
Regulation of the Receptor Tyrosine Kinase AXL in Response to Therapy and Its Role in Therapy Resistance in Glioblastoma

Abstract
:1. Introduction
2. Results
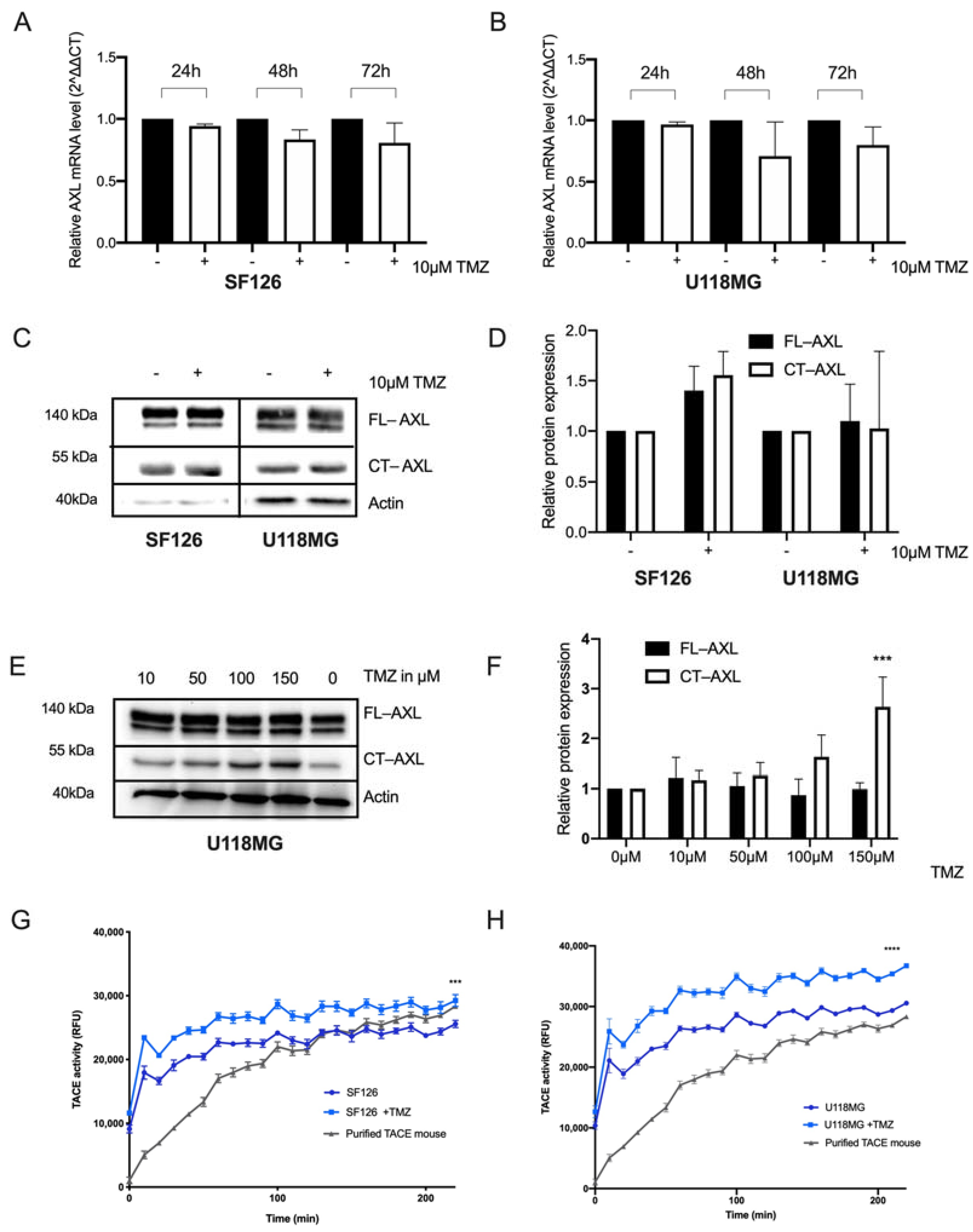
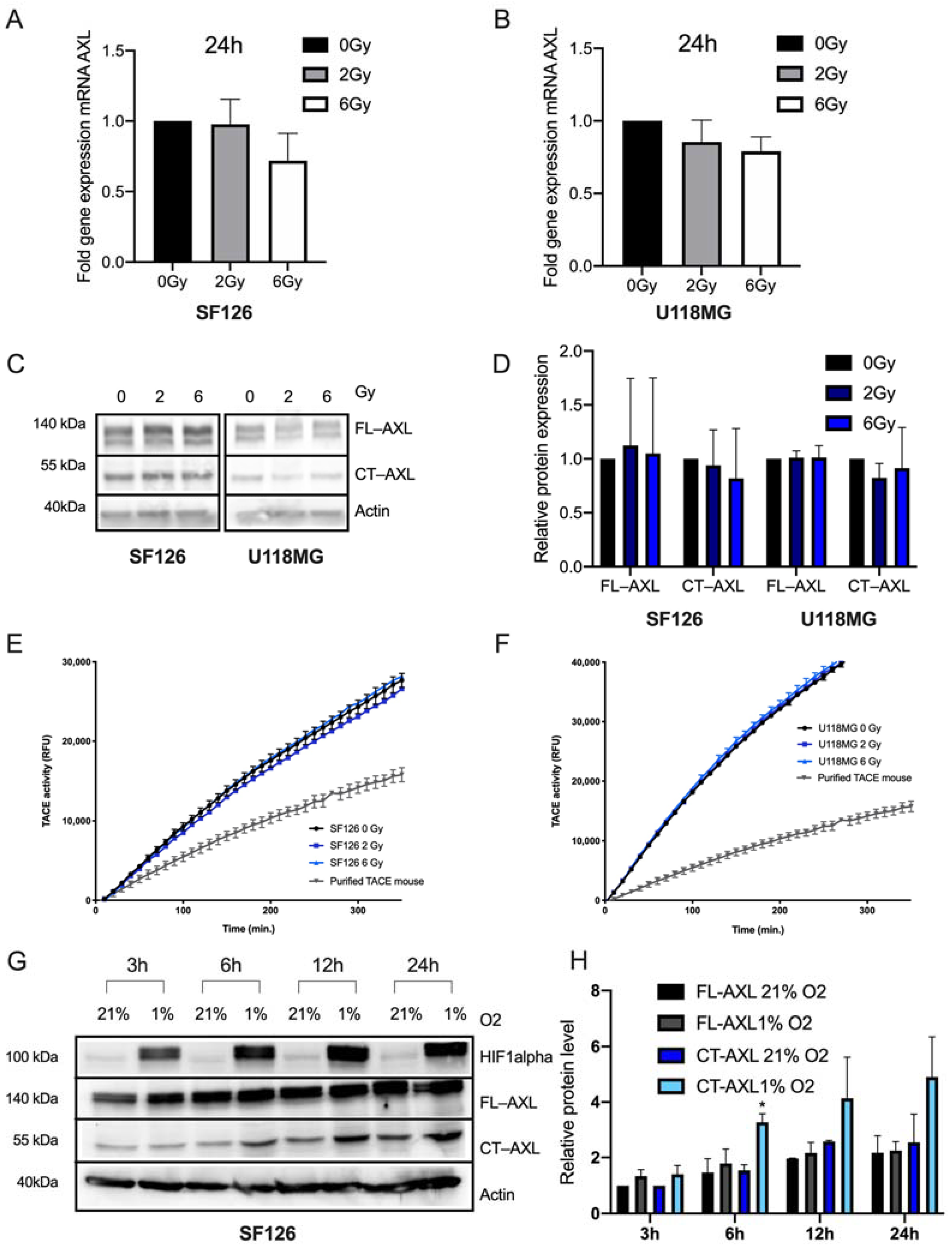
2.1. RTK-AXL Undergoes Posttranlational Receptor Modification in Response to Therapy
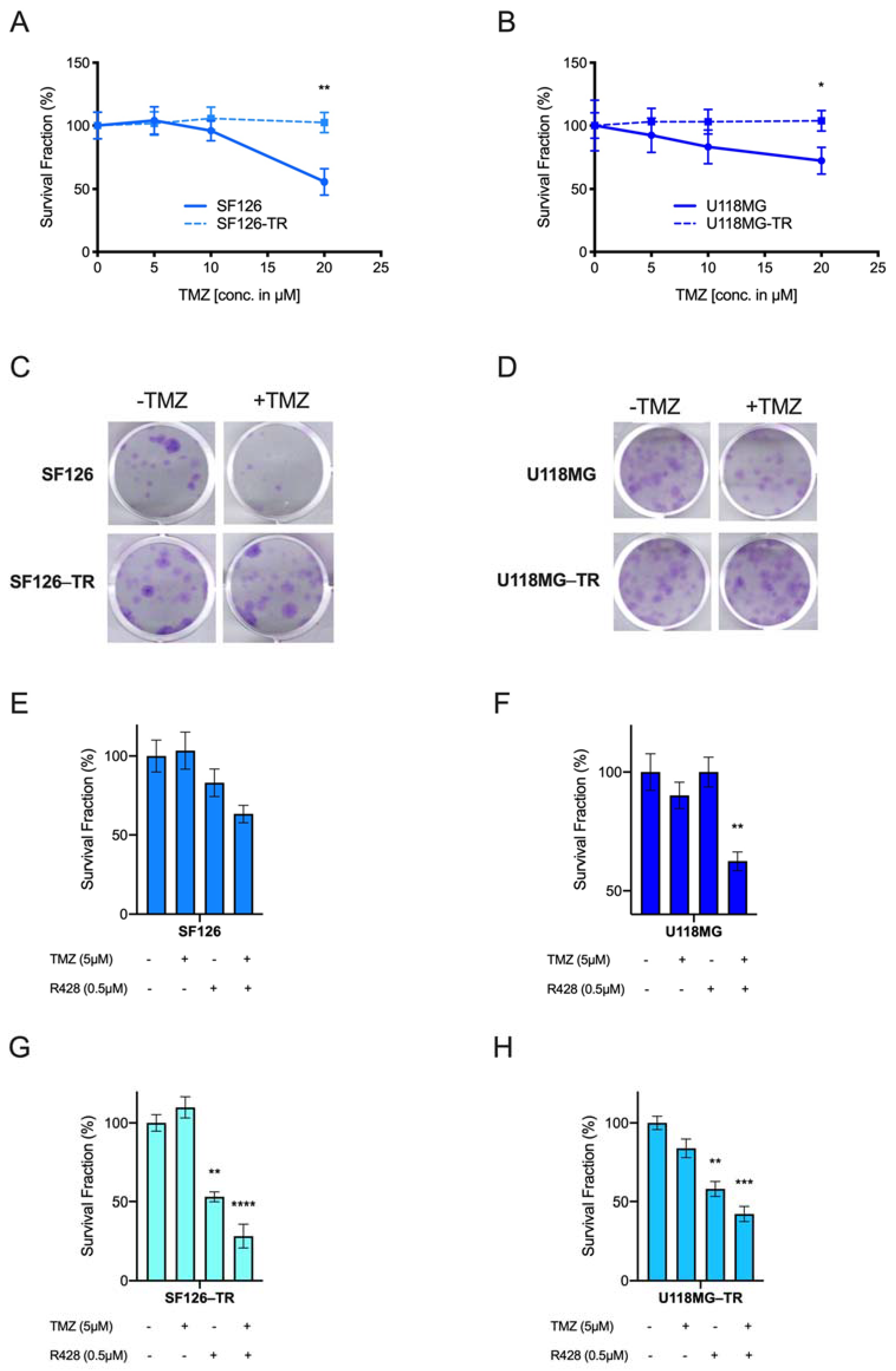
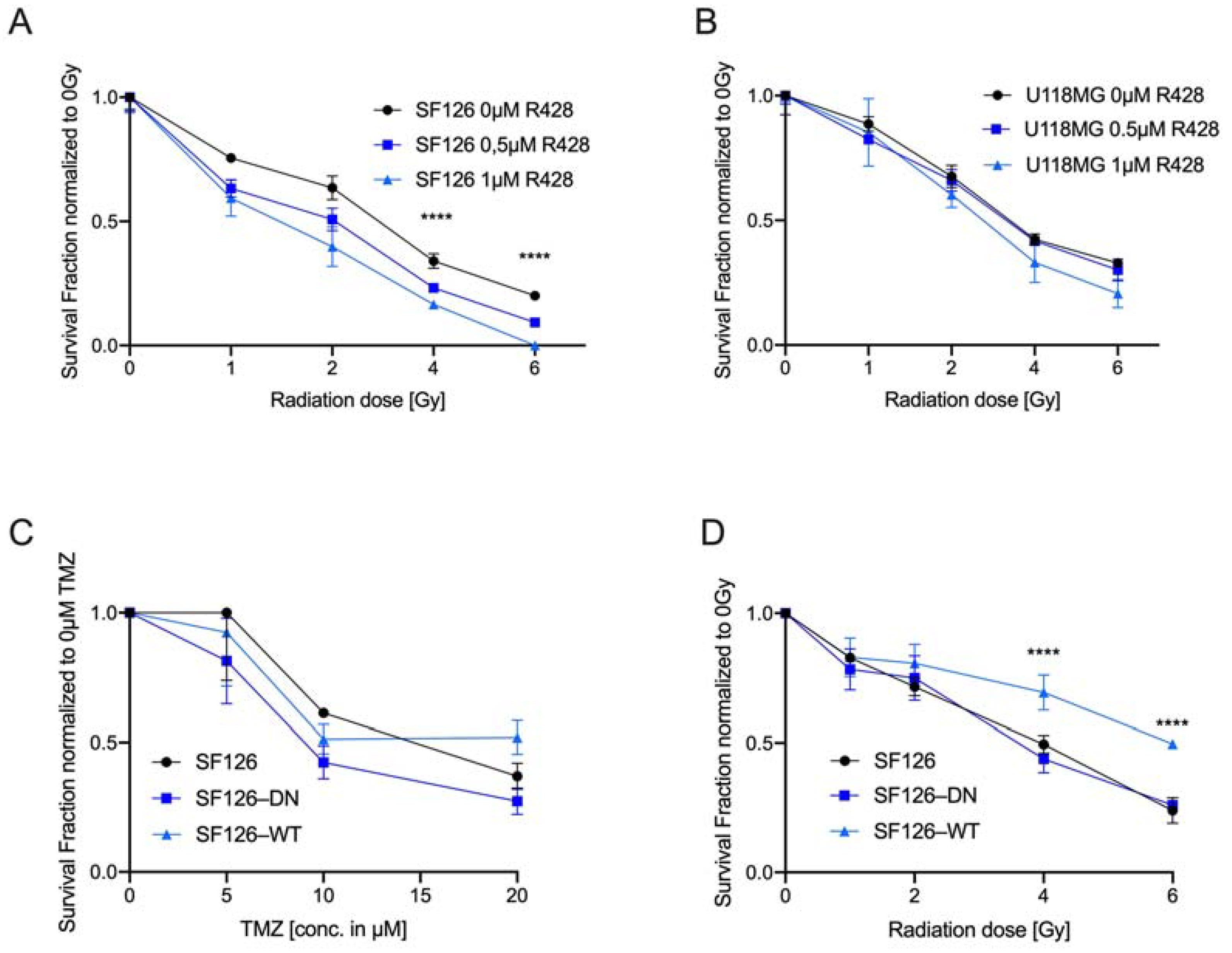
2.2. AXL TKI R428 Combined with Standard Therapy Increases Efficacy of TMZ and Radiation
2.3. Endogenous RTK-AXL Expression Is Related to Radiation Sensitivity
3. Discussion
4. Material and Methods
4.1. Cell Culture
4.2. Colony Formation Assay (CFA)
4.3. Quantitative Real-Time PCR
4.4. Western Blot
4.5. TACE Assay
4.6. Protein Phospho-Kinase Array
4.7. Temozolomide (TMZ) Treatment in Cell Culture
4.8. Radiation Protocol for Cell Culture
4.9. Tyrosine Kinase Inhibitor (TKI) R428 Treatment in Cell Culture
4.10. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.M.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma: A Randomized Clinical Trial. JAMA 2017, 318, 2306–2316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grisanti, S.; Ferrari, V.D.; Buglione, M.; Agazzi, G.M.; Liserre, R.; Poliani, L.; Buttolo, L.; Gipponi, S.; Pedersini, R.; Consoli, F.; et al. Second line treatment of recurrent glioblastoma with sunitinib: Results of a phase II study and systematic review of literature. J. Neurosurg. Sci. 2019, 63, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Liu, X.D.; Sun, M.; Zhang, X.; German, P.; Bai, S.; Ding, Z.; Tannir, N.M.; Wood, C.G.; Matin, S.F.; et al. Targeting MET and AXL overcomes resistance to sunitinib therapy in renal cell carcinoma. Oncogene 2016, 35, 2687–2697. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, G.; De Salvo, G.L.; Brandes, A.A.; Eoli, M.; Rudà, R.; Faedi, M.; Lolli, I.; Pace, A.; Daniele, B.; Pasqualetti, F.; et al. Regorafenib compared with lomustine in patients with relapsed glioblastoma (REGOMA): A multicentre, open-label, randomised, controlled, phase 2 trial. Lancet Oncol. 2019, 20, 110–119. [Google Scholar] [CrossRef]
- Eskilsson, E.; Røsland, G.V.; Solecki, G.; Wang, Q.; Harter, P.N.; Graziani, G.; Verhaak, R.G.W.; Winkler, F.; Bjerkvig, R.; Miletic, H. EGFR heterogeneity and implications for therapeutic intervention in glioblastoma. Neuro-Oncol. 2017, 20, 743–752. [Google Scholar] [CrossRef] [Green Version]
- Scaltriti, M.; Elkabets, M.; Baselga, J. Molecular Pathways: AXL, a Membrane Receptor Mediator of Resistance to Therapy. Clin. Cancer Res. 2016, 22, 1313–1317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardone, C.; Blauensteiner, B.; Moreno-Viedma, V.; Martini, G.; Simeon, V.; Vitiello, P.P.; Ciardiello, D.; Belli, V.; Matrone, N.; Troiani, T.; et al. AXL is a predictor of poor survival and of resistance to anti-EGFR therapy in RAS wild-type metastatic colorectal cancer. Eur. J. Cancer 2020, 138, 1–10. [Google Scholar] [CrossRef]
- Colavito, S.A. AXL as a Target in Breast Cancer Therapy. J. Oncol. 2020, 2020, 5291952. [Google Scholar] [CrossRef] [Green Version]
- Meyer, A.S.; Miller, M.A.; Gertler, F.B.; Lauffenburger, D.A. The Receptor AXL Diversifies EGFR Signaling and Limits the Response to EGFR-Targeted Inhibitors in Triple-Negative Breast Cancer Cells. Sci. Signal. 2013, 6, ra66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Lee, J.C.; Lin, L.; Olivas, V.; Au, V.; LaFramboise, T.; Abdel-Rahman, M.; Wang, X.; Levine, A.D.; Rho, J.K.; et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat. Genet. 2012, 44, 852–860. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Siemann, D.W. Gas6/Axl Signaling Pathway in the Tumor Immune Microenvironment. Cancers 2020, 12, 1850. [Google Scholar] [CrossRef]
- Nakada, M.; Kita, D.; Teng, L.; Pyko, I.V.; Watanabe, T.; Hayashi, Y.; Hamada, J.-I. Receptor Tyrosine Kinases: Principles and Functions in Glioma Invasion. Adv. Exp. Med. Biol. 2020, 1202, 151–178. [Google Scholar]
- Korshunov, V.A. Axl-dependent signalling: A clinical update. Clin. Sci. 2012, 122, 361–368. [Google Scholar] [CrossRef] [Green Version]
- Varnum, B.C.; Young, C.; Elliott, G.; Garcia, A.; Bartley, T.D.; Fridell, Y.-W.; Hunt, R.W.; Trail, G.; Clogston, C.; Toso, R.J.; et al. Axl receptor tyrosine kinase stimulated by the vitamin K-dependent protein encoded by growth-arrest-specific gene 6. Nature 1995, 373, 623–626. [Google Scholar] [CrossRef]
- Onken, J.; Vajkoczy, P.; Torka, R.; Hempt, C.; Patsouris, V.; Heppner, F.; Radke, J. Phospho-AXL is widely expressed in glioblastoma and associated with significant shorter overall survival. Oncotarget 2017, 8, 50403–50414. [Google Scholar] [CrossRef] [Green Version]
- Vajkoczy, P.; Knyazev, P.; Kunkel, A.; Capelle, H.-H.; Behrndt, S.; von Tengg-Kobligk, H.; Kiessling, F.; Eichelsbacher, U.; Essig, M.; Read, T.-A.; et al. Dominant-negative inhibition of the Axl receptor tyrosine kinase suppresses brain tumor cell growth and invasion and prolongs survival. Proc. Natl. Acad. Sci. USA 2006, 103, 5799–5804. [Google Scholar] [CrossRef] [Green Version]
- Onken, J.; Torka, R.; Korsing, S.; Radke, J.; Krementeskaia, I.; Nieminen, M.; Bai, X.; Ullrich, A.; Heppner, F.; Vajkoczy, P. Inhibiting receptor tyrosine kinase AXL with small molecule inhibitor BMS-777607 reduces glioblastoma growth, migration, and invasion in vitro and in vivo. Oncotarget 2016, 7, 9876–9889. [Google Scholar] [CrossRef] [Green Version]
- Hutterer, M.; Knyazev, P.; Abate, A.; Reschke, M.; Maier, H.; Stefanova, N.; Knyazeva, T.; Barbieri, V.; Reindl, M.; Muigg, A.; et al. Axl and Growth Arrest Specific Gene 6 Are Frequently Overexpressed in Human Gliomas and Predict Poor Prognosis in Patients with Glioblastoma Multiforme. Clin. Cancer Res. 2008, 14, 130–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Bailey, C.P.; Sadighi, Z.; Zaky, W.; Chandra, J. Pediatric high-grade glioma: Aberrant epigenetics and kinase signaling define emerging therapeutic opportunities. J. Neuro-Oncol. 2020, 150, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Song, Q.; Yu, Q. Axl inhibitor R428 induces apoptosis of cancer cells by blocking lysosomal acidification and recycling independent of Axl inhibition. Am. J. Cancer Res. 2018, 8, 1466–1482. [Google Scholar]
- Flem-Karlsen, K.; Nyakas, M.; Farstad, I.N.; McFadden, E.; Wernhoff, P.; Jacobsen, K.D.; Flørenes, V.A.; Mælandsmo, G.M. Soluble AXL as a marker of disease progression and survival in melanoma. PLoS ONE 2020, 15, e0227187. [Google Scholar] [CrossRef]
- Vouri, M.; An, Q.; Birt, M.; Pilkington, G.J.; Hafizi, S. Small molecule inhibition of Axl receptor tyrosine kinase potently suppresses multiple malignant properties of glioma cells. Oncotarget 2015, 6, 16183–16197. [Google Scholar] [CrossRef] [Green Version]
- Myers, S.H.; Brunton, V.G.; Unciti-Broceta, A. AXL Inhibitors in Cancer: A Medicinal Chemistry Perspective. J. Med. Chem. 2016, 59, 3593–3608. [Google Scholar] [CrossRef] [Green Version]
- Miller, M.A.; Oudin, M.J.; Sullivan, R.J.; Wang, S.J.; Meyer, A.S.; Im, H.; Frederick, D.T.; Tadros, J.; Griffith, L.G.; Lee, H.; et al. Reduced Proteolytic Shedding of Receptor Tyrosine Kinases Is a Post-Translational Mechanism of Kinase Inhibitor Resistance. Cancer Discov. 2016, 6, 382–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.-H.; Yoo, K.-C.; Cui, Y.-H.; Uddin, N.; Lim, E.-J.; Kim, M.-J.; Nam, S.-Y.; Kim, I.-G.; Suh, Y.; Lee, S.-J. Radiation promotes malignant progression of glioma cells through HIF-1alpha stabilization. Cancer Lett. 2014, 354, 132–141. [Google Scholar] [CrossRef]
- Aebersold, D.M.; Burri, P.; Beer, K.T.; Laissue, J.; Djonov, V.; Greiner, R.H.; Semenza, G.L. Expression of hypoxia-inducible factor-1alpha: A novel predictive and prognostic parameter in the radiotherapy of oropharyngeal cancer. Cancer Res. 2001, 61, 2911–2916. [Google Scholar] [PubMed]
- Jensen, R.L. Hypoxia in the tumorigenesis of gliomas and as a potential target for therapeutic measures. Neurosurg. Focus 2006, 20, E24. [Google Scholar] [CrossRef]
- Miller, M.A.; Sullivan, R.J.; Lauffenburger, D.A. Molecular Pathways: Receptor Ectodomain Shedding in Treatment, Resistance, and Monitoring of Cancer. Clin. Cancer Res. 2016, 23, 623–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horsman, M.R.; Overgaard, J. The impact of hypoxia and its modification of the outcome of radiotherapy. J. Radiat. Res. 2016, 57 (Suppl. S1), i90–i98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holstein, E.; Binder, M.; Mikulits, W. Dynamics of Axl Receptor Shedding in Hepatocellular Carcinoma and Its Implication for Theranostics. Int. J. Mol. Sci. 2018, 19, 4111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Tetering, G.; Vooijs, M. Proteolytic cleavage of Notch: “HIT and RUN”. Curr. Mol. Med. 2011, 11, 255–269. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.; Thorp, E.B.; Doran, A.C.; Subramanian, M.; Sansbury, B.E.; Lin, C.-S.; Spite, M.; Fredman, G.; Tabas, I. MerTK cleavage limits proresolving mediator biosynthesis and exacerbates tissue inflammation. Proc. Natl. Acad. Sci. USA 2016, 113, 6526–6531. [Google Scholar] [CrossRef] [Green Version]
- McDaniel, N.K.; Iida, M.; Nickel, K.P.; Longhurst, C.A.; Fischbach, S.R.; Rodems, T.; Kranjac, C.A.; Bo, A.Y.; Luo, Q.; Gallagher, M.M.; et al. AXL Mediates Cetuximab and Radiation Resistance Through Tyrosine 821 and the c-ABL Kinase Pathway in Head and Neck Cancer. Clin. Cancer Res. 2020, 26, 4349–4359. [Google Scholar] [CrossRef]
- Hong, J.; Peng, D.; Chen, Z.; Sehdev, V.; Belkhiri, A. ABL Regulation by AXL Promotes Cisplatin Resistance in Esophageal Cancer. Cancer Res. 2013, 73, 331–340. [Google Scholar] [CrossRef] [Green Version]
- Cho, C.-Y.; Huang, J.-S.; Shiah, S.-G.; Chung, S.-Y.; Lay, J.-D.; Yang, Y.-Y.; Lai, G.-M.; Cheng, A.-L.; Chen, L.-T.; Chuang, S.-E. Negative feedback regulation of AXL by miR-34a modulates apoptosis in lung cancer cells. RNA 2016, 22, 303–315. [Google Scholar] [CrossRef] [Green Version]
- Aguilera, T.A.; Rafat, M.; Castellini, L.; Shehade, H.; Kariolis, M.S.; Hui, A.B.-Y.; Stehr, H.; Von Eyben, R.; Jiang, D.; Ellies, L.G.; et al. Reprogramming the immunological microenvironment through radiation and targeting Axl. Nat. Commun. 2016, 7, 13898. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Bao, S.; Wu, Q.; Wang, H.; Eyler, C.; Sathornsumetee, S.; Shi, Q.; Cao, Y.; Lathia, J.; McLendon, R.E.; et al. Hypoxia-Inducible Factors Regulate Tumorigenic Capacity of Glioma Stem Cells. Cancer Cell 2009, 15, 501–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, E.; Zhang, C.; Polavaram, N.; Liu, F.; Wu, G.; Schroeder, M.A.; Lau, J.S.; Mukhopadhyay, D.; Jiang, S.-W.; O’Neill, B.P.; et al. The Role of Factor Inhibiting HIF (FIH-1) in Inhibiting HIF-1 Transcriptional Activity in Glioblastoma Multiforme. PLoS ONE 2014, 9, e86102. [Google Scholar] [CrossRef] [Green Version]
- Roux, K.H. Optimization and Troubleshooting in PCR. Cold Spring Harb. Protoc. 2009, 2009, pdb.ip66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinn, B.; Tallen, G.; Schroeder, G.; Grassl, B.; Schulze, J.; Budach, V.; Tinhofer, I. Caffeine Confers Radiosensitization of PTEN-Deficient Malignant Glioma Cells by Enhancing Ionizing Radiation–Induced G1 Arrest and Negatively Regulating Akt Phosphorylation. Mol. Cancer Ther. 2010, 9, 480–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Hyun, W.; Lamborn, K.; Deen, D.F. Measurement of radiation-induced damage in human glioma cells with flow cytometry. Cancer Res. 1996, 56, 154–157. [Google Scholar] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Forward Primer 5′-3′ | Reverse Primer 5′-3′ |
|---|---|---|
| hAXL | GTGGGCAACCCAGGGAATATC | GTACTGTCCCGTGTCGGAAAG |
| mAXL | ATGGCCGACATTGCCAGTG | CGGTAGTAATCCCCGTTGTAGA |
| h18s | CATGGCCGTTCTTAGTTGGT | CGCTGAGCCAGTCAGTGTAG |
| m18s | AACCCGTTGAACCCCATT | CCATCCAATCGGTAGTAGCG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scherschinski, L.; Prem, M.; Kremenetskaia, I.; Tinhofer, I.; Vajkoczy, P.; Karbe, A.-G.; Onken, J.S. Regulation of the Receptor Tyrosine Kinase AXL in Response to Therapy and Its Role in Therapy Resistance in Glioblastoma. Int. J. Mol. Sci. 2022, 23, 982. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23020982
Scherschinski L, Prem M, Kremenetskaia I, Tinhofer I, Vajkoczy P, Karbe A-G, Onken JS. Regulation of the Receptor Tyrosine Kinase AXL in Response to Therapy and Its Role in Therapy Resistance in Glioblastoma. International Journal of Molecular Sciences. 2022; 23(2):982. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23020982
Chicago/Turabian StyleScherschinski, Lea, Markus Prem, Irina Kremenetskaia, Ingeborg Tinhofer, Peter Vajkoczy, Anna-Gila Karbe, and Julia Sophie Onken. 2022. "Regulation of the Receptor Tyrosine Kinase AXL in Response to Therapy and Its Role in Therapy Resistance in Glioblastoma" International Journal of Molecular Sciences 23, no. 2: 982. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23020982