human melanoma gangliosides. Biochimie 2020, 177, 226–237. [Google Scholar] [CrossRef]
- Sarbu, M.; Raab, S.; Henderson, L.; Fabris, D.; Vukelić, Z.; Clemmer, D.E.; Zamfir, A.D. Cerebrospinal fluid: Profiling and fragmentation of gangliosides by ion mobility mass spectrometry. Biochimie 2020, 170, 36–48. [Google Scholar] [CrossRef]
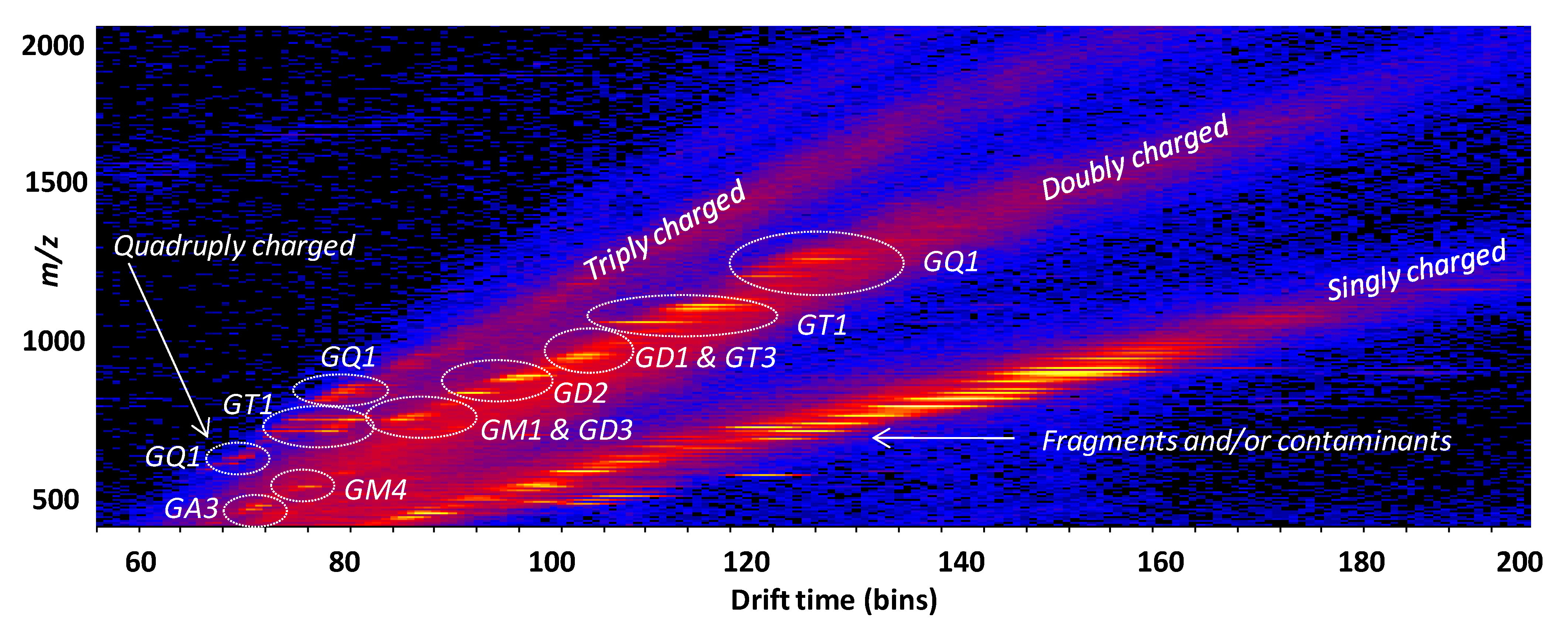
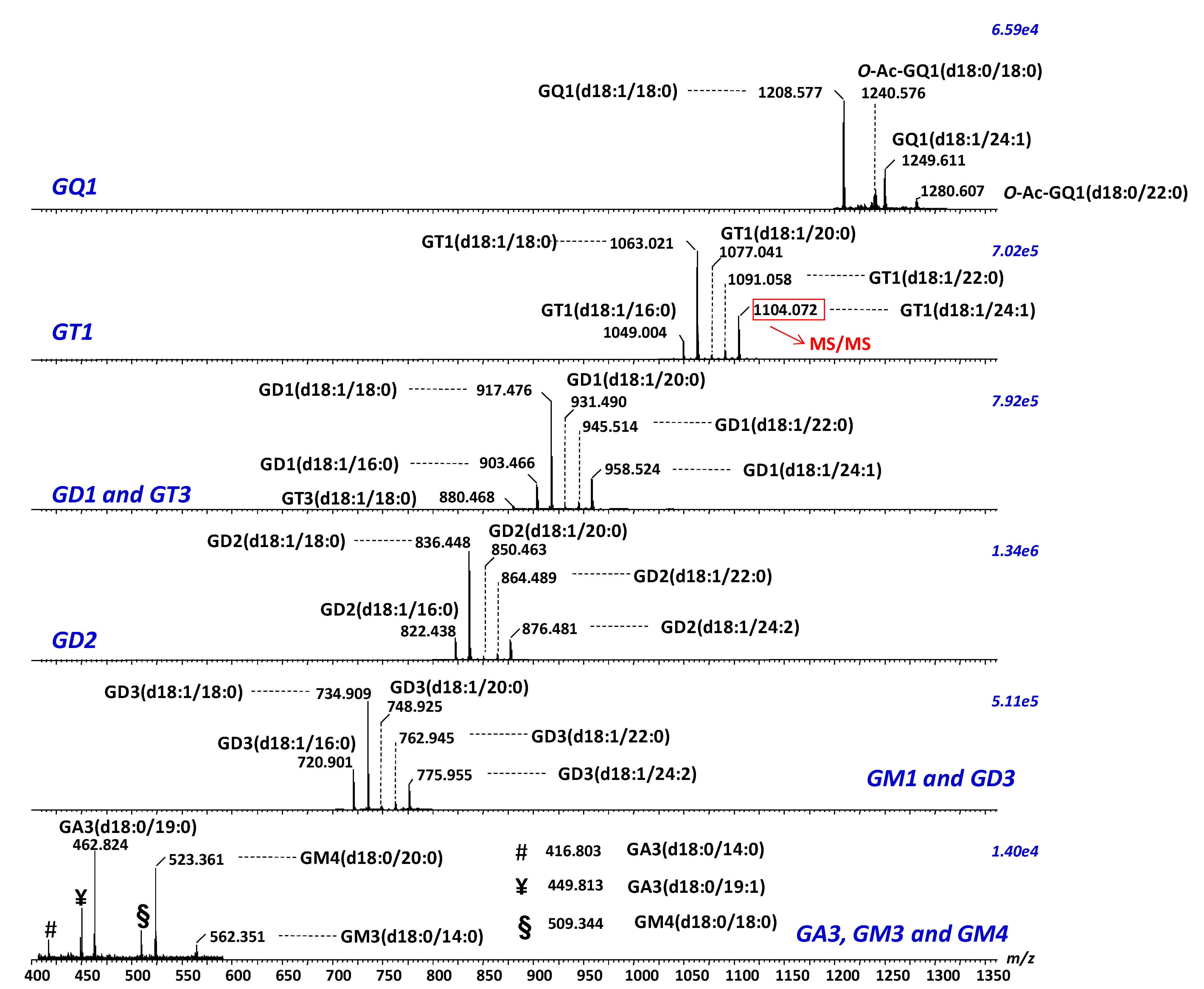
- Sarbu, M.; Vukelić, Z.; Clemmer, D.E.; Zamfir, A.D. Ion mobility mass spectrometry provides novel insights into the expression and structure of gangliosides in normal adult human hippocampus. Analyst 2018, 143, 5234–5246. [Google Scholar] [CrossRef]
- Sarbu, M.; Vukelić, Z.; Clemmer, D.E.; Zamfir, A.D. Electrospray ionization ion mobility mass spectrometry provides novel insights into the pattern and activity of fetal hippocampus gangliosides. Biochimie 2017, 139, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Sarbu, M.; Robu, A.C.; Ghiulai, R.M.; Vukelić, Ž.; Clemmer, D.E.; Zamfir, A.D. Electrospray ionization ion mobility mass spectrometry of human brain gangliosides. Anal. Chem. 2016, 88, 5166–5178. [Google Scholar] [CrossRef] [PubMed]
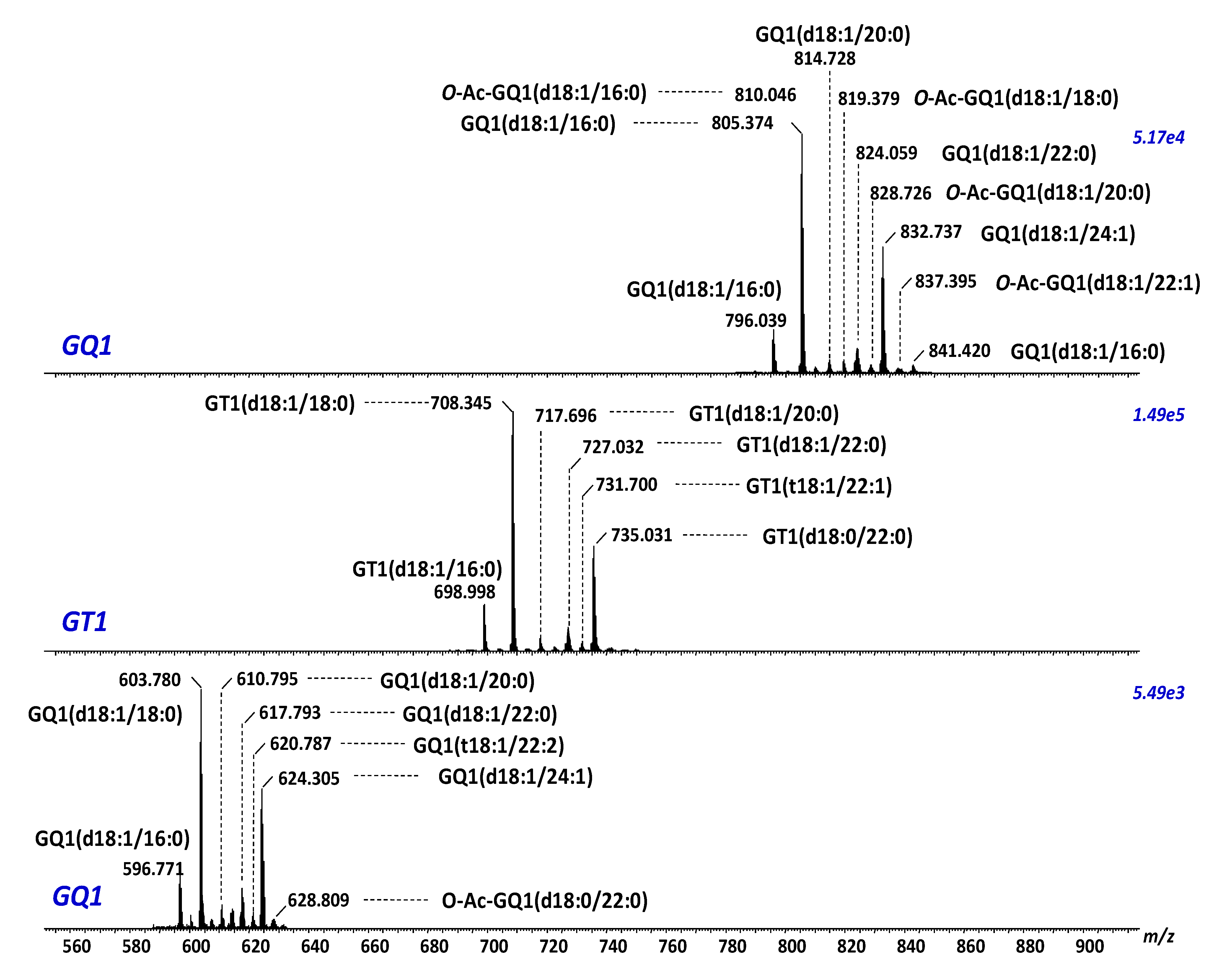
- Hamasaki, H.; Aoyagi, M.; Kasama, T.; Handa, S.; Hirakawa, K.; Taki, T. GT1b in human metastatic brain tumors: GT1b as a brain metastasis-associated ganglioside. Biochim. Biophys. Acta 1999, 1437, 93–99. [Google Scholar] [CrossRef]
- Morantz, R.A.; Feigin, I.; Ransohoff, J. Clinical and pathological study of 24 cases of gliosarcoma. J. Neurosurg. 1976, 45, 398–408. [Google Scholar] [CrossRef] [Green Version]
- Singh, G.; Das, K.K.; Sharma, P.; Guruprasad, B.; Jaiswal, S.; Mehrotra, A.; Srivastava, A.K.; Sahu, R.N.; Jaiswal, A.K.; Behari, S. Cerebral gliosarcoma: Analysis of 16 patients and review of literature. Asian J. Neurosurg. 2015, 10, 195–202. [Google Scholar]
- Meis, J.M.; Martz, K.L.; Nelson, J.S. Mixed glioblastoma multiforme and sarcoma. A clinicopathologic study of 26 Radiation Therapy Oncology Group cases. Cancer 1991, 67, 2342–2349. [Google Scholar] [CrossRef]
- Galanis, E.; Buckner, J.C.; Dinapoli, R.P.; Scheithauer, B.W.; Jenkins, R.B.; Wang, C.H.; O’Fallon, J.R.; Farr, G., Jr. Clinical outcome of gliosarcoma compared with glioblastoma multiforme: North central cancer treatment group results. J. Neurosurg. 1998, 89, 425–430. [Google Scholar] [CrossRef] [Green Version]
- Kozak, K.R.; Mahadevan, A.; Moody, J.S. Adult gliosarcoma: Epidemiology, natural history, and factors associated with outcome. Neuro. Oncol. 2009, 11, 183–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frandsen, S.; Broholm, H.; Larsen, V.A.; Grunnet, K.; Møller, S.; Poulsen, H.S.; Michaelsen, S.R. Clinical Characteristics of Gliosarcoma and Outcomes From Standardized Treatment Relative to Conventional Glioblastoma. Front. Oncol. 2019, 9, 1425. [Google Scholar] [CrossRef]
- Ohgaki, H.; Biernat, W.; Reis, R.; Hegi, M.; Kleihues, P. Gliosarcoma. In Pathology and Genetics of Tumors of the Nervous System; Kleihues, P., Cavenee, W.K., Eds.; IARC Press: Lyon, France, 2000; pp. 42–44. [Google Scholar]
- Shukla, G.S.; Krag, D.N. Selective delivery of therapeutic agents for the diagnosis and treatment of cancer. Expert Opin. Biol. Ther. 2006, 6, 39–54. [Google Scholar] [CrossRef] [PubMed]
- Kniep, B.; Kniep, E.; Ozkucur, N.; Barz, S.; Bachmann, M.; Malisan, F.; Testi, R.; Rieber, E.P. 9-O-acetyl GD3 protects tumor cells from apoptosis. Int. J. Cancer 2006, 119, 67–73. [Google Scholar] [CrossRef]
- Dewald, H.; Cavdarli, S.; Steenackers, A.; Delannoy, C.P.; Mortuaire, M.; Spriet, C.; Noël, M.; Groux-Degroote, S.; Delannoy, P. TNF differentially regulates ganglioside biosynthesis and expression in breast cancer cell lines. PLoS ONE 2018, 13, e0196369. [Google Scholar] [CrossRef] [Green Version]
- Robu, A.; Schiopu, C.; Capitan, F.; Zamfir, A.D. Mass spectrometry of gangliosides in extracranial tumors: Application to adrenal neuroblastoma. Anal. Biochem. 2016, 509, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Krengel, U.; Bousquet, P.A. Molecular recognition of gangliosides and their potential for cancer immunotherapies. Front. Immunol. 2014, 5, 325. [Google Scholar] [CrossRef] [Green Version]
- Sun, G.; Ding, X.; Bi, N.; Wang, Z.; Wu, L.; Zhou, W.; Zhao, Z.; Wang, J.; Zhang, W.; Fan, J.; et al. Molecular predictors of brain metastasis-related microRNAs in lung adenocarcinoma. PLoS Genet. 2019, 15, e1007888. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.W.; Dhahbi, J. Lung adenocarcinoma and lung squamous cell carcinoma cancer classification, biomarker identification, and gene expression analysis using overlapping feature selection methods. Sci. Rep. 2021, 11, 13323. [Google Scholar] [CrossRef]
- Herbst, R.S.; Morgensztern, D.; Boshof, C. The biology and management of non-small cell lung cancer. Nature 2018, 553, 446–454. [Google Scholar] [CrossRef]
- Ernani, V.; Stinchcombe, T.E. Management of brain metastases in non–small-cell lung cancer. J. Oncol. Pract. 2019, 15, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.; Kim, J.W.; Park, M.; Kim, J.W.; Kim, M.; Suh, S.H.; Chang, Y.S.; Ahn, S.J.; Lee, J.-M. Brain metastases from lung adenocarcinoma may preferentially involve the distal middle cerebral artery territory and cerebellum. Front. Oncol. 2020, 10, 1664–1674. [Google Scholar] [CrossRef] [PubMed]
- Groux-Degroote, S.; Delannoy, P. Cancer-associated glycosphingolipids as tumor markers and targets for cancer immunotherapy. Int. J. Mol. Sci. 2021, 22, 6145. [Google Scholar] [CrossRef] [PubMed]
- Sarbu, M.; Ica, R.; Zamfir, A.D. Developments and applications of separation and microfluidics methods coupled to electrospray mass spectrometry in glycomics of nervous system gangliosides. Electrophoresis 2021, 42, 429–449. [Google Scholar] [CrossRef] [PubMed]















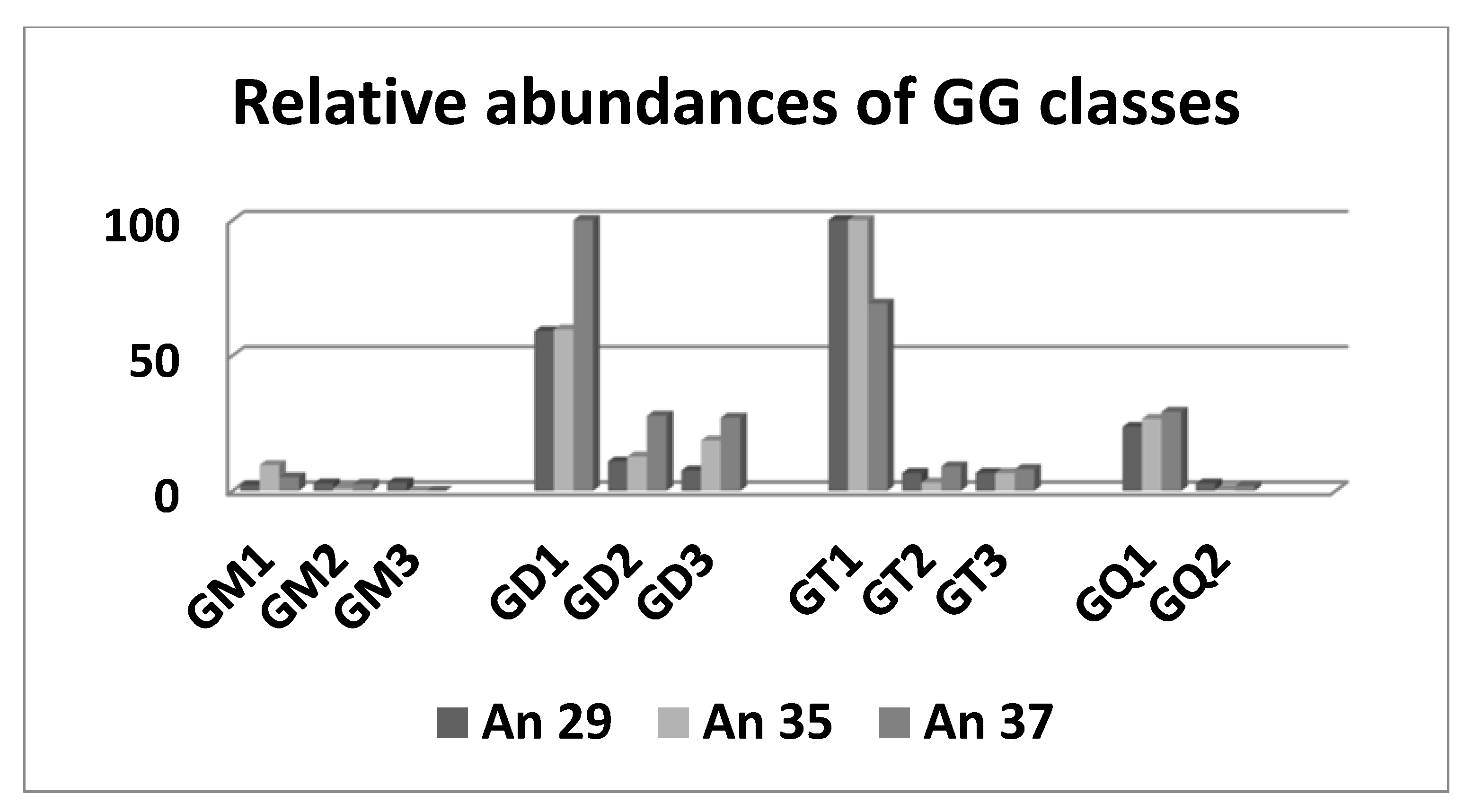{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | OMIM/Phenotype MIM Number | Mutated Gene or Pathway/Locus | Characterization | Symptoms | Prognosis | Incidence | |
|---|---|---|---|---|---|---|---|
| More common cephalic disorders | Anencephaly | 206500 619452 | NUAK12 NUAK2 | failure of neural tube closure at the cephalic end | blindness, deafness, unconscious, unable to feel pain | death before birth, although some may survive a few hours or a few days after birth [99] | 1:1000 pregnancies |
| Colpocephaly | - | COL4A1 | significant enlargement of the occipital horns | small head circumference, intellectual disability, movement abnormalities, muscle spasms and seizures, poor vision, speech and language difficulties, deafness, and chorioretinitis [100] | depends on the severity of the associated conditions and the degree of abnormal brain development | only 50 cases have been reported up to now | |
| Holoprosencephaly | 236100 | HPE1 | failure of the prosencephalon to develop | cyclopia, ethmocephaly, anophthalmi, microphthalmia, proboscis, hypotelorism, seizures, hydrocephalus, neural tube defects, pituitary dysfunction, short stature, feeding difficulties, instability of temperature, heart rate, and respiration, developmental delay, and intellectual disability [101] | stillbirth or up to maximum 12 months | 1:16,000 live births 1:5–10,000 (Europe) | |
| Ethmocephaly | - | - | failure of diverticularization and rotation of the prosencephalon into two normal cerebral hemispheres | proboscis situated above hypoteloric orbits in the midline, microphthalmos, absent nasal structures, low-set malformed ears [102] | incompatible with life | 1:15,000 live births 1:250 among abortuses | |
| Hydranencephaly | - | NDE1 | the cerebral hemispheres are absent and replaced by sacs filled with cerebrospinal fluid | hypertonia, seizures, hydrocephalus, visual impairment, lack of growth, deafness, blindness, paralysis, and intellectual disabilities [103] | several years or more | 1:5–10,000 worldwide | |
| Iniencephaly | - | - | combines extreme retroflexion of the head with severe defects of the spine | anencephaly, cephalocele, cyclopia, agnathia, cleft palate, arthrogryposis, clubfeet, holoprosencephaly, spina bifida, low-set ears, pulmonary hypoplasia, omphalocele, gastroschisis, cardiovascular disorders, diaphragmatic hernias, gastrointestinal atresia, single umbilical artery, renal abnormalities, genu recurvatum, hydramnios | spontaneous abortion or stillborn | 1:5–10,000 (Europe) | |
| Lissencephaly | 607432 | PAFAH1B1 DCX, TUBA3 ARX | smooth or nearly smooth cerebral surface due to the lack of normal convolutions in the brain | feeding and swallowing problems, seizures, severe psychomotor retardation and muscle spasticity or hypotonia [104] | up to the age of 10 | 1:100,000 births | |
| Megalencephaly | 155350 248000 | PI3K-AKT | abnormally large, heavy, and usually malfunctioning brain | autism, severe seizures, hemiparesis, and intellectual disability | do not survive through adulthood | 1:50 births | |
| Microcephaly | 251200 | MCPH1—MCPH12 | a much smaller head than normal because the brain has not developed properly | impaired cognitive development, delayed motor functions and speech, facial distortions, dwarfism or short stature, hyperactivity, seizures, difficulties with coordination and balance, feeding difficulties, and other brain or neurological abnormalities [105] | depends on associated medical conditions | 2–12:10,000 births | |
| Porencephaly | 175780 614483 | COLGALT1 [106] | a cyst or cavity filled with cerebrospinal fluid develops in the brain during fetal development or soon after birth | developmental delay, hypotonia, seizures, macrocephaly or microcephaly, spastic hemiplegia, speech problems, delayed growth, and intellectual disability, epilepsy, hydrocephalus, spastic contractures [107,108] | varies according to the location and extent of the cysts or cavities | 1:19,000 in Japan 1:28,500 in the USA | |
| Schizencephaly | 269160 | SIX3 SHH EMX2 COL4A1 | abnormal slits, or clefts, in the cerebral hemispheres | developmental delays, delays in speech and language skills, and problems with brain–spinal cord communication, abnormally small head, cognitive delay and impairment, partial or complete paralysis, or poor muscle tone, seizures [109] | varies depending on the size of the clefts and the degree of neurological deficit | 1:64,935 in USA 1:69,444 in UK | |
| Less common cephalies | Acephaly | - | DCTN5 LHX1 OTX2 [110] | manifests in a twin pregnancy where the head and heart fails to form on one of the embryos, absence of the head | body with no head and heart; the fetus’s neck is attached to the normal twin | not compatible with extrauterine survival | 1:34,600 deliveries [111] |
| Exencephaly | - | MSX2 | partial or complete absence of the calvaria with abnormal development of brain tissue | cranial vault is absent or poorly formed, cephalad to orbits, the nasal bone may be absent in most cases, brain tissue is herniating or dangling in amniotic fluid, brain tissue may be attached to the amniotic membrane [112] | incompatible with extrauterine life | 3:10,000 pregnancies [113] | |
| Otocephaly | 202650 | PRRX1 | mandibular hypoplasia/agnathia, melotia and/or synotia, microstomia with oroglossal hypoplasia/aglossia | holoprosencephaly, skeletal, genitourinary, cardiovascular anomalies, situs inversus, polyhydramnios | incompatible with life | 1:70,000 births [114] | |
| Craniosynostosis | 218500 | FGFR2 FGFR3 FGFR1 TWIST1 EFNB1 [115] | premature closure of cranial sutures, causing problems with normal brain and skull growth | abnormallyshapedskull/restricted brain growth, increased pressure in the skull, impaired brain development, seizures, blindness and/or intellectual disability, obstructive sleep apnea [116,117] | varies depending on whether single or multiple cranial sutures are involved or other abnormalities are present | 1:2100–2500 births | |
| Brachycephaly | 123100 615314 616602 | FGFR3 P250R | premature fusion of both coronal sutures leading to skull deformity with a broad flat forehead and palpable coronal ridges [118] | short skull | usually good | 1:20,000 births | |
| Oxycephaly | 123100 616602 | FGFR2 MEGF8 TWIST1 [119] | a premature closure of the bony vault of the skull | top of the skull is pointed/cone-shaped, blindness, syndactyly and aracluiodactyly, mental deficiency, elevated intracranial pressure [120] | complete recovery after cranial expansion surgery | <1:1,000,000 births | |
| Plagiocephaly | 123100 615314 616602 | NIPBL | an asymmetrical distortion of the skull | motor and language developmental delays, head deformities, increased pressure inside the head, seizures, developmental delay [121] | recovery with helmet treatment after surgery [122] | 46:100 births [123] | |
| Scaphocephaly | 123100 600775 615529 | TWIST1 | premature fusion of the sagittal suture | misshapen skull; development of a raised, hard ridge along affected sutures; slow/no growth of the head as the baby grows | recovery after surgery | 1:2000 live births [124] | |
| Trigonocephaly | 190440 | FGFR1 | premature closure of the metopic sutures | delay in language development, hyperactivity, autistic tendencies, and motor dysfunctions, palpable metopic ridge | recovery after surgery | 1:700 up to 1:15,000 newborns globally (differs per country) [125,126] |
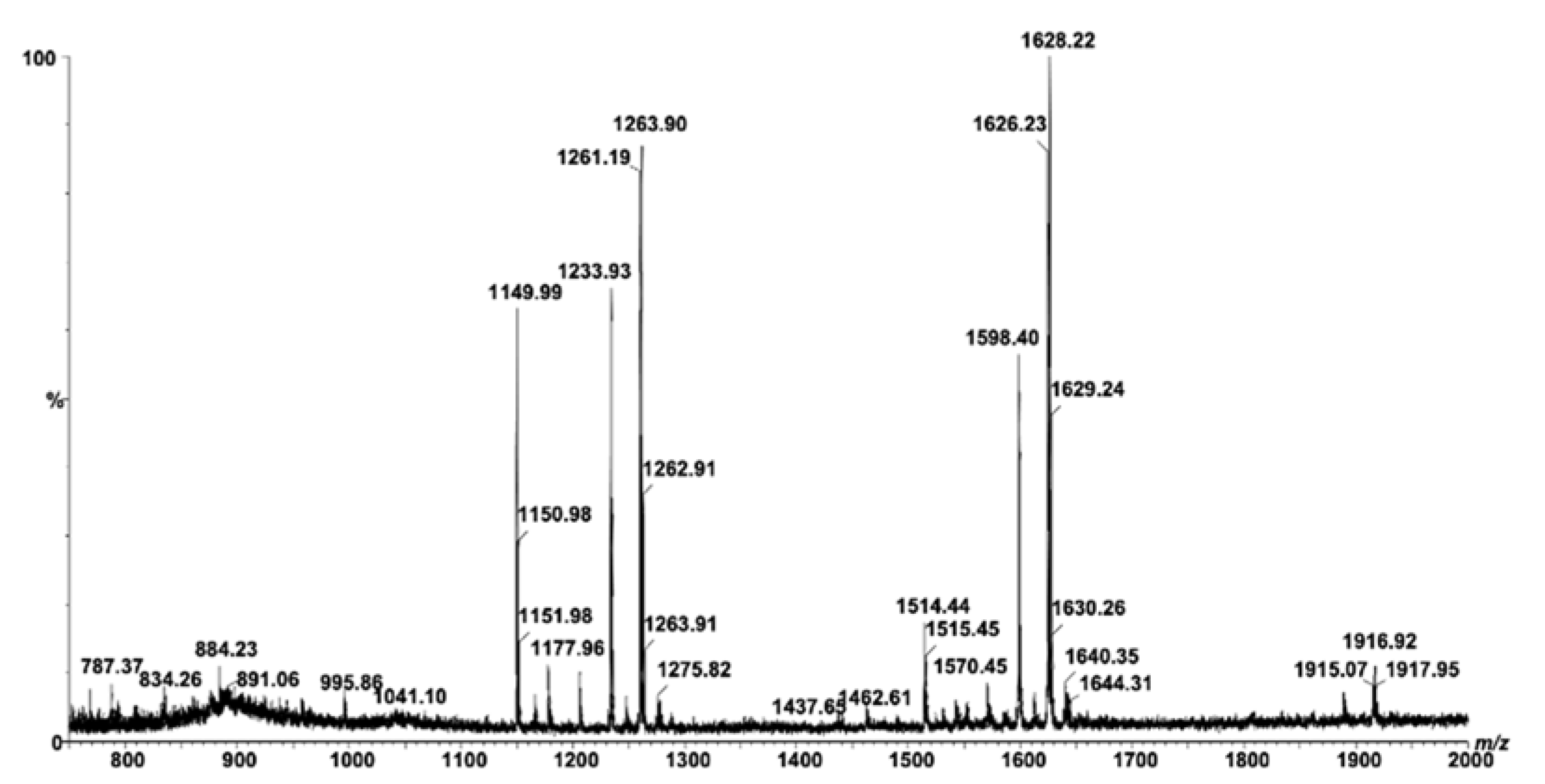
| m/z (Monoisotopic) | Molecular Ion | Proposed Structure |
|---|---|---|
| 787.37 | [M-H]− | LacCer(d18:1/11:2) |
| 834.26 | [M-H]− | LacCer(d18:1/14:0) |
| 884.32 | [M-H]− | LacCer(d18:1/18:2) |
| 891.06 | [M-H]− | LacCer(d18:0/18:0) |
| 995.86 | [M-H]− | GA2(d18:0/11:0) |
| 1041.10 | [M-H]− | GM4(d18:1/20:2) |
| 1149.99 | [M-H]− | GM3(d18:1/16:1) |
| 1151.98 | [M-H]− | GM3(d18:1/16:0) |
| 1167.70 | [M-H]− | GM3(t18:1/16:0) |
| 1177.96 | [M-H]− | GM3(d18:1/18:1) |
| 1233.93 | [M-H]− | GM3(d18:1/22:1) |
| 1259.90 | [M-H]− | GM3(d18:1/24:2) |
| 1261.91 | [M-H]− | GM3(d18:1/24:1) |
| 1275.82 | [M-H]− | GM3(t18:1/24:2) |
| 1277.80 | [M-H]− | GM3(t18:1/24:1) |
| 1437.65 | [M-H]− | GM2(d18:1/22:1) |
| 1462.61 | [M-H]− | GM2(d18:1/24:2) |
| 1489.55 | [M-H]− | GD3(t18:0/18:0) |
| 1514.44 | [M-H]− | GD3(d18:0/21:0) or O-Ac-GD3(d18:0/18:0) |
| 1542.41 | [M-H]− | GM1(d18:1/18:1) |
| 1570.45 | [M-H]− | GM1(d18:1/20:1) |
| 1598.40 | [M-H]− | GM1(d18:1/22:1) |
| 1626.23 | [M-H]− | GM1(d18:1/24:1) or GM1(d18:0/24:2) |
| 1628.22 | [M-H]− | GM1(d18:1/24:0) |
| 1640.35 | [M-H]− | GM1(d18:1/25:1) |
| 1644.31 | [M-H]− | GM1(d18:0/25:0) |
| 1659.32 | [M-H]− | GM1(t18:1/25:0) |
| 1792.69 | [M-H]− | GD1(d18:1/15:1) |
| 1833.98 | [M-H]− | GD1(d18:1/18:1) |
| 1861.95 | [M-H]− | GD1(d18:0/20:1) |
| 1915.07 | [M-H]− | GD1(t18:1/24:3) |
| 1916.92 | [M-H]− | GD1(d18:1/24:2) |
| 1919.86 | [M-H]− | GD1(d18:1/24:0) or GD1(t18:1/23:1) |
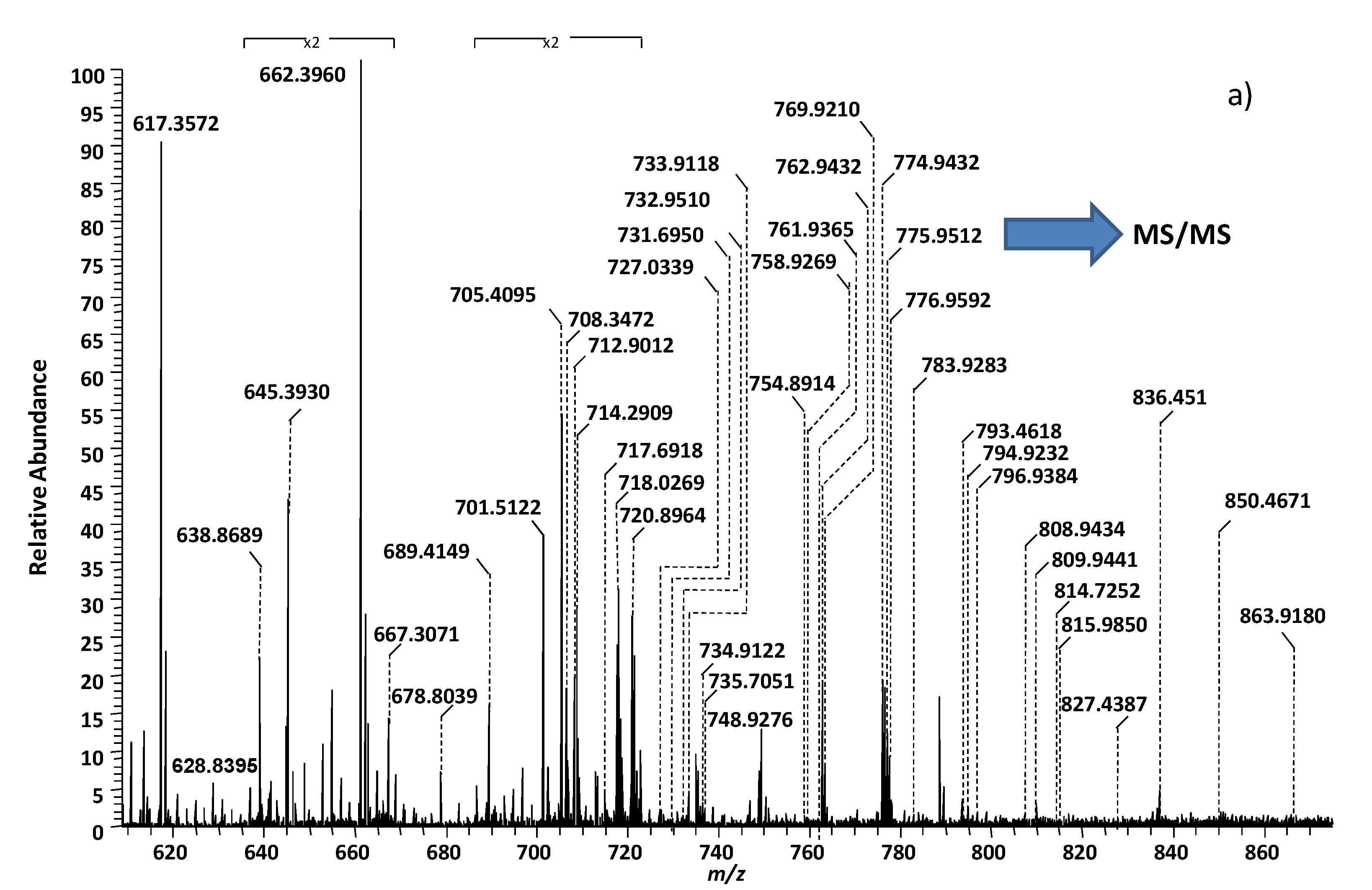
| Nr. crt. | Proposed Structure | m/zexp | Molecular Ion |
|---|---|---|---|
| 1 | GM3(d18:0/24:0) | 645.3930 | [M-4H++2Na+-H2O]2− |
| 2 | GM3(t18:0/24:0) | 662.3960 | [M-4H++2Na+]2− |
| 3 | GT1(d18:1/18:0) | 708.3472 | [M-3H+]3− |
| 4 | GD3(d18:0/16:0) | 712.9012 | [M-2H+-H2O]2− |
| 5 | GD3(d18:1/16:0) | 720.8964 | [M-2H+]2− |
| 6 | GT1(d18:1/20:0) | 717.6918 | [M-3H+]3− |
| 7 | GT1(d18:1/22:0) | 727.0339 | [M-3H+]3− |
| 8 | GT1(t18:1/22:1) | 731.6950 | [M-3H+]3− |
| 9 | GM2(d18:1/24:0) | 732.9510 | [M-2H+]2− |
| 10 | GD3(d18:1/18:1) | 733.9118 | [M-2H+]2− |
| 11 | GD3(d18:1/18:0) | 734.9122 | [M-2H+]2− |
| 12 | O-Ac-GT1(d18:0/22:0) | 735.7051 | [M-3H+-H2O]3− |
| 13 | GD3(d18:1/20:0) | 748.9276 | [M-2H+]2− |
| 14 | GM1(d18:1/14:0) | 754.8914 | [M-3H+ +Na+]2− |
| 15 | GM1(d18:0/16:0) | 758.9269 | [M-2H+]2− |
| 16 | GD3(d18:1/22:1) | 761.9365 | [M-2H+]2− |
| 17 | GD3(d18:1/22:0) | 762.9432 | [M-2H+]2− |
| 18 | GM1(d18:1/18:2) | 769.9210 | [M-2H+]2− |
| 19 | GD3(d18:1/24:2) | 774.9432 | [M-2H+]2− |
| 20 | GD3(d18:1/24:1) | 775.9512 | [M-2H+]2− |
| 21 | GD3(d18:1/24:0) | 776.9592 | [M-2H+]2− |
| 22 | GM1(d18:0/18:0) | 783.9283 | [M-3H++Na+]2− |
| 23 | GD2(d18:0/13:0) | 793.4618 | [M-2H+-H2O]2− |
| 24 | GM1(d18:0/18:0) | 794.9232 | [M-4H++2Na+]2− |
| 25 | GM1(d18:1/20:0) | 796.9384 | [M-3H++Na+]2− |
| 26 | GM1(d18:0/20:0) | 808.9434 | [M-4H++2Na+]2− |
| 27 | GM1(d18:0/20:0) | 809.9441 | [M-4H++2Na+]2− |
| 28 | GQ1(d18:1/20:0) | 814.7252 | [M-3H+]3− |
| 29 | GD3(d18:1/28:0) | 815.9850 | [M-3H++Na]2− |
| 30 | GD2(d18:1/18:0) | 827.4387 | [M-2H+ -H2O]2− |
| 31 | GD2(d18:1/18:0) | 836.4515 | [M-2H+]2− |
| 32 | GD2(d18:1/20:0) | 850.4671 | [M-2H+]2− |
| 33 | Fuc-GM1(d18:1/18:3) | 863.9180 | [M-4H++2Na+]2− |
| 34 | GD1(d18:1/18:0) | 917.4782 | [M-2H+]2− |
| 35 | GD1(d18:1/20:0) | 922.4835 | [M-2H+-H2O]2− |
| 36 | GT3(d18:1/25:1) | 928.5061 | [M-2H+]2− |
| 37 | GD1(d18:1/20:0) | 931.4938 | [M-2H+]2− |
| 38 | GD1(d18:1/22:0) | 945.5081 | [M-2H+]2− |
| 39 | O-Ac-GD1(d18:1/20:1) | 951.4899 | [M-2H+]2− |
| 40 | GD1(d18:1/24:1) | 958.5155 | [M-2H+]2− |
| 41 | Fuc-GT3(d18:1/20:2) | 965.4874 | [M-2H+]2− |
| 42 | Fuc-GT3(d18:1/20:1) | 966.4971 | [M-2H+]2− |
| O-Ac- GD1(d18:1/22:0) | 966.4971 | [M-2H+]2− | |
| 43 | Fuc-GD1(d18:0/18:0) | 991.5089 | [M-2H+]2− |
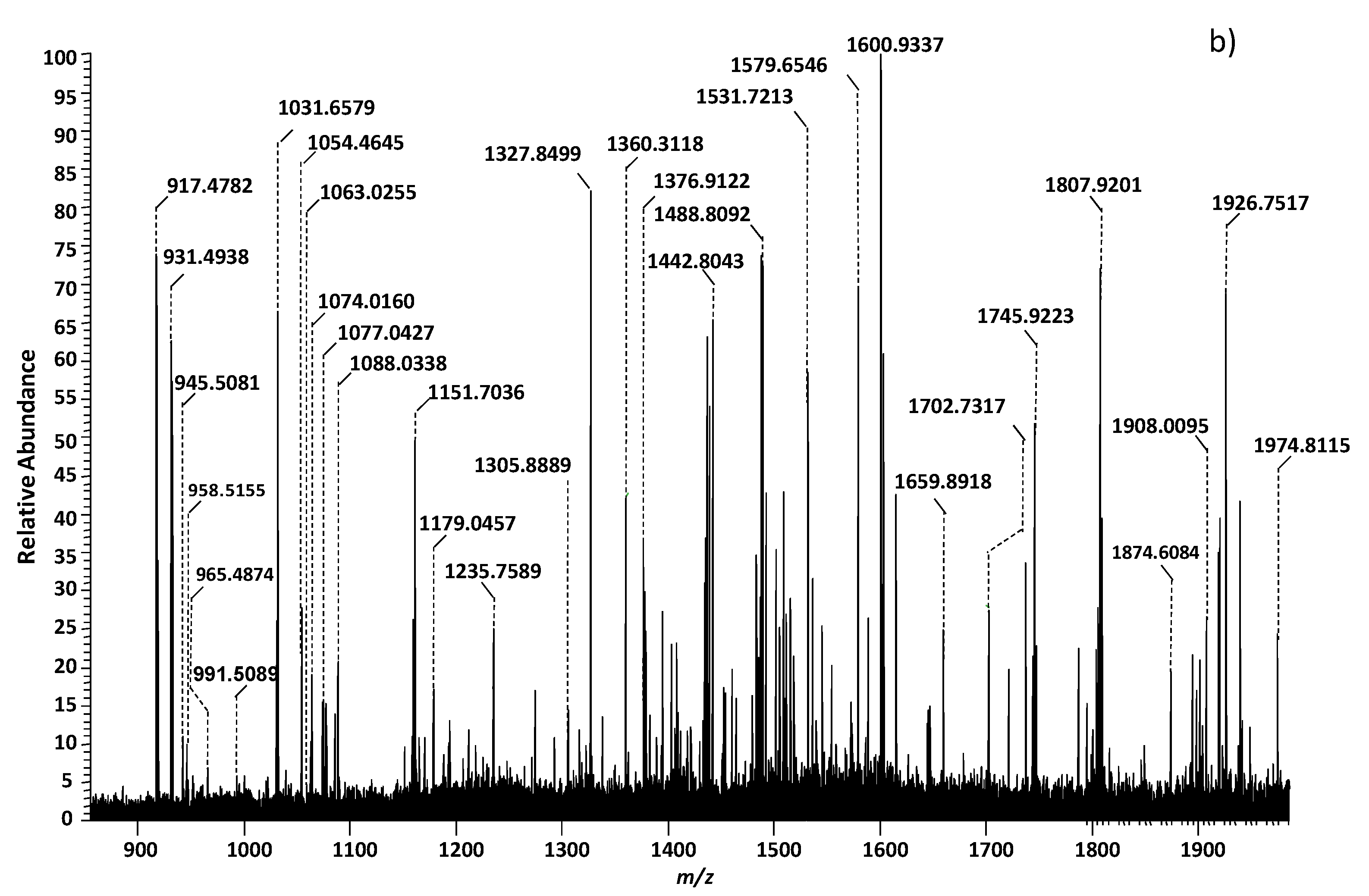
| 44 | O-Ac-GM4(d18:1/16:0) | 1031.6579 | [M-H+]− |
| 45 | GT1(d18:1/14:0) | 1034.9885 | [M-2H+]2− |
| 46 | GT1(d18:1/18:0) | 1063.0255 | [M-2H+]2− |
| 47 | GT1(d18:1/20:3) | 1074.0160 | [M-2H+]2− |
| GT1(d18:1/18:0) | 1074.0160 | [M-3H++Na+]2− | |
| 48 | GT1(d18:1/20:0) | 1077.0427 | [M-2H+]2− |
| 49 | GT1(d18:1/22:3) | 1088.0338 | [M-2H+]2− |
| GT1(d18:1/20:0) | 1088.0338 | [M-3H++Na+]2− | |
| 50 | GT1(d18:1/22:0) | 1091.0479 | [M-2H+]2− |
| 51 | GT1(d18:1/24:1) | 1115.0584 | [M-3H++Na+]2− |
| 52 | GM3(d18:0/16:0) | 1151.7036 | [M-H+]− |
| 53 | (CH3COO−)GalNAc-GT1(d18:1/16:2) | 1179.0457 | [M−-H+]2− |
| 54 | GA1(d18:1/18:0) | 1235.7589 | [M-H+ -H2O]− |
| 55 | GM3(d18:1/27:0) | 1305.8889 | [M-H+]− |
| 56 | GM3(d18:1/27:0) | 1327.8499 | [M-2H++Na+]− |
| 57 | GD3(d18:1/16:0) | 1442.8043 | [M-H+]− |
| 58 | GM1(d18:1/14:0) | 1488.8092 | [M-H+]− |
| 59 | GM1(d18:1/22:0) | 1600.9337 | [M-H+]− |
| 60 | GD2(d18:1/17:0) | 1659.8918 | [M-H+]− |
| 61 | GalNAc-GM1(d18:1/8:1) | 1745.9223 | [M-H+]− |
| 62 | GD1(d18:1/16:0) | 1807.9201 | [M-H+]− |
| m/z (Monoisotopic) Experimental | Molecular Ion | Proposed Structure |
|---|---|---|
| 874.91 | [M-H]− | LacCer(d18:1/17:0) |
| 932.99 | [M-H]− | LacCer(d18:0/21:0) |
| 947.19 | [M-H]− | LacCer(d18:0/22:0) |
| 949.24 | [M+2Na-3H]− | LacCer(d18:0/19:0) |
| 963.90 | [M-H]− | GM4(d18:0/14:0) |
| 981.94 | [M+Na-2H]− | GM4(d18:1/14:1) |
| 983.87 | [M+Na-2H]− | GM4(d18:1/14:0) or GM4(d18:0/14:1) |
| 1122.23 | [M-H]− | GA2(d18:0/20:0) |
| 1138.15 | [M-H]− | Fuc-GM4(d18:0/16:0) GA2(t18:0/20:0) |
| 1150.17 | [M-H]− | GA2(d18:1/21:0) GM3(d18:1/16:1) |
| 1168.01 | [M-H]− | GM3(t18:0/16:0) |
| 1178.14 | [M-H]− | GM3(d18:1/18:1) |
| 1180.10 | [M-H]− | GM3(d18:1/18:0) |
| 1182.21 | [M-H]− | GM3(d18:0/18:0) |
| 1184.08 | [M-H]− | O-Ac-GA1(d18:1/10:0) |
| 1194.15 | [M-H]− | GM3(d18:1/19:0) or GM3(d18:0/19:1) |
| 1206.33 | [M-H]− | GM3(d18:1/20:1) GA2(d18:0/26:0) |
| 1222.19 | [M-H]− | O-Ac-GM3(d18:1/18:0) GM3(d18:0/21:1) or GM3(d18:1/21:0) |
| 1234.22 | [M-H]− | GM3(d18:1/22:1) O-Ac- GM3(d18:1/19:1) |
| 1248.18 | [M-H]− | O-Ac-GM3(d18:1/20:1) GM3(d18:1/23:1) |
| 1249.02 | [M-H]− | GM3(d18:1/23:0) |
| 1260.33 | [M-H]− | GM3(d18:1/24:2) |
| 1262.35 | [M-H]− | GM3(d18:1/24:1) |
| 1264.19 | [M-H]− | GM3(d18:1/24:0) |
| 1277.01 | [M-H]− [M-H]− [M-H]−(-H2O) | O-Ac-GM3(d18:1/22:0) GM3(d20:1/23:1) GM3(d18:0/26:0) |
| 1278.21 | [M-H]− | GM3(d20:1/23:0) O-Ac-GM3(d18:1/22:0) or O-Ac-GM3(d18:0/22:1) |
| 1289. 04 | [M-H]− [M-H]− | GM3(d18:1/26:2) or GM3(d18:2/26:1) GM3(d20:1/24:2) |
| 1292.23 | [M-H]− | GM3(d18:1/26:0) or GM3(d18:0/26:1) |
| 1296.24 | [M-H]− | Fuc-GM3(d18:1/16:1) O-Ac-GA1(d18:1/18:0) GA1(d18:0/21:0) or GA1(d18:0/21:0) |
| 1405.21 | [M+Na-2H]− | GM2(d18:1/18:0) |
| 1420.80 | [M-H]− | O-Ac-GM2(d18:2/18:2) |
| 1435.21 | [M+Na-2H]− | GD3(d18:1/14:1) or GD3(d18:0/14:2) or GD3(d18:2/14:0) |
| 1441.19 | [M-H]− | GD3(d18:1/16:1) or GD3(d18:0/16:2) or GD3(d18:2/16:0) |
| 1471.28 | [M-H]− | GD3(d18:1/18:0) |
| 1493.23 | [M+Na-2H]− | GD3(d18:1/18:0) |
| 1515.29 | [M+2Na-3H]− [M-H]− | GD3(d18:1/18:0) or GD3(d18:0/18:1) GM1(d18:1/16:1) or GM1(d18:0/16:2) or GM1(d18:2/16:0) O-Ac-GD3(d18:0/18:0) |
| 1516.01 | [M+Na-2H]− [M-H]− | GD3(d18:1/20:2) or GD3(d18:0/20:3) or GD3(d18:2/20:1) GM1(d18:2/16:0) or GM1(d18:1/16:1) GM1(d18:0/16:2) |
| 1528. 16 | [M-H]− | GD3(d18:0/22:0) |
| 1542. 19 | [M-H]− | GM1(d18:1/18:2) or GM1(d18:2/18:1) or GM1(d18:0/18:3) |
| 1570.29 | [M+2Na-3H]− [M-H]− [M+Na-2H]− | GD3(d18:1/22:0) or GD3(d18:0/22:1) GM1(d18:1/20:1) or GM1(d18:0/20:2) or GM1(d18:2/20:0) GD3(d18:0/24:2) or GD3(d18:1/24:1) or GD3(d18:2/24:0) |
| 1598.09 | [M-H]− | GM1(d18:0/22:2) or GM1(d18:1/22:1) or GM1(d18:2/22:0) |
| 1612.17 | [M+2Na-3H]− | GM1(d18:1/20:2) |
| 1625.40 | [M+2Na-3H]− [M-H]− | GD3(d18:1/26:1) or GD3(d18:0/26:2) or GD3(d18:2/26:0) GM1(d18:1/24:2) |
| 1627.41 | [M+2Na-3H]− [M-H]− | GD3(d18:0/26:1) or GD3(d18:1/26:0) GM1(d18:0/24:2) or GM1(d18:1/24:1) or GM1(d18:2/24:0) |
| 1629.42 | [M-H]− [M-H]− | GM1(d18:0/24:1) or GM1(d18:1/24:0) di-O-Ac-GM1(d18:1/18:0) |
| 1660.18 | [M+3Na-4H]− | GM1(d18:1/22:3) or GM1(d18:0/22:4) or GM1(d18:2/22:2) |
| 1675.23 | [M+Na-2H]−(-H2O) | GD2 (d18:1/18:2) |
| 1749.39 | [M+Na-2H]− | GD2 (d18:1/22:1) |
| 1767.28 | [M-H]−(-H2O) | GT3 (d18:1/20:1) |
| 1785.37 | [M-H]− | O-Ac-GD2(d18:1/23:0) or O-Ac-GD2(d18:0/23:1) |
| 1833.28 | [M-H]− [M-H]− | GT3(d18:0/23:0) O-Ac-GT3 (d18:0/20:0) |
| 1861.24 | [M-H]− [M-H] (-H2O) | O-Ac-GT3-lactone(d18:0/22:0) O-Ac-GT3(d18:0/22:0) |
| 1879.39 | [M+Na-2H]− [M-H]−(-H2O) [M-H]− | O-Ac-GT3 (d18:2/22:1) Fuc-GT3(d18:0/17:0) GT2(d18:1/12:1) or GT2(d18:2/12:0) |
| 1909.03 | [M-H]− | GD1 (d18:1/22:0) |
| 1959.84 | [M-H]−(-2H2O) [M-H]− | GT2(d18:0/20:0) GT2(d18:1/18:3) or GT2(d18:2/18:2) |
| 1989.78 | [M+Na-2H]− [M-H]− | GT2(d18:0/18:0) GT2(d18:0/20:3) or GT2(d18:1/20:2) or GT2(d18:2/20:1) |
| 1990.83 | [M-H]− | GT2(d18:1/20:1) or GT2(d18:0/20:2) or GT2(d18:2/20:0) |
| 2005.63 | [M-H]− | Fuc-GD1(d18:1/20:2) O-Ac-GT2(d18:1/18:1) |
| 2048.80 | [M-H]− [M-H]−(-H2O) | di-O-Ac-GT2(d18:0/18:0) GT1(d18:2/14:2) or GT1(d18:3/14:1) |
| m/z (Monoisotopic) Experimental | Molecular Ion | Proposed Structure |
|---|---|---|
| 708.38 | [M-3H]3− | GT1(d18:1/18:0) |
| 714.40 | [M-3H]3− [M+Na-4H]3− | GT1(t18:0/18:0) GT1(d18:1/18:2) |
| 717.54 | [M-3H]3− | GT1(d18:1/20:0) or GT1(d18:0/20:1) |
| 735.12 | [M-2H]2− | GD3(d18:1/18:0) or GD3(d18:0/18:1) |
| 756.30 | [M-2H]2− | O-Ac-GD3(d18:1/18:0) |
| 771.93 | [M-2H]2− | GM1(d18:0/18:1) or GM1(d18:1/18:0) |
| 822.06 | [M+Na-4H]3− | GQ1(d18:1/20:0) or GQ1(d18:0/20:1) |
| 835.69 | [M-2H] 2− | GD2(d18:1/18:1) or GD2(d18:0/18:2) or GD2(d18:2/18:0) |
| 844.69 | [M-2H]2− | O-Ac-GD2(d18:0/16:0) |
| 850.22 | [M-2H]2− | GD2 (d18:1/20:0) |
| 863.21 | [M-2H]2− [M-2H]2− [M+Na-3H]2− | Fuc-GM1(d18:1/22:2) or Fuc-GM1(d18:0/22:3) or Fuc-GM1(d18:2/22:1) GD2(d18:0/22:3) or GD2(d18:1/22:2) or GD2(d18:2/22:1) GD2(d18:0/20:0) |
| 877.88 | [M-2H]2– | GD2(d18:1/24:1) |
| 885.78 | [M-2H]2− | O-Ac-GD2(d18:1/22:0) or O-Ac-GD2(d18:0/22:1) |
| 890.76 | [M+Na-3H]2− | GT3(d18:1/18:1) |
| 905.11 | [M+Na-3H]2− | GT3(d18:1/20:0) |
| 917.44 | [M-2H]2− | GD1(d18:1/18:0) or GD1(d18:0/18:1) |
| 924.76 | [M-2H]2– [M+2Na-4H]2− | GD1(d18:1/19:0) or O-Ac-GT3(t18:1/20:0) or O-Ac-GT3(d18:0/20:1) |
| 931.45 | [M-2H]2− | GD1(d18:1/20:0) or GD1(d18:0/20:1) |
| 940.46 | [M-2H]2– [M+2Na-4H]2− | GD1(t18:0/20:0) GD1 (d18:1/18:0) or GD1(d18:0/18:1) |
| 945.50 | [M-2H]2− | GD1(d18:1/22:0) |
| 952.50 | [M-2H]2− | O-Ac-GD1 (d18:1/20:0) or O-Ac-GD1 (d18:0/20:1) |
| 968.34 | [M-3H]3− | GH2(d18:1/24:0) or GH2(d18:0/24:1) |
| 991.27 | [M+Na-3H]2− | GT2(d18:1/18:2) or GT2(d18:0/18:3) or GT2(d18:2/18:3) |
| 1005.28 | [M+2Na-4H]2− | GT2 (d18:0/18:0) |
| 1019.36 | [M-2H]2– | GalNAc-GD1(d18:0/18:0) |
| 1024.68 | [M-2H]2– [M+2Na-4H]2− | di-O-Ac-GT2 (d18:1/18:0) O-Ac-GT2 (d18:1/18:1) |
| 1033.94 | [M+2Na-3H]− | GM4 (d18:1/16:0) or GM4 (d18:0/16:1) |
| 1042.51 | [M-2H]2 | GT1 (t18:1/14:1) or GalNAc-GD1(t18:0/20:0) |
| 1046.46 | [M+Na-3H]2− | GT1(d18:1/14:0) or GT1(d18:0/14:1) |
| 1049.51 | [M-2H]2− | GT1(d18:1/16:0) or GT1(d18:0/16:1) |
| 1059.28 | [M+Na-3H]2− | GT1(d18:1/16:1) |
| 1063. 35 | [M-2H]2− | GT1(d18:1/18:0) or GT1(d18:0/18:1) |
| 1074.05 | [M+Na-3H]2− | GT1(d18:1/18:0) |
| 1077.37 | [M-2H]2− | GT1(d18:1/20:0) |
| 1096.81 | M-2H]2− [M+Na-3H]2− | O-Ac-GT1(d18:1/20:1) or O-Ac-GT1(d18:0/20:2) or O-Ac-GT1(d18:2/20:0) GT1(t18:0/20:0) |
| 1110.36 | [M-2H]2− | O-Ac-GT1(d18:1/22:2) |
| 1118.56 | [M-H]− | GM4(t18:1/24:0) |
| 1180.09 | [M-H]− | GM3(d18:1/18:0) or GM3(d18:0/18:1) |
| 1228.61 | [M-H]− [M+Na-2H]− | GA1(d18:0/16:0) GM3(d18:1/20:1) or GM3(d18:0/20:2) |
| 1232.12 | [M-H]− [M+Na-2H]− | GM3(d18:1/22:2) or GM3(d18:0/22:3) or GM3(d18:2/22:1) GM3(d18:0/20:0) |
| 1240.86 | [M+Na-3H]2− | O-Ac-GQ1(d18:1/18:0) |
| 1252.19 | [M-H]− | O-Ac-GM3(d18:0/20:0) GM3 (d18:0/23:0) |
| 1264.12 | [M-H]− | di-O-Ac-GM3(d18:1/18:0) |
| 1284.36 | [M+Na-2H]− | GM3(d18:1/24:1) |
| 1382.87 | [M-H]− | GM2(d18:1/18:0) or GM2(d18:0/18:1) |
| 1410.19 | [M-H]− | GM2(d18:1/20:0) or GM2(d18:0/20:1) |
| 1467.86 | [M-H]− [M+Na-2H]− | GD3(d18:1/18:2) GD3(d18:0/16:0) |
| 1492.89 | [M+Na-2H]− | GD3(d18:1/18:0) |
| 1509.71 | [M-H]− | O-Ac-GD3(d18:1/18:1) Fuc-GM2-lactone (d18:1/18:1) |
| 1513.29 | [M-H]− | O-Ac-GD3(d18:1/18:0) |
| 1517.30 | [M-H]− [M+Na-2H]− [M+Na-2H]− | GM1(d18:1/16:0) GD3(d18:2/20:2) Fuc-GM2(d18:2/16:2) |
| 1537.26 | [M+Na-2H]− | GM1(d18:1/16:1) |
| 1541.21 | [M+2Na-3H]− | GD3(d18:1/20:0) |
| 1544.92 | [M-H]− | GM1(d18:1/18:0) or GM1(d18:0/18:1) |
| 1561.36 | [M-H]− | O-Ac-GM1(d18:0/16:0) |
| 1566.68 | [M+Na-2H]− | GM1(d18:1/18:0) or GM1(d18:0/18:1) |
| 1572.92 | [M-H]− | GM1(d18:1/20:0) or GM1(d18:0/20:1) |
| 1589.28 | [M-H]− | Fuc-GD3(d18:1/16:0) |
| 1594.16 | [M+Na-2H]− | GM1(d18:1/20:0) |
| 1600.23 | [M-H]− | GM1(d18:0/22:0) |
| 1629.32 | [M-H]− | di-O-Ac-GM1(d18:1/18:0) |
| 1648.32 | [M-H]− | GD2(d18:0/16:0) |
| 1656.88 | [M-H]− | GD2-lactone (d18:1/18:0) |
| 1663.16 | [M+Na-2H]− | GD2(d18:2/16:2) |
| 1674.33 | [M-H]− | GD2(d18:1/18:0) or GD2(d18:0/18:1) |
| 1690.95 | [M-H]− | Fuc-GM1(d18:1/18:0) |
| 1701.42 | [M-H]− | GD2(d18:1/20:0) or GD2(d18:0/20:1) |
| 1709.36 | [M+Na-2H]− | O-Ac-GD2 (d18:1/16:1) |
| 1716.91 | [M-H]− | Fuc-GM1 (d18:1/20:1) |
| 1718.48 | [M-H]− | Fuc-GM1 (d18:1/20:0) |
| 1729.62 | [M-H]− | GD2(d18:1/22:0) or GD2(d18:0/22:1) |
| 1741.27 | [M+Na-2H]− | Fuc-GM1 (d18:0/20:0) |
| 1746.38 | [M+2Na-3H]− | GD2(d18:0/20:0) |
| 1775.57 | [M+Na-2H]− | GD2(d18:2/24:3) |
| 1787.76 | [M+2Na-3H]− | Fuc-GM1(d18:1/22:1) |
| 1796.50 | [M+Na-2H]− | Fuc-GM1(d18:1/24:0) |
| 1803.64 | [M-H]− | O-Ac-GT3(d18:1/18:1) |
| 1835.91 | [M-H]− | GD1(d18:1/18:0) or GD1(d18:0/18:1) |
| 1857.59 | [M+Na-2H]− | GD1(d18:0/18:0) |
| 1863.61 | [M-H]− | GD1(d18:1/20:0) |
| 1872.68 | [M+Na-2H]− | GD1(d18:1/19:0) |
| 1879.91 | [M+2Na-3H]− | GD1(d18:1/18:0) |
| 1886.98 | [M-H]− | Fuc-GT3-lactone(d18:1/18:2) Fuc-GT3-lactone(d18:0/18:3) Fuc-GT3-lactone(d18:2/18:1) |
| 1896.06 | [M+2Na-3H]− | GD1(d18:0/19:0) |
| 1901.71 | [M+2Na-3H]− | O-Ac-GT3(d18:1/22:1) |
| 1909.43 | [M-H]− | GT2(d18:0/14:0) or GT2(d18:0/14:1) |
| 1915.78 | [M-H]− | GD1(d18:1/24:2) |
| 1925.77 | [M+Na-2H]− | GD1(d18:1/23:1) |
| 1937.80 | [M+Na-2H]− | GD1(d18:1/24:2) or GD1(d18:0/24:3) or GD1(d18:2/24:1) |
| 1964.84 | [M-H]− | GT2(d18:0/18:0) |
| 1982.55 | [M-H]− | Fuc-GD1(d18:1/18:0) |
| 2004.61 | [M-H]− | Fuc-GD1(d18:1/20:2) |
| 2010.78 | [M+Na-2H]− | GT2(d18:1/20:2) or GT2(d18:0/20:3) or GT2(d18:2/20:1) |
| 2032.63 | [M-H]− [M+2Na-3H]− | O-Ac-GT2(d18:1/20:1) or O-Ac-GT2(d18:0/20:2) or O-Ac-GT2(d18:2/20:0) GT2(d18:1/20:2) or GT2(d18:0/20:3) or GT2(d18:2/20:1) |
| 2049.75 | [M-H]− | di-O-Ac-GT2 (d18:1/18:0) |
| 2059.78 | [M+Na-2H]− | Fuc-GD1(d18:1/22:0) |
| 2076.85 | [M-H]− | GQ3(d18:1/20:2) or GQ3(d18:0/20:3) or GQ3(d18:2/20:1) |
| 2105.78 | [M-H]−(-H2O) | GT1(d18:1/18:2) |
| 2165.56 | [M-H]− [M+Na-2H]− | O-Ac-GT1(d18:1/18:2) or O-Ac-GT1(d18:0/18:3) or O-Ac-GT1(d18:2/18:1) GT1(t18:1/18:0) O-Ac-GT1(d18:1/16:0) |
| 2172.04 | [M+2Na-3H]− | O-Ac-GT1(t18:1/14:1) |
| 2187.62 | [M+Na-2H]−(-H2O) [M+Na-2H]− | GT1(d18:1/22:0) GT1(d18:1/21:1) |
| 2198.08 | [M+2Na-3H]− | GT1(d18:1/20:0) |
| 2215.78 | [M+Na-2H]− | O-Ac-GT1(d18:1/20:2) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sarbu, M.; Ica, R.; Zamfir, A.D. Gangliosides as Biomarkers of Human Brain Diseases: Trends in Discovery and Characterization by High-Performance Mass Spectrometry. Int. J. Mol. Sci. 2022, 23, 693. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23020693
Sarbu M, Ica R, Zamfir AD. Gangliosides as Biomarkers of Human Brain Diseases: Trends in Discovery and Characterization by High-Performance Mass Spectrometry. International Journal of Molecular Sciences. 2022; 23(2):693. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23020693
Chicago/Turabian StyleSarbu, Mirela, Raluca Ica, and Alina D. Zamfir. 2022. "Gangliosides as Biomarkers of Human Brain Diseases: Trends in Discovery and Characterization by High-Performance Mass Spectrometry" International Journal of Molecular Sciences 23, no. 2: 693. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23020693