
Allosteric Modulation of Adenosine A2A Receptors as a New Therapeutic Avenue


1
Department of Neurology, Division of Sleep Medicine, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA 02215, USA
2
Molecular Neuroscience Unit, Okinawa Institute of Science and Technology Graduate University, Kunigami-gun, Onna 904-0412, Japan
3
International Institute for Integrative Sleep Medicine (WPI-IIIS), University of Tsukuba, Tsukuba 305-8575, Japan
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2022, 23(4), 2101; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23042101
Submission received: 18 January 2022
/
Revised: 11 February 2022
/
Accepted: 11 February 2022
/
Published: 14 February 2022
(This article belongs to the Special Issue Molecular Circuits Regulating Sleep and Wakeful Consciousness)
Abstract
:The therapeutic potential of targeting adenosine A2A receptors (A2ARs) is immense due to their broad expression in the body and central nervous system. The role of A2ARs in cardiovascular function, inflammation, sleep/wake behaviors, cognition, and other primary nervous system functions has been extensively studied. Numerous A2AR agonist and antagonist molecules are reported, many of which are currently in clinical trials or have already been approved for treatment. Allosteric modulators can selectively elicit a physiologic response only where and when the orthosteric ligand is released, which reduces the risk of an adverse effect resulting from A2AR activation. Thus, these allosteric modulators have a potential therapeutic advantage over classical agonist and antagonist molecules. This review focuses on the recent developments regarding allosteric A2AR modulation, which is a promising area for future pharmaceutical research because the list of existing allosteric A2AR modulators and their physiologic effects is still short.
1. Introduction
Adenosine is a naturally occurring purine nucleoside that regulates various physiologic functions, including inflammation and wound healing, cardiac contraction, blood vessel formation, vasodilation, learning, memory, sleep, and arousal [1,2,3,4,5,6,7]. Adenosine is released by neurons and glial cells [8]. Extracellular adenosine modulates neuronal excitability, synaptic plasticity, and the release and reuptake of several neurotransmitters [9,10,11,12]. The effects of extracellular adenosine are modulated via four subtypes of G-protein coupled adenosine receptors (GPCRs), denoted A1, A2A, A2B, and A3 [13]. Adenosine A2A receptors (A2ARs) are broadly expressed in the brain, cardiovascular system, blood vessels, spleen, thymus, leukocytes, and lung, making them an important drug target [14]. This review focuses on allosteric A2AR modulation and the latest developments in this emerging field.
The therapeutic potential of targeting A2ARs has prompted the development of numerous antagonist and agonist molecules to selectively control A2AR function. The myriad A2AR agonists and antagonists are considered potential therapeutic agents for inflammation, sickle cell disease, ischemia-reperfusion injury, and central nervous system (CNS) diseases [15,16]. The A2AR agonist regadenoson was approved by the US Food and Drug Administration to boost blood flow during cardiac stress tests [16]. Many other agonists and antagonists are undergoing clinical trials.
Medicinal chemists have made many efforts to develop small molecules as allosteric modulators in recent years. Unlike agonist and antagonist molecules, allosteric modulators evoke a selective physiologic response only where and when the orthosteric ligand is released [17]. GPCRs, including adenosine receptors, are allosterically regulated [17,18]. The list of existing allosteric A2AR modulators is short, however, and the physiologic opportunities for modulators are just emerging, making allosteric A2AR modulation a promising area for future research.
2. Adenosine and Its Receptors
Adenosine was initially recognized as a physiologic regulator of coronary vascular tone; since then, a growing body of reports indicates that adenosine regulates cellular functions through specific receptors present on the cell surface [19,20,21]. Adenosine is an endogenous purine nucleoside consisting of adenine and D-ribose, and is formed through hydrolysis of S-adenosylhomocysteine or adenosine monophosphate [22,23]. Adenosine formation from S-adenosylhomocysteine relies on the intracellular activity of the enzyme S-adenosylhomocysteine hydrolase, which bi-directionally assures the constant occupancy of a bound adenosine concentration in the cells [24]. Different enzymes mediate the formation of adenosine from adenosine monophosphate at both intracellular and extracellular levels.
Although adenosine does not exclusively act on synapses and is not stored in synaptic vesicles, it has a direct role in synaptic processes and the regulation of various neurotransmitters in the CNS. Nucleoside transporters mediate adenosine release and reuptake mechanisms through a concentration gradient between the intracellular and extracellular spaces. Therefore, adenosine is postulated as a modulator that affects neurotransmitter release and neuronal hyper- or depolarization and regulates glial cells [25]. Despite the modulatory role of adenosine, neurotransmitter properties are also observed for adenosine, which is due to the presence of the adenosine-producing enzyme in synapses. Extracellular adenosine acts on neurons through specific adenosine receptors [26].
Purinergic receptors are the natural target of purine molecules such as adenosine and adenosine triphosphate. These receptors were recognized for the first time in 1978 [27]. Two types of purinergic receptors, P1 and P2, were subsequently identified based on their pharmacologic profile [28]. P1 receptors recognize adenosine as a primary natural ligand and are therefore also called adenosine receptors. Each of the four types of adenosine receptors, A1R, A2AR, A2BR, or A3R, is characterized by a distinct pharmacologic profile. These receptors are members of the GPCR superfamily [17]. A2ARs and A2BRs are Gs-coupled receptors, and their activation increases the activity of adenylyl cyclase, the enzyme that initiates cyclic AMP (cAMP) synthesis in the cells. A1Rs and A3Rs are Gi/q coupled receptors, and their activation through adenosine or agonist molecules inhibits the activity of adenylyl cyclase, which suppresses cAMP synthesis in the cells.
3. A2AR and Its Physiologic Roles
The four types of adenosine receptors, A1R, A2AR, A2BR, or A3R, react with extracellular adenosine [13]. The activation of A2BRs reportedly requires a high adenosine concentration. Unlike A2BRs, adenosine levels under basal physiologic conditions are adequate to activate A1Rs, A2ARs, and A3Rs with relatively equal potency. The pharmacologic strength of an endogenous ligand or agonist at its receptor, however, relies on the number of receptors on the cells. Higher concentrations of adenosine are needed to show an effect in the presence of only a few receptors. Local expression of the A1Rs and A2ARs in the brain is suggested to be relatively higher than that of the other two adenosine receptors [6,29].
A2ARs were first identified by Libert and colleagues when they cloned several orphan GPCRs from the dog thyroid [30]. Afterward, A2ARs were cloned from other species, including guinea pigs, mice, rats, and humans [31,32,33,34]. As with the other GPCRs, A2ARs induce classical secondary messenger pathways. The A2AR signaling pathway may vary depending on the cell and tissue type in which the receptors occur. For example, Gs is the major G-protein associated with A2ARs in the peripheral system. On the other hand, A2ARs in the striatum, where they are highly expressed, mediate their effects mainly through Golf activation in the rat. Active Gs and Golf proteins stimulate adenylyl cyclase (Figure 1) which increases cellular cAMP levels and activates protein kinase A (PKA) which then phosphorylates and promotes cAMP-responsive element-binding protein 1 (CREB1) [16,35]. Activation of A2ARs also activates extracellular signal-regulated kinases (ERK) and several other kinases of the mitogen-activated protein kinase (MAPK) family which trigger specific cellular responses [36]. A2ARs form heterodimer structures with other GPCRs (e.g., metabotropic glutamate type 5 receptor (mGluR5)/A2AR, cannabinoid receptor type 1 (CB1)/A2AR, dopamine D2 receptor (D2R)/A2AR, dopamine D3 receptor/A2AR), and even CB1/A2AR/D2R heterotrimers [37,38,39,40,41].
The development of electron microscopy, selective radioligands, and antibodies has greatly contributed to A2AR distribution mapping. Furthermore, advancements in electron microscopy helped to determine the interactions of agonists and antagonists with their receptors and receptor density in particular regions. A2ARs are concentrated on GABAergic medium-sized spiny neurons of the striatum, core and shell regions of the nucleus accumbens (NAc), olfactory tubercule, and dopamine-rich areas of the brain [16].
A2ARs play a significant role in regulating the indirect pathways of the basal ganglia in the brain (Figure 2) [16]. The basal ganglia have an evolutionarily conserved essential role in learned habits, goal-directed movements, and locomotion [42]. The basal ganglia carry out their functions through direct and indirect circuits, originating in conspicuous populations of striatal medium spiny neurons that project to different output structures. Direct pathway neurons express excitatory dopamine D1 receptors (D1Rs) and inhibitory A1Rs, whereas indirect pathway neurons express inhibitory D2Rs and excitatory A2ARs [43]. Studies in mice revealed that both direct- and indirect pathway medium spiny neurons are active during mouse locomotion but quiescent during inactive phases [44], and chemogenetic activation of direct and indirect pathways neurons increases and decreases locomotor activity, respectively [45]. Moreover, recent findings indicate that optogenetic activation of indirect pathway neurons in the NAc, a part of the brain that is associated with motivation and pleasure, induces slow-wave sleep, whereas inhibition suppresses slow-wave sleep [46]. Other observations show that when an action does not result in a reward, increased activity of indirect pathways occurs, suggesting a role of the indirect pathways in controlling goal-directed behavior [47].
Apart from medium-sized spiny neurons in the basal ganglia, A2ARs are expressed in various other tissues, including smooth muscle cells, thymus, blood platelets, endothelial and lymphoid cells, leukocytes, spleen, blood vessels, lung, heart, and neurons in sympathetic and parasympathetic nervous systems [14,48] (Figure 2). A2ARs have a wide range of physiologic functions in the body, such as protecting tissues from inflammatory damage, mediating vasodilation, and supporting the formation of new blood vessels.
Several A2AR agonists and antagonists are currently in clinical trials. The selective A2AR agonist regadenoson was approved by the US Food and Drug Administration to increase blood flow in cardiac nuclear stress tests. On the other hand, the effects of A2AR antagonists for the treatment of Parkinson’s disease (PD) are promising. Other trials have been conducted with several agonists and antagonists aimed at treating infectious disease, ischemia-reperfusion injury, cancer, inflammation, sickle cell disease, diabetic nephropathy, and other CNS disorders. The increasing number of reports and patents demonstrates the growing interest in targeting the A2AR [16].
4. The Concept of Allosteric Modulation
The most common method to stimulate receptors in pharmacology and biochemistry is to target orthosteric sites with their endogenous ligand, agonists, or antagonists. On the other hand, studies show that receptor activity can be altered by small molecules that bind to an allosteric site different from the site where the endogenous ligand, agonists, or antagonists would bind [49]. The small molecules that bind to the allosteric sites of the receptors are termed allosteric modulators. Unlike endogenous ligands, agonists, or antagonists, an allosteric modulator cannot itself activate or inactivate receptors but alters the receptor’s response to substrates that bind to orthosteric sites in two ways: (1) increase or decrease affinity, i.e., the ability of orthosteric substances to bind receptors, and (2) increase/decrease efficacy, i.e., the ability of orthosteric substances to activate receptors [50]. Allosteric modulators reportedly change the receptor conformation, which alters the effect of the endogenous ligand, agonist, and antagonist binding [51]. The concept of receptor modulation is not straightforward with respect to practical implementation. Allosteric modulators do not necessarily equally alter the affinity and efficacy of endogenous ligands, agonists, or antagonists of the receptors. An allosteric modulator may alter the efficacy or affinity of the endogenous ligand, but not that of the agonist or antagonist of the receptors or vice versa [52].
The term ‘allostery’ was first used in enzymology studies in the early 1960s [53,54,55]. Subsequently, allosteric modulation has been identified for all receptor superfamilies, including GPCRs, nuclear hormone receptors [56,57], receptor tyrosine kinases [58,59], and ligand/voltage-gated ion channels [60,61,62,63,64]. The term “allosteric” began to be used increasingly in the literature, and a broad spectrum of allosteric modulators was described. Consequently, the classification of allosteric modulators was necessary to avoid possible confusion [65,66,67]. Three properties are considered in the classification of allosteric modulators: (1) affinity modulation of the orthosteric ligand, (2) modulation of the signaling effect of the orthosteric ligand, and (3) direct effects of the allosteric modulator in the absence of the orthosteric ligand. Moreover, allosteric modulators are classified in terms of their effects on orthosteric ligands as positive allosteric modulators (PAM), negative allosteric modulators (NAM), or silent allosteric modulators, also known as neutral allosteric ligands [68]. PAMs enhance the agonist/antagonist affinity and efficacy, whereas NAMs decrease orthosteric ligand affinity and efficacy. Unlike PAMs and NAMs, silent allosteric modulators do not affect the agonist or antagonist activity of orthosteric ligands, but bind to the allosteric site of the receptors and prevent PAMs or NAMs from binding to the same site, thereby inhibiting the activity of positive/negative allosteric modulators [52]. It is important to note that activities of allosteric modulators are therefore limited by where and when the orthosteric ligand is released. Thus, in contrast to agonists or antagonists, allosteric modulators promise greater safety and fewer side effects in therapeutic applications.
5. Allosteric A2AR Modulation
Adenosine receptors are among the first known allosterically regulated GPCRs. Early studies demonstrated that amiloride and its analogs are allosteric A2AR inhibitors [17,18,69]. Subsequent studies revealed that the amiloride analog 5-(N,N-hexamethylene)-amiloride (HMA) is a potent allosteric A2AR inhibitor. The other amiloride analogs, benzamil, 5-(N-methyl-N-isobutyl)amiloride (MIBA), 5-(N-methyl- N-guanidinocarbonyl-methyl)amiloride (MCGMA), and phenamil, were found to be more effective allosteric inhibitors than amiloride at rat A2ARs [17,70]. Moreover, amiloride and its analogues do not affect the dissociation rate of the agonist [3H]CGS21680 (3-{4-[2-({6-amino-9-[(2R,3R,4S,5S)-5-(ethylcarbamoyl)-3,4-dihydroxyoxolan-2-yl]-9H-purin-2-yl}amino) ethyl]phenyl}propanoic acid), but increase the dissociation rate of the antagonist [3H]ZM241385 (4-(2-{[7-amino-2-(furan-2-yl)[1,2,4]triazolo[1,5-a] [1,3,5] triazin-5-yl]amino}ethyl)phenol) from A2ARs [71]. By contrast, sodium ions, for example, deteriorate the dissociation rate of the antagonist [3H]ZM241385 from A2ARs in a dose-dependent manner [17]. It is important to note that other adenosine receptor agonists and antagonists are differentially affected by amilorides [70]. A new approach specifically targeting the sodium ion pocket, known as fragment-screening based on affinity mass spectrometry, led to the discovery of fragment Fg754 as a new A2AR NAM carrying a novel azetidine moiety and exhibiting inhibitory potency comparable to HMA. Subsequent simulations of the molecular dynamics, structure-activity relationship studies of the ligand, and nuclear magnetic resonance analyses in solution revealed the unique binding mode and antagonistic properties of Fg754, which is distinctly different from HMA [72]. In addition, cholesterol is reported to be a weak PAM of A2ARs [73].
Identification of binding sites for allosteric modulators on A2ARs based on the crystal structure of the receptor is critical for the development of new allosteric modulators. Two tightly linked residues, histidine residue number 278 (His278) in transmembrane domain 1 and glutamic acid13 in transmembrane domain 7 of the human A2AR, are reported to be the most crucial components for agonist recognition and play a partial role in the allosteric regulation by sodium ions [70,71,74,75,76,77]. Studies of the crystal structure of the antagonist-bound adenosine A2AR revealed that a highly conserved aspartate (Asp) residue in the second transmembrane domain is involved in sodium modulation of GPCRs [78]. Comparative studies of crystal structures in which a sodium ion bound in the allosteric site of human protease-activated receptor 1 [79], the β1-adrenergic receptor [80,81], the human δ-opioid receptor [82], and the human adenosine A2AR [78] show that sodium ions interact with the common residues Asp2.50 (superscript numbers refer to the Ballesteros and Weinstein residue numbering system [83]) serine3.39, tryptophan6.48, asparagine (Asn)7.45, and Asn7.49, either directly or through water-mediated hydrogen bonding [83]. Pre-crystal structure studies revealed that the positively charged sodium ion forms a permanent salt bridge with the negatively charged amino acid Asp2.50, suggesting that this residue represents the most conserved sodium ion binding site among GPCRs [84].
Subsequent studies on the crystal structure of the A2AR at 1.8 Å resolution provided sufficient resolution to confirm that Asp2.50 interacts directly with sodium ions via the salt bridge [78]. The crystal structures of agonist complexes for two variants in the first sodium coordination shell of the human A2A adenosine receptor have also been reported [85]. A fluorine-19 nuclear magnetic resonance spectroscopy study suggested that A2ARs have four distinct activation states; a partial agonist that favors the population of an active state (S3), an active state induced by full agonists (S3′), and two inactive states (S1–2); this study also demonstrated that sodium ions enhance the inactive states of A2ARs [86]. In contrast, partial agonists and HMA induce active states, indicating that HMA competes with sodium ions for interaction with A2ARs [84]. Moreover, all-atom simulations of molecular dynamics have shown that Fg754 can steadily enter the transmembrane domain core and form contacts with transmembrane helices 2, 3, 6, and 7, and extracellular loop 2. Particularly, the azetidine moiety of Fg754 may occupy the sodium ion-binding site by forming a salt bridge [72]. Another molecular dynamics simulation study described the allosteric effects of a mini-Gs protein on A2ARs [87].
In conclusion, the effects of amiloride and its derivatives on A2ARs are well studied. While the findings indicate that amiloride competes with sodium ions at the allosteric site of the A2AR, with Asp being the crucial amino acid, the allosteric binding site(s) of other small molecules selective for A2ARs remain unknown.
6. Allosteric A2AR Modulators and Their Potential Clinical Application
Allosteric A2AR modulation could be a new target for drug discovery [88]. Allosteric modulators can selectively elicit a physiologic response where and when the orthosteric ligand is released, thereby reducing the risk of an adverse effect of A2AR activation. Moreover, the possibility of saturating allosteric effects offers greater potential for fine-tuning the physiologic response in a positive or negative direction. As allosteric modulators have no pharmacologic effect beyond the saturation dose, these molecules are associated with a lower risk for adverse effects than orthosteric ligands, giving them a potential therapeutic advantage over classical agonists and antagonists [18,89].
Some compounds act as allosteric A2AR modulators, such as sodium ions, amiloride, and potassium-sparing diuretics, that also modulate other GPCRs than A2ARs [90]. For example, PD120918 is reported to enhance the activity of A2AR agonists in the rat striatum [91]. In contrast, thiadiazoles such as SCH-202676 alter the binding characteristics A2AR agonists and antagonists [92]. Some studies, however, suggest that thiadiazoles act as binding or oxidizing agents for SH groups rather than as allosteric modulators [92]. To date, only a relatively small number of selective allosteric A2AR modulators have been reported (Table 1) [93].
6.1. Allosteric A2AR Modulation Related to Inflammation
Adenosine is present in high concentrations in inflamed areas due to cell activation and breakdown [98,99,100]. The intracellular concentration of cAMP has a regulatory role in immune and inflammatory cells [101] and specifically, A2ARs are responsible for the anti-inflammatory effects of adenosine [102,103]. The anti-inflammatory effects of A2AR agonists are well known. Their therapeutic benefit, however, is not a given due to the potential adverse effects of A2AR agonists following systemic administration [7].
AEA061, which has an undisclosed structure, promotes the anti-inflammatory effects of adenosine by allosterically enhancing the activity of endogenous adenosine at A2ARs [96]. AEA061, which has no activity at a rat or human A2ARs in the absence of adenosine, inhibits the production of cytokines such as interleukin-1α, macrophage inflammatory protein-1α, 1β, and 2, keratinocyte chemokine, RANTES (regulated upon activation, normal T cell expressed and presumably secreted), and tumor necrosis factor-α in monocytes and splenocytes in a mouse model of lipopolysaccharide-induced inflammation. Therefore, positive allosteric modulators of A2ARs may represent a potential therapeutic approach to inflammation.
Inosine and inosine analog 6-S-[(4-nitrophenyl)methyl]-6-thioinosine (NBMPR) selectively and dose-dependently activate human A2ARs. NBMPR and inosine inhibit the production of pro-inflammatory cytokines and chemokines in splenic monocytes of wild-type mice, but not A2AR knockout mice. The positive allosteric A2AR modulator AEA061 enhances inosine-mediated A2AR activation, inosine-mediated inhibition of pro-inflammatory cytokines, and chemokine production by splenic monocytes [97].
6.2. Allosteric A2AR Modulation Related to Sleep and Neurologic Disorders
A2ARs are also expressed in the CNS, with the highest levels in the ventral and dorsal striatum [104]. A2ARs are present in the pre/postsynaptic compartment of neurons and microglia, oligodendrocytes, astrocytes, and capillary endothelial cells [12,105,106,107,108,109,110]. A growing number of reports illustrate that A2ARs play a critical role in emotional and cognitive processes, motivation, and voluntary movements [111]. Moreover, A2AR-expressing neurons in the NAc regulate sleep [8,47,112]. Therefore, A2AR stimulation should be considered a potential treatment approach for insomnia. Insomnia is a sleep disorder that affects millions of people worldwide and frequently co-occurs with a wide range of psychiatric disorders [113,114,115] Although A2AR agonists have strong sleep-inducing effects [116,117,118,119], they also have adverse cardiovascular effects and thus cannot be used clinically to treat sleep disorders. Moreover, the development of adenosine analogs to treat CNS disorders, including insomnia, is hampered by the poor transport of these drugs across the blood–brain barrier. In mice, a small blood–brain barrier-permeable monocarboxylate (3,4-difluoro-2-((2-fluoro-4-iodophenyl)amino) benzoic acid, denoted as A2AR PAM-1, was recently found to induce sleep by enhancing A2AR signaling in the brain (Figure 3) but, surprisingly, did not exhibit the typical unwanted cardiovascular and body temperature effects of A2AR agonists [94,95]. More specifically, A2AR PAM-1 dose-dependently enhanced A2AR signaling in A2AR-expressing Chinese hamster ovary (CHO) cells but not in CHO cells lacking A2AR expression or in the absence of adenosine (Figure 3). The A2AR PAM-1 did not alter the activity of the A2AR agonist CGS 21680 [120]. Intracerebroventricular infusion and intraperitoneal injection of A2AR PAM-1 induced prolonged slow-wave sleep, but not rapid-eye-movement sleep, in wild-type mice, but not A2AR knockout mice. Further testing revealed that A2AR PAM-1, unlike A2AR agonists, had no effects on blood pressure, cardiac function, or body temperature, suggesting that adenosine or A2AR expression levels in the cardiovascular system are insufficient to elicit an A2AR PAM-1 response under normal physiologic conditions. Therefore, molecules that allosterically enhance A2AR signaling may be developed to help people with insomnia fall asleep more easily. Moreover, A1Rs play a crucial role in the resolution of sleep need by modulating slow-wave activity, a slow, oscillatory neocortical activity that intensifies in correlation with wake duration and declines during sleep [121]. Slow-wave activity is widely used as a marker of mammalian sleep homeostasis and is necessary for sleep function. Therefore, dual allosteric A1R/A2AR modulators may be useful for improving not only the maintenance of sleep but also its function.
A2ARs have roles in neurodegenerative, neurodevelopmental, and psychiatric diseases. The potential therapeutic use of A2AR agonists and antagonists for specific conditions such as Niemann Pick disease, schizophrenia, autism-spectrum disorders, depression, anxiety, Alzheimer’s disease, attention-deficit hyperactivity disorder, PD, and fragile X syndrome is comprehensively discussed in the literature [122]. Allosteric A2AR modulators may provide alternative therapeutic options for neurologic disorders to circumvent the complexity of central and peripheral adenosine signaling. For example, dopamine-replacement therapy in PD is potentiated by blocking A2ARs due to the adenosine-dopamine antagonism in the striatum [123]. Decade-long preclinical studies of A2AR antagonists in PD models led to clinical trials of the A2AR antagonist istradefylline, which confirmed its clinically significant motor benefit in advanced PD patients and resulted in the approval of istradefylline for the treatment of PD patients in Japan and the US. The complexity of adenosine signaling contributed at least partially to the debilitating side effects and suspension of the clinical phase III trial of the A2AR antagonist tozadenant for PD, which resulted in the death of five patients due to inflammatory complications. Thus, there is also a critical need to develop safer and more effective means of suppressing A2AR signaling; for example, by negative allosteric modulation. Whereas the most potent PD medication is levodopa (l-3,4-dihydroxyphenylalanine), clinicians try to limit levodopa doses to the extent possible to avoid various adverse effects occurring with chronic use, such as dyskinesia and dopamine dysregulation. A2AR PAM, when administered concomitantly with levodopa, may mitigate some of these side effects, but strong evidence is currently lacking.
In addition, positive allosteric modulators of A2ARs may also alleviate various symptoms in neuropsychologic disorders. For example, psychotic symptoms such as delusions are caused by impaired discrimination of environmental stimuli. Recent evidence shows that D2Rs mediate discrimination learning in the NAc, but A2ARs expressed together with D2Rs in the NAc are required for discrimination learning. While normal mice can discriminate between reward-predictive and non-reward-predictive tones several days after generalized reward conditioning (when any tone is reward-predictive), mice in which A2ARs are blocked in the NAc do not show this ability [124]. In addition, hypofunction of NMDA-type glutamate receptors is thought to be involved in schizophrenia, as NMDA receptor antagonists such as phencyclidine and dizocilpine (MK-801) cause psychotic and cognitive disorders in humans and animals [125]. Deleting A2ARs in NAc astrocytes leads to motor and memory impairments relevant to schizophrenia, namely exacerbation of the MK-801-induced psychomotor response and impaired working memory [126]. Thus, the enhancement of A2AR signaling may be helpful to treat sleep disorders as well as schizophrenia and other psychotic disorders by overcoming dopaminergic hyperactivity or glutamatergic hypoactivity.
7. Concluding Remarks
Here, we discussed recent developments regarding allosteric A2AR modulation. Although numerous allosteric modulators of A2ARs have been identified, the physiologic functions of only a few of them have been established. The sleep-promoting effects and inflammatory process-modulating roles of allosteric A2AR modulators open the doors for the potential therapeutic use of these molecules for treating diseases. Allosteric modulators exert their effects only where and when the orthosteric ligand is released, conferring a potential therapeutic advantage over classical antagonists and agonist molecules. Thus, allosteric A2AR modulation could provide patients with an effective and safe treatment for various diseases.
Author Contributions
M.K. collected the data and drafted the manuscript. L.A. and M.L. helped in the literature survey and manuscript writing. All the authors provided substantial contributions to the discussion of its content and editing. M.K., L.A., and M.L. prepared all the graphical illustrations in the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Japan Society for the Promotion of Science [Grant-in-Aid for Scientific Research B (grant number 21H02802) to M.L.]; the Japan Science and Technology Agency [CREST (grant number JPMJCR1655) to M.L.]; Japan Agency for Medical Research and Development (AMED) [Moonshot Research and Development Program (grant number JP21zf0127005) to M.L.]; and the World Premier International Research Center Initiative (WPI) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT; to M.L.).
Conflicts of Interest
The authors declare no competing interest associated with the manuscript.
References
- Adair, T.H. Growth Regulation of the Vascular System: An Emerging Role for Adenosine. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 289, R283–R296. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-F. Adenosine Receptor Control of Cognition in Normal and Disease. Int. Rev. Neurobiol. 2014, 119, 257–307. [Google Scholar] [CrossRef] [PubMed]
- Feoktistov, I.; Biaggioni, I.; Cronstein, B.N. Adenosine Receptors in Wound Healing, Fibrosis and Angiogenesis. Handb. Exp. Pharmacol. 2009, 383–397. [Google Scholar] [CrossRef] [Green Version]
- Headrick, J.P.; Ashton, K.J.; Rose’meyer, R.B.; Peart, J.N. Cardiovascular Adenosine Receptors: Expression, Actions and Interactions. Pharmacol. Ther. 2013, 140, 92–111. [Google Scholar] [CrossRef] [PubMed]
- Hein, T.W.; Wang, W.; Zoghi, B.; Muthuchamy, M.; Kuo, L. Functional and Molecular Characterization of Receptor Subtypes Mediating Coronary Microvascular Dilation to Adenosine. J. Mol. Cell. Cardiol. 2001, 33, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Lazarus, M.; Chen, J.-F.; Huang, Z.-L.; Urade, Y.; Fredholm, B.B. Adenosine and Sleep. Handb. Exp. Pharmacol. 2019, 253, 359–381. [Google Scholar] [CrossRef]
- Ohta, A.; Sitkovsky, M. Role of G-Protein-Coupled Adenosine Receptors in Downregulation of Inflammation and Protection from Tissue Damage. Nature 2001, 414, 916–920. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Oishi, Y.; Cherasse, Y.; Korkutata, M.; Fujii, S.; Lee, C.-Y.; Lazarus, M. Extracellular Adenosine and Slow-Wave Sleep Are Increased after Ablation of Nucleus Accumbens Core Astrocytes and Neurons in Mice. Neurochem. Int. 2019, 124, 256–263. [Google Scholar] [CrossRef] [Green Version]
- Chin, J.H. Adenosine Receptors in Brain: Neuromodulation and Role in Epilepsy. Ann. Neurol. 1989, 26, 695–698. [Google Scholar] [CrossRef]
- Ciruela, F.; Casadó, V.; Rodrigues, R.J.; Luján, R.; Burgueño, J.; Canals, M.; Borycz, J.; Rebola, N.; Goldberg, S.R.; Mallol, J.; et al. Presynaptic Control of Striatal Glutamatergic Neurotransmission by Adenosine A1-A2A Receptor Heteromers. J. Neurosci. 2006, 26, 2080–2087. [Google Scholar] [CrossRef] [Green Version]
- Kamikubo, Y.; Shimomura, T.; Fujita, Y.; Tabata, T.; Kashiyama, T.; Sakurai, T.; Fukurotani, K.; Kano, M. Functional Cooperation of Metabotropic Adenosine and Glutamate Receptors Regulates Postsynaptic Plasticity in the Cerebellum. J. Neurosci. 2013, 33, 18661–18671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matos, M.; Augusto, E.; Santos-Rodrigues, A.D.; Schwarzschild, M.A.; Chen, J.-F.; Cunha, R.A.; Agostinho, P. Adenosine A2A Receptors Modulate Glutamate Uptake in Cultured Astrocytes and Gliosomes. Glia 2012, 60, 702–716. [Google Scholar] [CrossRef] [PubMed]
- Fredholm, B.B.; Ijzerman, A.P.; Jacobson, K.A.; Linden, J.; Müller, C.E. International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and Classification of Adenosine Receptors—An Update. Pharmacol. Rev. 2011, 63, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Fredholm, B.B.; Ijzerman, A.P.; Jacobson, K.A.; Klotz, K.N.; Linden, J. International Union of Pharmacology. XXV. Nomenclature and Classification of Adenosine Receptors. Pharmacol. Rev. 2001, 53, 527–552. [Google Scholar]
- Chen, J.-F.; Eltzschig, H.K.; Fredholm, B.B. Adenosine Receptors as Drug Targets—What Are the Challenges? Nat. Rev. Drug Discov. 2013, 12, 265–286. [Google Scholar] [CrossRef] [Green Version]
- de Lera Ruiz, M.; Lim, Y.-H.; Zheng, J. Adenosine A2A Receptor as a Drug Discovery Target. J. Med. Chem. 2014, 57, 3623–3650. [Google Scholar] [CrossRef]
- Göblyös, A.; Ijzerman, A.P. Allosteric Modulation of Adenosine Receptors. Purinergic Signal. 2009, 5, 51–61. [Google Scholar] [CrossRef] [Green Version]
- Gao, Z.-G.; Kim, S.-K.; Ijzerman, A.P.; Jacobson, K.A. Allosteric Modulation of the Adenosine Family of Receptors. Mini Rev. Med. Chem. 2005, 5, 545–553. [Google Scholar] [CrossRef] [Green Version]
- Drury, A.N.; Szent-Györgyi, A. The Physiological Activity of Adenine Compounds with Especial Reference to their Action upon the Mammalian Heart. J. Physiol. 1929, 68, 213–237. [Google Scholar] [CrossRef]
- Haskó, G.; Pacher, P.; Deitch, E.A.; Vizi, E.S. Shaping of Monocyte and Macrophage Function by Adenosine Receptors. Pharmacol. Ther. 2007, 113, 264–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sattin, A.; Rall, T.W. The Effect of Adenosine and Adenine Nucleotides on the Cyclic Adenosine 3′, 5′-Phosphate Content of Guinea Pig Cerebral Cortex Slices. Mol. Pharmacol. 1970, 6, 13–23. [Google Scholar]
- Fredholm, B.B. Adenosine, an Endogenous Distress Signal, Modulates Tissue Damage and Repair. Cell Death Differ. 2007, 14, 1315–1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrader, J. Metabolism of Adenosine and Sites of Production in the Heart. In Regulatory Function of Adenosine, Proceedings of the International Symposium on Adenosine, Charlottesville, Virginia, 7–11 June 1982; Berne, R.M., Rall, T.W., Rubio, R., Eds.; Developments in Pharmacology 2; Springer: Boston, MA, USA, 1983; pp. 133–156. ISBN 978-1-4613-3909-0. [Google Scholar]
- Ballarín, M.; Fredholm, B.B.; Ambrosio, S.; Mahy, N. Extracellular Levels of Adenosine and its Metabolites in the Striatum of Awake Rats: Inhibition of Uptake and Metabolism. Acta Physiol. Scand. 1991, 142, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Boison, D.; Singer, P.; Shen, H.-Y.; Feldon, J.; Yee, B.K. Adenosine Hypothesis of Schizophrenia—Opportunities for Pharmacotherapy. Neuropharmacology 2012, 62, 1527–1543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cacciari, B.; Pastorin, G.; Bolcato, C.; Spalluto, G.; Bacilieri, M.; Moro, S. A2B Adenosine Receptor Antagonists: Recent Developments. Mini Rev. Med. Chem. 2005, 5, 1053–1060. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G.; Cocks, T.; Crowe, R.; Kasakov, L. Purinergic Innervation of the Guinea-Pig Urinary Bladder. Br. J. Pharmacol. 1978, 63, 125–138. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, T.; Tostes, R.C.; Webb, R.C. Alterations in Vasoconstrictor Responses to the Endothelium-Derived Contracting Factor Uridine Adenosine Tetraphosphate Are Region Specific in DOCA-Salt Hypertensive Rats. Pharmacol. Res. 2012, 65, 81–90. [Google Scholar] [CrossRef] [Green Version]
- Fredholm, B.B.; Chen, J.-F.; Cunha, R.A.; Svenningsson, P.; Vaugeois, J.-M. Adenosine and Brain Function. Int. Rev. Neurobiol. 2005, 63, 191–270. [Google Scholar] [CrossRef]
- Maenhaut, C.; Van Sande, J.; Libert, F.; Abramowicz, M.; Parmentier, M.; Vanderhaegen, J.J.; Dumont, J.E.; Vassart, G.; Schiffmann, S. RDC8 Codes for an Adenosine A2 Receptor with Physiological Constitutive Activity. Biochem. Biophys. Res. Commun. 1990, 173, 1169–1178. [Google Scholar] [CrossRef]
- Chern, Y.; King, K.; Lai, H.L.; Lai, H.T. Molecular Cloning of a Novel Adenosine Receptor Gene from Rat Brain. Biochem. Biophys. Res. Commun. 1992, 185, 304–309. [Google Scholar] [CrossRef]
- Furlong, T.J.; Pierce, K.D.; Selbie, L.A.; Shine, J. Molecular Characterization of a Human Brain Adenosine A2 Receptor. Brain Res. Mol. Brain Res. 1992, 15, 62–66. [Google Scholar] [CrossRef]
- Ledent, C.; Vaugeois, J.M.; Schiffmann, S.N.; Pedrazzini, T.; El Yacoubi, M.; Vanderhaeghen, J.J.; Costentin, J.; Heath, J.K.; Vassart, G.; Parmentier, M. Aggressiveness, Hypoalgesia and High Blood Pressure in Mice Lacking the Adenosine A2a Receptor. Nature 1997, 388, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Xie, G.X.; Chalmers, D.; Morgan, C.; Watson, S.J.; Akil, H. Cloning and Expression of the A2a Adenosine Receptor from Guinea Pig Brain. Neurochem. Res. 1994, 19, 613–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kull, B.; Svenningsson, P.; Fredholm, B.B. Adenosine A2A Receptors Are Colocalized with and Activate Golf in Rat Striatum. Mol. Pharmacol. 2000, 58, 771–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulte, G.; Fredholm, B.B. Human Adenosine A(1), A(2A), A(2B), and A(3) Receptors Expressed in Chinese Hamster Ovary Cells All Mediate the Phosphorylation of Extracellular-Regulated Kinase 1/2. Mol. Pharmacol. 2000, 58, 477–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferré, S.; Goldberg, S.R.; Lluis, C.; Franco, R. Looking for the Role of Cannabinoid Receptor Heteromers in Striatal Function. Neuropharmacology 2009, 56 (Suppl. S1), 226–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferré, S.; Karcz-Kubicha, M.; Hope, B.T.; Popoli, P.; Burgueño, J.; Gutiérrez, M.A.; Casadó, V.; Fuxe, K.; Goldberg, S.R.; Lluis, C.; et al. Synergistic Interaction between Adenosine A2A and Glutamate MGlu5 Receptors: Implications for Striatal Neuronal Function. Proc. Natl. Acad. Sci. USA 2002, 99, 11940–11945. [Google Scholar] [CrossRef] [Green Version]
- Fuxe, K.; Ferré, S.; Canals, M.; Torvinen, M.; Terasmaa, A.; Marcellino, D.; Goldberg, S.R.; Staines, W.; Jacobsen, K.X.; Lluis, C.; et al. Adenosine A2A and Dopamine D2 Heteromeric Receptor Complexes and Their Function. J. Mol. Neurosci. 2005, 26, 209–220. [Google Scholar] [CrossRef]
- Navarro, G.; Carriba, P.; Gandía, J.; Ciruela, F.; Casadó, V.; Cortés, A.; Mallol, J.; Canela, E.I.; Lluis, C.; Franco, R. Detection of Heteromers Formed by Cannabinoid CB1, Dopamine D2, and Adenosine A2A G-Protein-Coupled Receptors by Combining Bimolecular Fluorescence Complementation and Bioluminescence Energy Transfer. Sci. World J. 2008, 8, 1088–1097. [Google Scholar] [CrossRef] [Green Version]
- Torvinen, M.; Marcellino, D.; Canals, M.; Agnati, L.F.; Lluis, C.; Franco, R.; Fuxe, K. Adenosine A2A Receptor and Dopamine D3 Receptor Interactions: Evidence of Functional A2A/D3 Heteromeric Complexes. Mol. Pharmacol. 2005, 67, 400–407. [Google Scholar] [CrossRef]
- Grillner, S.; Robertson, B. The Basal Ganglia Over 500 Million Years. Curr. Biol. 2016, 26, R1088–R1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oishi, Y.; Lazarus, M. The Control of Sleep and Wakefulness by Mesolimbic Dopamine Systems. Neurosci. Res. 2017, 118, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Cui, G.; Jun, S.B.; Jin, X.; Pham, M.D.; Vogel, S.S.; Lovinger, D.M.; Costa, R.M. Concurrent Activation of Striatal Direct and Indirect Pathways during Action Initiation. Nature 2013, 494, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Ottenheimer, D.; DiLeone, R.J. Activity of D1/2 Receptor Expressing Neurons in the Nucleus Accumbens Regulates Running, Locomotion, and Food Intake. Front. Behav. Neurosci. 2016, 10, 66. [Google Scholar] [CrossRef] [Green Version]
- Oishi, Y.; Xu, Q.; Wang, L.; Zhang, B.-J.; Takahashi, K.; Takata, Y.; Luo, Y.-J.; Cherasse, Y.; Schiffmann, S.N.; de Kerchove d’Exaerde, A.; et al. Slow-Wave Sleep Is Controlled by a Subset of Nucleus Accumbens Core Neurons in Mice. Nat. Commun. 2017, 8, 734. [Google Scholar] [CrossRef] [Green Version]
- Nonomura, S.; Nishizawa, K.; Sakai, Y.; Kawaguchi, Y.; Kato, S.; Uchigashima, M.; Watanabe, M.; Yamanaka, K.; Enomoto, K.; Chiken, S.; et al. Monitoring and Updating of Action Selection for Goal-Directed Behavior through the Striatal Direct and Indirect Pathways. Neuron 2018, 99, 1302–1314. [Google Scholar] [CrossRef] [Green Version]
- Fredholm, B.B.; Cunha, R.A.; Svenningsson, P. Pharmacology of Adenosine A2A Receptors and Therapeutic Applications. Curr. Top. Med. Chem. 2003, 3, 413–426. [Google Scholar] [CrossRef]
- Ritter, J.; Flower, R.; Henderson, G.; Loke, Y.K.; MacEwan, D.; Rang, H. Rang & Dale’s Pharmacology, 9th ed.; Elsevier: Amsterdam, The Netherlands, 2018. [Google Scholar]
- Neubig, R.R.; Spedding, M.; Kenakin, T.; Christopoulos, A. International Union of Pharmacology Committee on Receptor Nomenclature and Drug Classification International Union of Pharmacology Committee on Receptor Nomenclature and Drug Classification. XXXVIII. Update on Terms and Symbols in Quantitative Pharmacology. Pharmacol. Rev. 2003, 55, 597–606. [Google Scholar] [CrossRef] [Green Version]
- Hu, J. Allosteric Modulators of the Human Calcium-Sensing Receptor: Structures, Sites of Action, and Therapeutic Potentials. Endocr. Metab. Immune Disord. Drug Targets 2008, 8, 192–197. [Google Scholar] [CrossRef]
- Kenakin, T. Pharmacology in Drug Discovery and Development, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2016. [Google Scholar]
- Koshland, D.E.; Némethy, G.; Filmer, D. Comparison of Experimental Binding Data and Theoretical Models in Proteins Containing Subunits. Biochemistry 1966, 5, 365–385. [Google Scholar] [CrossRef]
- Monod, J.; Wyman, J.; Changeux, J.-P. On the Nature of Allosteric Transitions: A Plausible Model. J. Mol. Biol. 1965, 12, 88–118. [Google Scholar] [CrossRef]
- Monod, J.; Changeux, J.-P.; Jacob, F. Allosteric Proteins and Cellular Control Systems. J. Mol. Biol. 1963, 6, 306–329. [Google Scholar] [CrossRef]
- Estébanez-Perpiñá, E.; Arnold, L.A.; Nguyen, P.; Rodrigues, E.D.; Mar, E.; Bateman, R.; Pallai, P.; Shokat, K.M.; Baxter, J.D.; Guy, R.K.; et al. A Surface on the Androgen Receptor That Allosterically Regulates Coactivator Binding. Proc. Natl. Acad. Sci. USA 2007, 104, 16074–16079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, T.S.; Giri, P.K.; de Vera, I.M.S.; Marciano, D.P.; Kuruvilla, D.S.; Shin, Y.; Blayo, A.-L.; Kamenecka, T.M.; Burris, T.P.; Griffin, P.R.; et al. An Alternate Binding Site for PPARγ Ligands. Nat. Commun. 2014, 5, 3571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bono, F.; De Smet, F.; Herbert, C.; De Bock, K.; Georgiadou, M.; Fons, P.; Tjwa, M.; Alcouffe, C.; Ny, A.; Bianciotto, M.; et al. Inhibition of Tumor Angiogenesis and Growth by a Small-Molecule Multi-FGF Receptor Blocker with Allosteric Properties. Cancer Cell 2013, 23, 477–488. [Google Scholar] [CrossRef] [Green Version]
- De Smet, F.; Christopoulos, A.; Carmeliet, P. Allosteric Targeting of Receptor Tyrosine Kinases. Nat. Biotechnol. 2014, 32, 1113–1120. [Google Scholar] [CrossRef]
- Catterall, W.A.; Cestèle, S.; Yarov-Yarovoy, V.; Yu, F.H.; Konoki, K.; Scheuer, T. Voltage-Gated Ion Channels and Gating Modifier Toxins. Toxicon 2007, 49, 124–141. [Google Scholar] [CrossRef] [Green Version]
- Olsen, R.W.; Chang, C.-S.S.; Li, G.; Hanchar, H.J.; Wallner, M. Fishing for Allosteric Sites on GABA(A) Receptors. Biochem. Pharmacol. 2004, 68, 1675–1684. [Google Scholar] [CrossRef]
- Spedding, M.; Kenny, B.; Chatelain, P. New Drug Binding Sites in Ca2+ Channels. Trends Pharmacol. Sci. 1995, 16, 139–142. [Google Scholar] [CrossRef]
- Taly, A.; Corringer, P.-J.; Guedin, D.; Lestage, P.; Changeux, J.-P. Nicotinic Receptors: Allosteric Transitions and Therapeutic Targets in the Nervous System. Nat. Rev. Drug Discov. 2009, 8, 733–750. [Google Scholar] [CrossRef]
- Traynelis, S.F.; Wollmuth, L.P.; McBain, C.J.; Menniti, F.S.; Vance, K.M.; Ogden, K.K.; Hansen, K.B.; Yuan, H.; Myers, S.J.; Dingledine, R. Glutamate Receptor Ion Channels: Structure, Regulation, and Function. Pharmacol. Rev. 2010, 62, 405–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colquhoun, D. Binding, Gating, Affinity and Efficacy: The Interpretation of Structure-Activity Relationships for Agonists and of the Effects of Mutating Receptors. Br. J. Pharmacol. 1998, 125, 924–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fenton, A.W. Allostery: An Illustrated Definition for the “Second Secret of Life”. Trends Biochem. Sci. 2008, 33, 420–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nussinov, R.; Tsai, C.-J. Allostery in Disease and in Drug Discovery. Cell 2013, 153, 293–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christopoulos, A.; Changeux, J.-P.; Catterall, W.A.; Fabbro, D.; Burris, T.P.; Cidlowski, J.A.; Olsen, R.W.; Peters, J.A.; Neubig, R.R.; Pin, J.-P.; et al. International Union of Basic and Clinical Pharmacology. XC. Multisite Pharmacology: Recommendations for the Nomenclature of Receptor Allosterism and Allosteric Ligands. Pharmacol. Rev. 2014, 66, 918–947. [Google Scholar] [CrossRef] [Green Version]
- Jacobson, K.A.; Gao, Z.-G. Adenosine Receptors as Therapeutic Targets. Nat. Rev. Drug Discov. 2006, 5, 247–264. [Google Scholar] [CrossRef] [Green Version]
- Gao, Z.-G.; Melman, N.; Erdmann, A.; Kim, S.G.; Müller, C.E.; IJzerman, A.P.; Jacobson, K.A. Differential Allosteric Modulation by Amiloride Analogues of Agonist and Antagonist Binding at A(1) and A(3) Adenosine Receptors. Biochem. Pharmacol. 2003, 65, 525–534. [Google Scholar] [CrossRef]
- Gao, Z.G.; Ijzerman, A.P. Allosteric Modulation of A(2A) Adenosine Receptors by Amiloride Analogues and Sodium Ions. Biochem. Pharmacol. 2000, 60, 669–676. [Google Scholar] [CrossRef]
- Lu, Y.; Liu, H.; Yang, D.; Zhong, L.; Xin, Y.; Zhao, S.; Wang, M.-W.; Zhou, Q.; Shui, W. Affinity Mass Spectrometry-Based Fragment Screening Identified a New Negative Allosteric Modulator of the Adenosine A2A Receptor Targeting the Sodium Ion Pocket. ACS Chem. Biol. 2021, 16, 991–1002. [Google Scholar] [CrossRef]
- Huang, S.K.; Almurad, O.; Pejana, R.J.; Morrison, Z.A.; Pandey, A.; Picard, L.-P.; Nitz, M.; Sljoka, A.; Prosser, R.S. Allosteric Modulation of the Adenosine A2A Receptor by Cholesterol. eLife 2022, 11, e73901. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Youkey, R.L.; Ghartey, K.; Leonard, M.; Linden, J.; Tucker, A.L. The Allosteric Enhancer PD81,723 Increases Chimaeric A1/A2A Adenosine Receptor Coupling with Gs. Biochem. J. 2006, 396, 139–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Z.-G.; Kim, S.G.; Soltysiak, K.A.; Melman, N.; IJzerman, A.P.; Jacobson, K.A. Selective Allosteric Enhancement of Agonist Binding and Function at Human A3 Adenosine Receptors by a Series of Imidazoquinoline Derivatives. Mol. Pharmacol. 2002, 62, 81–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Z.G.; Van Muijlwijk-Koezen, J.E.; Chen, A.; Müller, C.E.; Ijzerman, A.P.; Jacobson, K.A. Allosteric Modulation of A(3) Adenosine Receptors by a Series of 3-(2-Pyridinyl)Isoquinoline Derivatives. Mol. Pharmacol. 2001, 60, 1057–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Galen, P.J.; Nissen, P.; van Wijngaarden, I.; IJzerman, A.P.; Soudijn, W. 1H-Imidazo[4,5-c]Quinolin-4-Amines: Novel Non-Xanthine Adenosine Antagonists. J. Med. Chem. 1991, 34, 1202–1206. [Google Scholar] [CrossRef]
- Liu, W.; Chun, E.; Thompson, A.A.; Chubukov, P.; Xu, F.; Katritch, V.; Han, G.W.; Roth, C.B.; Heitman, L.H.; IJzerman, A.P.; et al. Structural Basis for Allosteric Regulation of GPCRs by Sodium Ions. Science 2012, 337, 232–236. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Srinivasan, Y.; Arlow, D.H.; Fung, J.J.; Palmer, D.; Zheng, Y.; Green, H.F.; Pandey, A.; Dror, R.O.; Shaw, D.E.; et al. High-Resolution Crystal Structure of Human Protease-Activated Receptor 1. Nature 2012, 492, 387–392. [Google Scholar] [CrossRef] [Green Version]
- Christopher, J.A.; Brown, J.; Doré, A.S.; Errey, J.C.; Koglin, M.; Marshall, F.H.; Myszka, D.G.; Rich, R.L.; Tate, C.G.; Tehan, B.; et al. Biophysical Fragment Screening of the Β1-Adrenergic Receptor: Identification of High Affinity Arylpiperazine Leads Using Structure-Based Drug Design. J. Med. Chem. 2013, 56, 3446–3455. [Google Scholar] [CrossRef]
- Miller-Gallacher, J.L.; Nehmé, R.; Warne, T.; Edwards, P.C.; Schertler, G.F.X.; Leslie, A.G.W.; Tate, C.G. The 2.1 Å Resolution Structure of Cyanopindolol-Bound Β1-Adrenoceptor Identifies an Intramembrane Na+ Ion That Stabilises the Ligand-Free Receptor. PLoS ONE 2014, 9, e92727. [Google Scholar] [CrossRef] [Green Version]
- Fenalti, G.; Giguere, P.M.; Katritch, V.; Huang, X.-P.; Thompson, A.A.; Cherezov, V.; Roth, B.L.; Stevens, R.C. Molecular Control of δ-Opioid Receptor Signalling. Nature 2014, 506, 191–196. [Google Scholar] [CrossRef] [Green Version]
- Ballesteros, J.A.; Weinstein, H. Integrated Methods for the Construction of Three-Dimensional Models and Computational Probing of Structure-Function Relations in G Protein-Coupled Receptors. In Receptor Molecular Biology; Methods in Neurosciences 25; Sealfon, S.C., Ed.; Academic Press: Cambridge, MA, USA, 1995; pp. 366–428. [Google Scholar]
- Horstman, D.A.; Brandon, S.; Wilson, A.L.; Guyer, C.A.; Cragoe, E.J.; Limbird, L.E. An Aspartate Conserved among G-Protein Receptors Confers Allosteric Regulation of Alpha 2-Adrenergic Receptors by Sodium. J. Biol. Chem. 1990, 265, 21590–21595. [Google Scholar] [CrossRef]
- White, K.L.; Eddy, M.T.; Gao, Z.-G.; Han, G.W.; Lian, T.; Deary, A.; Patel, N.; Jacobson, K.A.; Katritch, V.; Stevens, R.C. Structural Connection between Activation Microswitch and Allosteric Sodium Site in GPCR Signaling. Structure 2018, 26, 259–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, L.; Van Eps, N.; Zimmer, M.; Ernst, O.P.; Prosser, R.S. Activation of the A2A Adenosine G-Protein-Coupled Receptor by Conformational Selection. Nature 2016, 533, 265–268. [Google Scholar] [CrossRef] [PubMed]
- Renault, P.; Louet, M.; Marie, J.; Labesse, G.; Floquet, N. Molecular Dynamics Simulations of the Allosteric Modulation of the Adenosine A2A Receptor by a Mini-G Protein. Sci. Rep. 2019, 9, 5495. [Google Scholar] [CrossRef] [PubMed]
- Christopoulos, A. Allosteric Binding Sites on Cell-Surface Receptors: Novel Targets for Drug Discovery. Nat. Rev. Drug Discov. 2002, 1, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Christopoulos, A. Advances in G Protein-Coupled Receptor Allostery: From Function to Structure. Mol. Pharmacol. 2014, 86, 463–478. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Q.; Lee, B.X.; Glashofer, M.; van Rhee, A.M.; Jacobson, K.A. Mutagenesis Reveals Structure-Activity Parallels between Human A2A Adenosine Receptors and Biogenic Amine G Protein-Coupled Receptors. J. Med. Chem. 1997, 40, 2588–2595. [Google Scholar] [CrossRef] [Green Version]
- Bruns, R.F.; Fergus, J.H. Allosteric Enhancement of Adenosine A1 Receptor Binding and Function by 2-Amino-3-Benzoylthiophenes. Mol. Pharmacol. 1990, 38, 939–949. [Google Scholar]
- Göblyös, A.; de Vries, H.; Brussee, J.; Ijzerman, A.P. Synthesis and Biological Evaluation of a New Series of 2,3,5-Substituted [1,2,4]-Thiadiazoles as Modulators of Adenosine A1 Receptors and Their Molecular Mechanism of Action. J. Med. Chem. 2005, 48, 1145–1151. [Google Scholar] [CrossRef]
- Giorgi, I.; Biagi, G.; Bianucci, A.M.; Borghini, A.; Livi, O.; Leonardi, M.; Pietra, D.; Calderone, V.; Martelli, A. N6-1,3-Diphenylurea Derivatives of 2-Phenyl-9-Benzyladenines and 8-Azaadenines: Synthesis and Biological Evaluation as Allosteric Modulators of A2A Adenosine Receptors. Eur. J. Med. Chem. 2008, 43, 1639–1647. [Google Scholar] [CrossRef]
- Korkutata, M.; Saitoh, T.; Cherasse, Y.; Ioka, S.; Duo, F.; Qin, R.; Murakoshi, N.; Fujii, S.; Zhou, X.; Sugiyama, F.; et al. Enhancing Endogenous Adenosine A2A Receptor Signaling Induces Slow-Wave Sleep without Affecting Body Temperature and Cardiovascular Function. Neuropharmacology 2019, 144, 122–132. [Google Scholar] [CrossRef] [Green Version]
- Korkutata, M.; Saitoh, T.; Feng, D.; Murakoshi, N.; Sugiyama, F.; Cherasse, Y.; Nagase, H.; Lazarus, M. Allosteric Modulation of Adenosine A2A Receptors in Mice Induces Slow-Wave Sleep without Cardiovascular Effects. Sleep Med. 2017, 40, e181. [Google Scholar] [CrossRef]
- Welihinda, A.A.; Amento, E.P. Positive Allosteric Modulation of the Adenosine A2a Receptor Attenuates Inflammation. J. Inflamm. 2014, 11, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welihinda, A.A.; Kaur, M.; Raveendran, K.S.; Amento, E.P. Enhancement of Inosine-Mediated A2AR Signaling through Positive Allosteric Modulation. Cell. Signal. 2018, 42, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.; Leone, M.; Viviand, X.; Ayem, M.L.; Guieu, R. High Adenosine Plasma Concentration as a Prognostic Index for Outcome in Patients with Septic Shock. Crit. Care Med. 2000, 28, 3198–3202. [Google Scholar] [CrossRef] [PubMed]
- Sottofattori, E.; Anzaldi, M.; Ottonello, L. HPLC Determination of Adenosine in Human Synovial Fluid. J. Pharm. Biomed. Anal. 2001, 24, 1143–1146. [Google Scholar] [CrossRef]
- Sperlágh, B.; Dóda, M.; Baranyi, M.; Haskó, G. Ischemic-like Condition Releases Norepinephrine and Purines from Different Sources in Superfused Rat Spleen Strips. J. Neuroimmunol. 2000, 111, 45–54. [Google Scholar] [CrossRef]
- Livingston, M.; Heaney, L.G.; Ennis, M. Adenosine, Inflammation and Asthma—A Review. Inflamm. Res. 2004, 53, 171–178. [Google Scholar] [CrossRef]
- Haskó, G.; Cronstein, B.N. Adenosine: An Endogenous Regulator of Innate Immunity. Trends Immunol. 2004, 25, 33–39. [Google Scholar] [CrossRef]
- Sitkovsky, M.V. Use of the A(2A) Adenosine Receptor as a Physiological Immunosuppressor and to Engineer Inflammation In Vivo. Biochem. Pharmacol. 2003, 65, 493–501. [Google Scholar] [CrossRef]
- Rosin, D.L.; Hettinger, B.D.; Lee, A.; Linden, J. Anatomy of Adenosine A2A Receptors in Brain: Morphological Substrates for Integration of Striatal Function. Neurology 2003, 61, S12–S18. [Google Scholar] [CrossRef]
- Carman, A.J.; Mills, J.H.; Krenz, A.; Kim, D.-G.; Bynoe, M.S. Adenosine Receptor Signaling Modulates Permeability of the Blood–Brain Barrier. J. Neurosci. 2011, 31, 13272–13280. [Google Scholar] [CrossRef] [PubMed]
- Melani, A.; Cipriani, S.; Vannucchi, M.G.; Nosi, D.; Donati, C.; Bruni, P.; Giovannini, M.G.; Pedata, F. Selective Adenosine A2a Receptor Antagonism Reduces JNK Activation in Oligodendrocytes after Cerebral Ischaemia. Brain J. Neurol. 2009, 132, 1480–1495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mills, J.H.; Alabanza, L.; Weksler, B.B.; Couraud, P.-O.; Romero, I.A.; Bynoe, M.S. Human Brain Endothelial Cells Are Responsive to Adenosine Receptor Activation. Purinergic Signal. 2011, 7, 265–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishizaki, T.; Nagai, K.; Nomura, T.; Tada, H.; Kanno, T.; Tozaki, H.; Li, X.X.; Kondoh, T.; Kodama, N.; Takahashi, E.; et al. A New Neuromodulatory Pathway with a Glial Contribution Mediated via A(2a) Adenosine Receptors. Glia 2002, 39, 133–147. [Google Scholar] [CrossRef]
- Saura, J.; Angulo, E.; Ejarque, A.; Casadó, V.; Tusell, J.M.; Moratalla, R.; Chen, J.-F.; Schwarzschild, M.A.; Lluis, C.; Franco, R.; et al. Adenosine A2A Receptor Stimulation Potentiates Nitric Oxide Release by Activated Microglia. J. Neurochem. 2005, 95, 919–929. [Google Scholar] [CrossRef]
- Yu, L.; Shen, H.-Y.; Coelho, J.E.; Araújo, I.M.; Huang, Q.-Y.; Day, Y.-J.; Rebola, N.; Canas, P.M.; Rapp, E.K.; Ferrara, J.; et al. Adenosine A2A Receptor Antagonists Exert Motor and Neuroprotective Effects by Distinct Cellular Mechanisms. Ann. Neurol. 2008, 63, 338–346. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.-F.; Lee, C.; Chern, Y. Adenosine Receptor Neurobiology: Overview. Int. Rev. Neurobiol. 2014, 119, 1–49. [Google Scholar] [CrossRef]
- Lazarus, M.; Shen, H.-Y.; Cherasse, Y.; Qu, W.-M.; Huang, Z.-L.; Bass, C.E.; Winsky-Sommerer, R.; Semba, K.; Fredholm, B.B.; Boison, D.; et al. Arousal Effect of Caffeine Depends on Adenosine A2A Receptors in the Shell of the Nucleus Accumbens. J. Neurosci. 2011, 31, 10067–10075. [Google Scholar] [CrossRef] [Green Version]
- Roth, T. Insomnia: Definition, Prevalence, Etiology, and Consequences. J. Clin. Sleep Med. 2007, 3, S7–S10. [Google Scholar] [CrossRef] [Green Version]
- de Zambotti, M.; Goldstone, A.; Colrain, I.M.; Baker, F.C. Insomnia Disorder in Adolescence: Diagnosis, Impact, and Treatment. Sleep Med. Rev. 2018, 39, 12–24. [Google Scholar] [CrossRef]
- Seow, L.S.E.; Abdin, E.; Chang, S.; Chong, S.A.; Subramaniam, M. Identifying the Best Sleep Measure to Screen Clinical Insomnia in a Psychiatric Population. Sleep Med. 2018, 41, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Methippara, M.M.; Kumar, S.; Alam, M.N.; Szymusiak, R.; McGinty, D. Effects on Sleep of Microdialysis of Adenosine A1 and A2a Receptor Analogs into the Lateral Preoptic Area of Rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 289, R1715–R1723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satoh, S.; Matsumura, H.; Koike, N.; Tokunaga, Y.; Maeda, T.; Hayaishi, O. Region-Dependent Difference in the Sleep-Promoting Potency of an Adenosine A2A Receptor Agonist. Eur. J. Neurosci. 1999, 11, 1587–1597. [Google Scholar] [CrossRef] [PubMed]
- Scammell, T.E.; Gerashchenko, D.Y.; Mochizuki, T.; McCarthy, M.T.; Estabrooke, I.V.; Sears, C.A.; Saper, C.B.; Urade, Y.; Hayaishi, O. An Adenosine A2a Agonist Increases Sleep and Induces Fos in Ventrolateral Preoptic Neurons. Neuroscience 2001, 107, 653–663. [Google Scholar] [CrossRef]
- Urade, Y.; Eguchi, N.; Qu, W.-M.; Sakata, M.; Huang, Z.-L.; Chen, J.-F.; Schwarzschild, M.A.; Fink, J.S.; Hayaishi, O. Sleep Regulation in Adenosine A2A Receptor-Deficient Mice. Neurology 2003, 61, S94–S96. [Google Scholar] [CrossRef]
- Mustafa, K. A Potential Treatment for Insomnia by Positive Allosteric Modulation of Adenosine A2A Receptors. Ph.D. Thesis, University of Tsukuba, Tsukuba, Japan, 2019. [Google Scholar]
- Lazarus, M.; Oishi, Y.; Bjorness, T.E.; Greene, R.W. Gating and the Need for Sleep: Dissociable Effects of Adenosine A1 and A2A Receptors. Front. Neurosci. 2019, 13, 740. [Google Scholar] [CrossRef]
- Domenici, M.R.; Ferrante, A.; Martire, A.; Chiodi, V.; Pepponi, R.; Tebano, M.T.; Popoli, P. Adenosine A2A Receptor as Potential Therapeutic Target in Neuropsychiatric Disorders. Pharmacol. Res. 2019, 147, 104338. [Google Scholar] [CrossRef]
- Franco, R.; Navarro, G. Adenosine A2A Receptor Antagonists in Neurodegenerative Diseases: Huge Potential and Huge Challenges. Front. Psychiatry 2018, 9, 68. [Google Scholar] [CrossRef] [Green Version]
- Iino, Y.; Sawada, T.; Yamaguchi, K.; Tajiri, M.; Ishii, S.; Kasai, H.; Yagishita, S. Dopamine D2 Receptors in Discrimination Learning and Spine Enlargement. Nature 2020, 579, 555–560. [Google Scholar] [CrossRef]
- Field, J.R.; Walker, A.G.; Conn, P.J. Targeting Glutamate Synapses in Schizophrenia. Trends Mol. Med. 2011, 17, 689–698. [Google Scholar] [CrossRef] [Green Version]
- Matos, M.; Shen, H.-Y.; Augusto, E.; Wang, Y.; Wei, C.J.; Wang, Y.T.; Agostinho, P.; Boison, D.; Cunha, R.A.; Chen, J.-F. Deletion of Adenosine A2A Receptors from Astrocytes Disrupts Glutamate Homeostasis Leading to Psychomotor and Cognitive Impairment: Relevance to Schizophrenia. Biol. Psychiatry 2015, 78, 763–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferre, S.; Ciruela, F.; Borycz, J.; Solinas, M.; Quarta, D.; Antoniou, K.; Quiroz, C.; Justinova, Z.; Lluis, C.; Franco, R.; et al. Adenosine A1-A2A Receptor Heteromers: New Targets for Caffeine in the Brain. Front. Biosci. J. Virtual Libr. 2008, 13, 2391–2399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferré, S.; Díaz-Ríos, M.; Salamone, J.D.; Prediger, R.D. New Developments on the Adenosine Mechanisms of the Central Effects of Caffeine and Their Implications for Neuropsychiatric Disorders. J. Caffeine Adenosine Res. 2018, 8, 121–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferré, S.; Bonaventura, J.; Tomasi, D.; Navarro, G.; Moreno, E.; Cortés, A.; Lluís, C.; Casadó, V.; Volkow, N.D. Allosteric Mechanisms within the Adenosine A2A-Dopamine D2 Receptor Heterotetramer. Neuropharmacology 2016, 104, 154–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuxe, K.; Marcellino, D.; Leo, G.; Agnati, L.F. Molecular Integration via Allosteric Interactions in Receptor Heteromers. A Working Hypothesis. Curr. Opin. Pharmacol. 2010, 10, 14–22. [Google Scholar] [CrossRef]
- Moreno, E.; Chiarlone, A.; Medrano, M.; Puigdellívol, M.; Bibic, L.; Howell, L.A.; Resel, E.; Puente, N.; Casarejos, M.J.; Perucho, J.; et al. Singular Location and Signaling Profile of Adenosine A2A-Cannabinoid CB 1 Receptor Heteromers in the Dorsal Striatum. Neuropsychopharmacology 2018, 43, 964–977. [Google Scholar] [CrossRef] [Green Version]
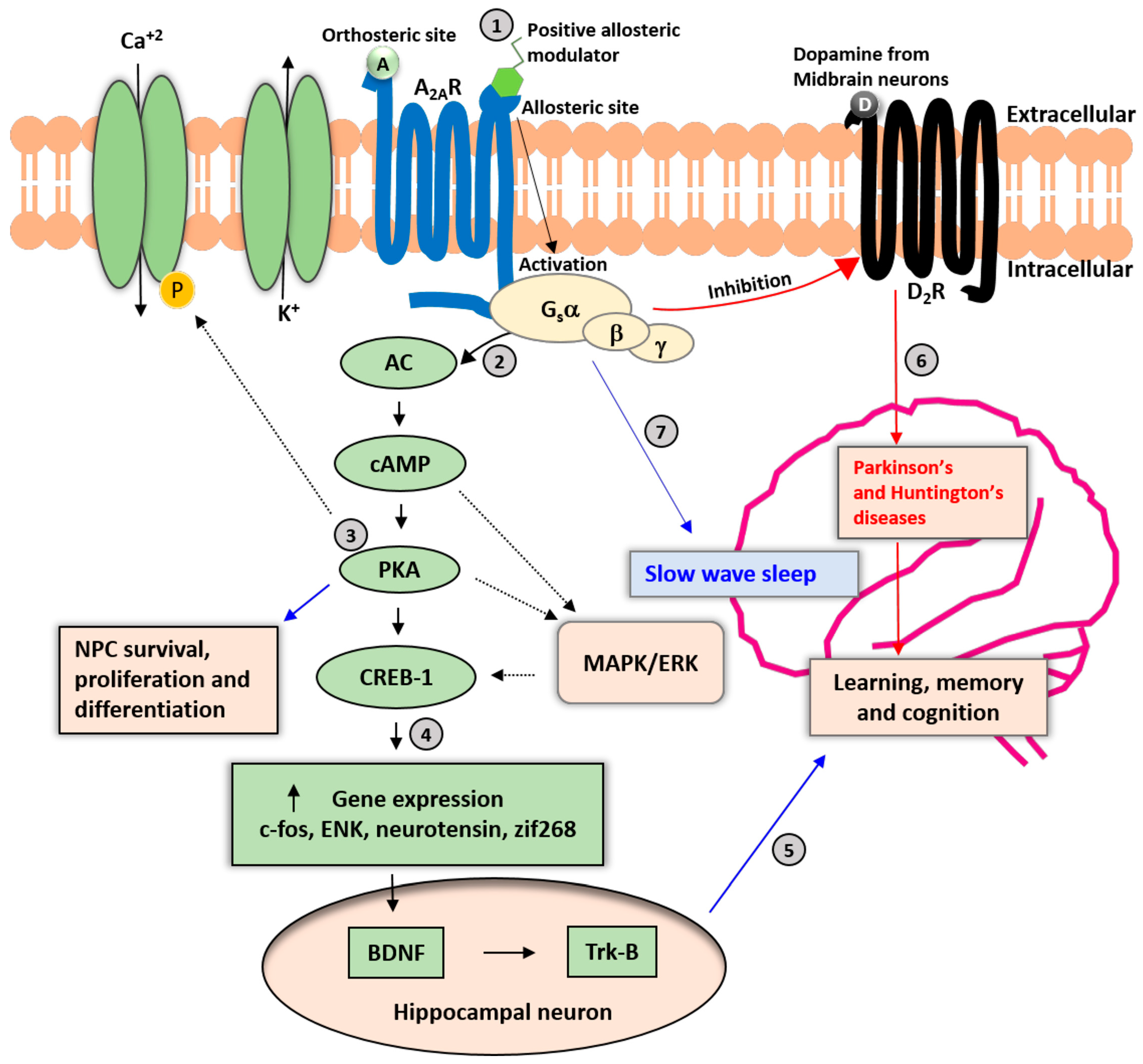
Figure 1.
Neuronal A2AR signaling cascades. A2AR is a Gs(olf)-protein-coupled receptor involved in various physiologic processes. (1) The allosteric modulation sites may be pharmacologically relevant for avoiding adverse effects on the cardiovascular and other peripheral systems. (2) Binding of adenosine and an allosteric modulator to A2ARs enhances the activation of cyclic adenosine monophosphate (cAMP) and protein kinase A (PKA), resulting in the phosphorylation of calcium ion channels and increased influx of Ca+2 into the cytoplasm. (3) The PKA pathway also promotes neural progenitor cell (NPC) survival, proliferation, and differentiation; and activation of the mitogen-activated protein (MAP)-kinase pathway. (4) PKA-mediated phosphorylation of the cAMP-responsive element binding protein 1 (CREB-1) regulates the expression of genes such as c-fos, enkephalin (ENK), neurotensin, and zinc finger protein 268 (zif268). (5) The secretion of brain-derived neurotrophic factor (BDNF) and activation of tropomyosin receptor kinase B (TrkB) receptors in response to A2AR activation in hippocampal neurons may be relevant for cognitive functions such as learning and memory. (6) A2AR activation may be a counter mechanism to control the activation and expression of dopamine D2 receptors (D2Rs). Long-term imbalance of D2R signaling leads to impairments in cognitive and motor functions and the development of Parkinson’s and Huntington’s diseases. (7) Activation of A2AR in the nucleus accumbens increases slow-wave sleep in mice. Solid black arrows represent the primary signaling pathway of A2ARs, and dashed black arrows represent secondary signaling pathways. A: Adenosine; D: Dopamine.
Figure 1.
Neuronal A2AR signaling cascades. A2AR is a Gs(olf)-protein-coupled receptor involved in various physiologic processes. (1) The allosteric modulation sites may be pharmacologically relevant for avoiding adverse effects on the cardiovascular and other peripheral systems. (2) Binding of adenosine and an allosteric modulator to A2ARs enhances the activation of cyclic adenosine monophosphate (cAMP) and protein kinase A (PKA), resulting in the phosphorylation of calcium ion channels and increased influx of Ca+2 into the cytoplasm. (3) The PKA pathway also promotes neural progenitor cell (NPC) survival, proliferation, and differentiation; and activation of the mitogen-activated protein (MAP)-kinase pathway. (4) PKA-mediated phosphorylation of the cAMP-responsive element binding protein 1 (CREB-1) regulates the expression of genes such as c-fos, enkephalin (ENK), neurotensin, and zinc finger protein 268 (zif268). (5) The secretion of brain-derived neurotrophic factor (BDNF) and activation of tropomyosin receptor kinase B (TrkB) receptors in response to A2AR activation in hippocampal neurons may be relevant for cognitive functions such as learning and memory. (6) A2AR activation may be a counter mechanism to control the activation and expression of dopamine D2 receptors (D2Rs). Long-term imbalance of D2R signaling leads to impairments in cognitive and motor functions and the development of Parkinson’s and Huntington’s diseases. (7) Activation of A2AR in the nucleus accumbens increases slow-wave sleep in mice. Solid black arrows represent the primary signaling pathway of A2ARs, and dashed black arrows represent secondary signaling pathways. A: Adenosine; D: Dopamine.

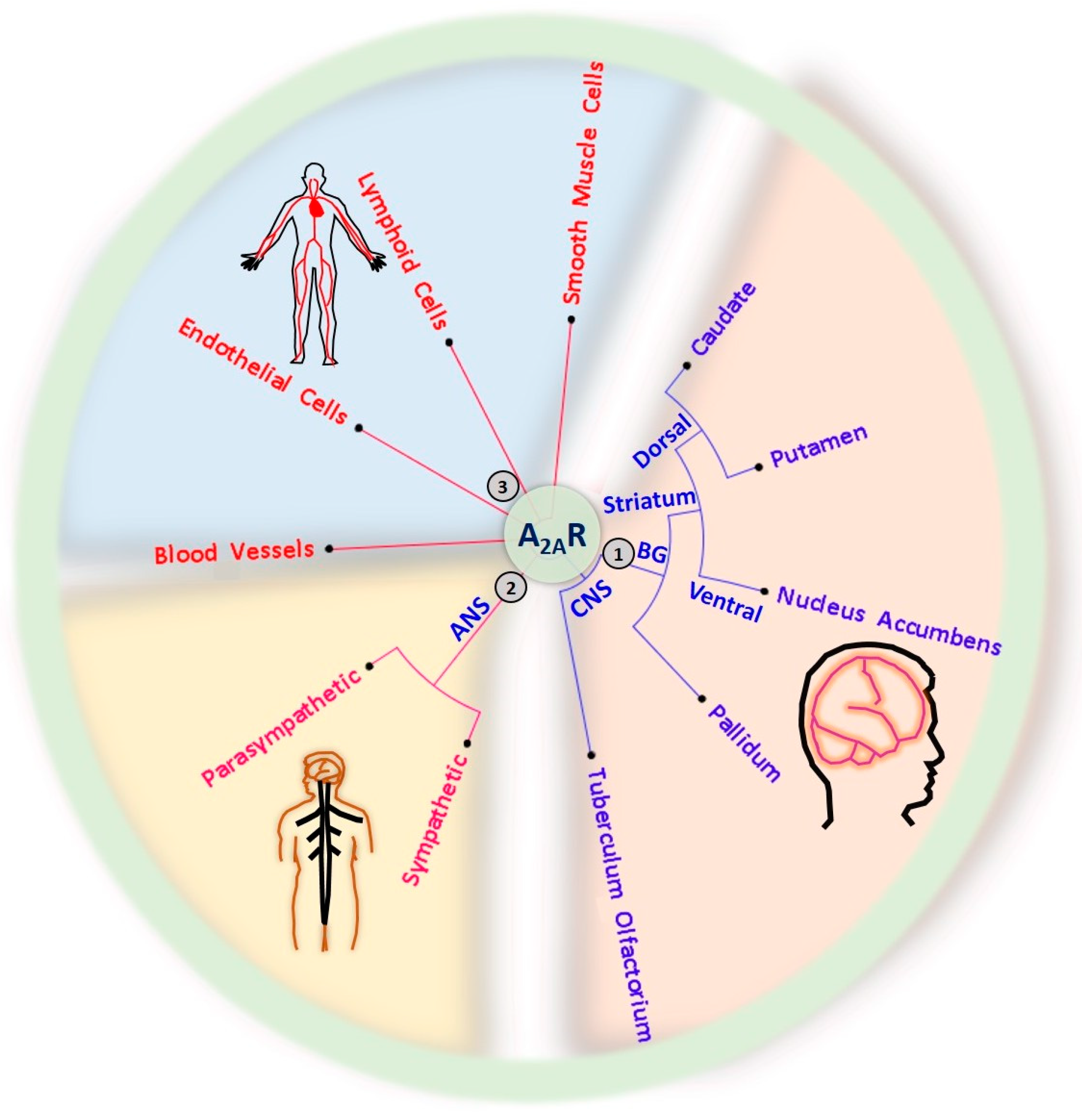
Figure 2.
Expression of A2ARs in the central nervous system (CNS), autonomic nervous system (ANS), circulatory system, and musculoskeletal system. (1) CNS A2ARs are mainly expressed in the basal ganglia (BG), including the dorsal pallidum, the nucleus accumbens in the ventral part of the striatum, and the dorsal striatum comprising the caudate and putamen. (2) A2ARs are also expressed in the sympathetic and parasympathetic ANS. (3) The distribution of A2ARs is not limited to the nervous system; A2ARs are also found in the circulation system, including heart, blood vessels, lymphoid cells (immune cells), and smooth muscle cells of the musculoskeletal system.
Figure 2.
Expression of A2ARs in the central nervous system (CNS), autonomic nervous system (ANS), circulatory system, and musculoskeletal system. (1) CNS A2ARs are mainly expressed in the basal ganglia (BG), including the dorsal pallidum, the nucleus accumbens in the ventral part of the striatum, and the dorsal striatum comprising the caudate and putamen. (2) A2ARs are also expressed in the sympathetic and parasympathetic ANS. (3) The distribution of A2ARs is not limited to the nervous system; A2ARs are also found in the circulation system, including heart, blood vessels, lymphoid cells (immune cells), and smooth muscle cells of the musculoskeletal system.

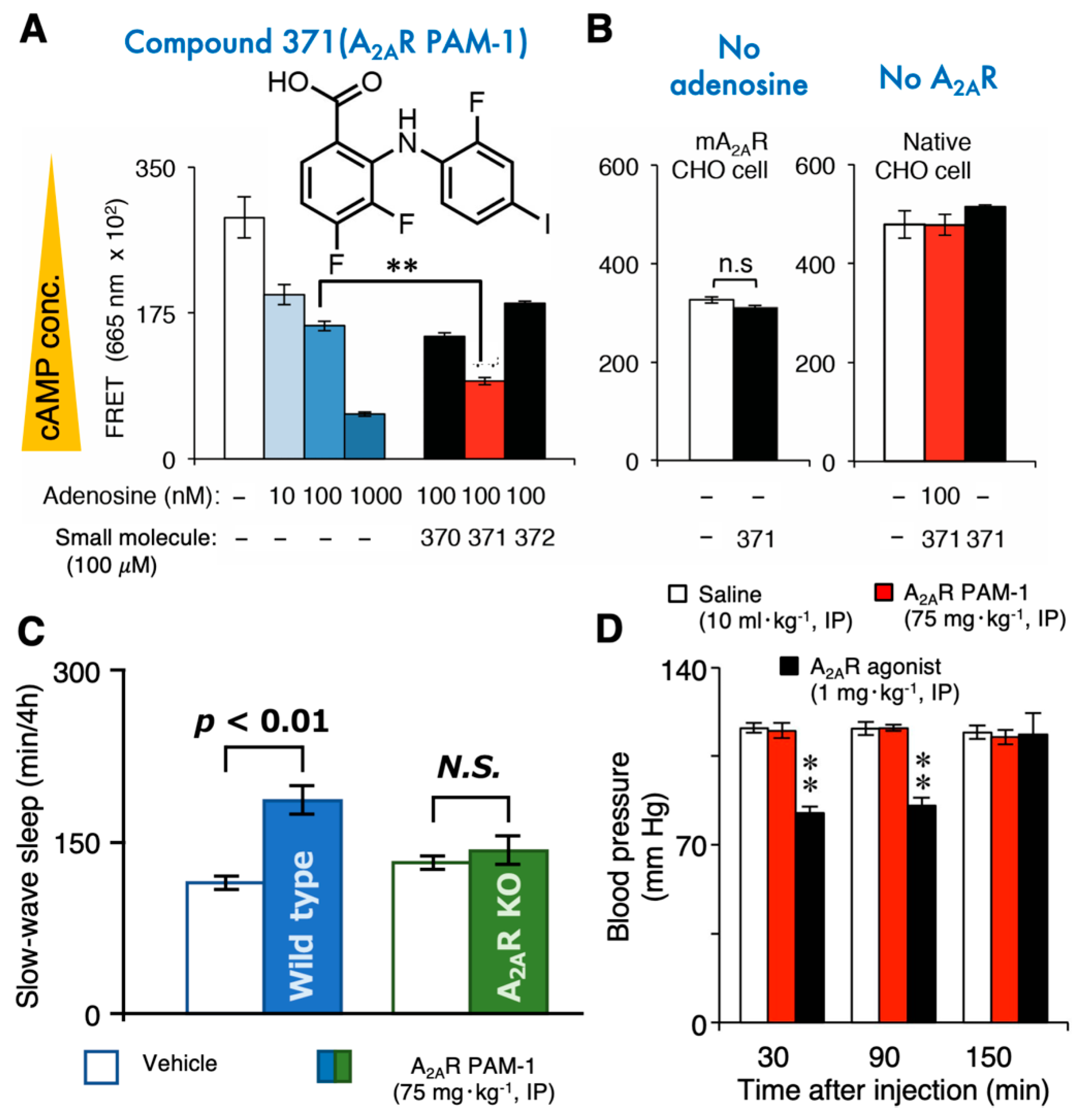
Figure 3.
The A2AR positive allosteric modulator (PAM)-1 induces sleep without cardiovascular effects. (A,B) A2AR PAM-1 enhanced the activity of adenosine on A2AR-expressing Chinese Hamster Ovary (CHO) cells when cAMP was measured by a fluorescence energy transfer (FRET) immunoassay (A), whereas A2AR PAM-1 did not enhance cAMP production without adenosine or in native CHO cells without A2AR expression (B). (C) Intraperitoneal (IP) injection of A2AR PAM-1 increased slow-wave sleep in wild-type mice, but not in A2AR-knockout (KO) mice. (D) A2AR PAM-1 did not affect cardiovascular functions (e.g., blood pressure), unlike a classic A2AR agonist (CGS 21680) [94,95]. ** p < 0.01.
Figure 3.
The A2AR positive allosteric modulator (PAM)-1 induces sleep without cardiovascular effects. (A,B) A2AR PAM-1 enhanced the activity of adenosine on A2AR-expressing Chinese Hamster Ovary (CHO) cells when cAMP was measured by a fluorescence energy transfer (FRET) immunoassay (A), whereas A2AR PAM-1 did not enhance cAMP production without adenosine or in native CHO cells without A2AR expression (B). (C) Intraperitoneal (IP) injection of A2AR PAM-1 increased slow-wave sleep in wild-type mice, but not in A2AR-knockout (KO) mice. (D) A2AR PAM-1 did not affect cardiovascular functions (e.g., blood pressure), unlike a classic A2AR agonist (CGS 21680) [94,95]. ** p < 0.01.


{kind=link}
{kind=link}
{kind=link}
Table 1.
Allosteric A2AR modulators and their functions.
| Name | Type | Pharmacology | Structure | Physiologic Effects |
|---|---|---|---|---|
| 3,4-Difluoro-2-((2-fluoro-4-iodophenyl)amino)benzoic acid | Allosteric enhancer/modulator | Enhanced adenosine signaling at mouse A2ARs. |  | Induced slow wave sleep without affecting cardiovascular function or body temperature in wild-type male mice [94,95]. |
| AEA061 | Allosteric enhancer/modulator | Enhanced adenosine and inosine signaling and increased effect of the A2AR agonist CGS 21680. | Not disclosed | Inhibited the production of tumor necrosis factor-α, macrophage inflammatory protein-1α, 1β, and 2, interleukin-1α, keratinocyte chemokine, and RANTES (regulated upon activation, normal T cell expressed and presumably secreted) in macrophages and splenocytes, reduced circulating plasma tumor necrosis factor-α and monocyte chemoattractant protein-1 levels, and increased plasma interleukin-10 during lipopolysaccharide-induced endotoxemia [96,97]. |
| N-(3-Benzyl-5-phenyl-3H-[1,2,3]triazolo[4,5-d]- pyrimidin-7yl-)-(4-aminophenyl)-amine | Allosteric modulator | Inhibited the binding of antagonists and agonists at the A2AR orthosteric site [93]. |  | Unknown |
| N6-[(4-Nitro)-phenyl]-9-benzyl-2-phenyladenine | Allosteric modulator | Inhibited the binding of antagonists and agonists at the A2AR orthosteric site [93]. |  | Unknown |
| N6-[(4-Amino)-phenyl]-9-benzyl-2-phenyladenine | Allosteric modulator | Inhibited the binding of antagonists and agonists at the A2AR orthosteric site [93]. |  | Unknown |
| 1-[4-(3-Benzyl-5-phenyl-3H-[1,2,3]triazolo[4,5-d]-pyrimidin-7-ylamino)-phenyl]-3-(4-fluorophenyl)-urea | Allosteric modulator | Modulated the binding of antagonist and agonist at the A2AR orthosteric site [93]. |  | Unknown |
| 1-[4-(3-Benzyl-5-phenyl-3H-[1,2,3]triazolo[4,5-d]-pyrimidin-7-ylamino)-phenyl]-3-(4-trifluoromethylphenyl)- urea | Allosteric modulator | Modulated the binding of antagonist and agonist at the A2AR orthosteric site [93]. |  | Unknown |
| 1-[4-(9-Benzyl-2-phenyl-9H-purin-6-ylamino)- phenyl]-3-(4-methoxyphenyl-urea | Allosteric modulator | Modulated the binding of antagonist and agonist at the A2AR orthosteric site [93]. |  | Unknown |
| Amiloride | Allosteric modulator | Increased the dissociation rate of the antagonist ZM-241,385 at rat A2ARs [18,71]. |  | Unknown |
| Benzamil | Allosteric modulator | Increased the dissociation rate of the antagonist ZM-241,385 at rat A2ARs [71]. |  | Unknown |
| HMA; 5-(N,N-hexamethylene)amiloride | Allosteric modulator | Increased the dissociation rate of the antagonist ZM-241,385 at rat A2ARs [71]. |  | Unknown |
| MGCMA; 5-(N-methyl-N-guanidinocarbonyl-methyl)amiloride | Allosteric modulator | Increased the dissociation rate of the antagonist ZM-241,385 at rat A2ARs [71]. |  | Unknown |
| MIBA; 5-(N-methyl-N-isobutyl)amiloride | Allosteric modulator | Increased the dissociation rate of the antagonist ZM-241,385 at rat A2ARs [71]. |  | Unknown |
| Phenamil | Allosteric modulator | Increased the dissociation rate of the antagonist ZM-241,385 at rat A2ARs [71]. |  | Unknown |
| Sodium Ion | Allosteric modulator | Positively modulated A2ARs [71]. | Na+ | Unknown |
| PD120918 {4-methyl-7-[(methyl- amino)carbonyl]oxy}-2H-1-benzopyran-2-one} | Allosteric modulator | Enhanced agonist radioligand binding to rat striatal A2ARs without functional enhancement [18,91]. |  | Unknown |
| Fg754 | Allosteric modulator | Increased the dissociation rate of the agonist CGS21680 at A2ARs expressing HEK-293 cells [72]. |  | Unknown |
| Cholesterol | Allosteric modulator | Decreased the dissociation rate of the agonist NECA at A2ARs-embedded nanodiscs [73]. |  | Unknown |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Korkutata, M.; Agrawal, L.; Lazarus, M. Allosteric Modulation of Adenosine A2A Receptors as a New Therapeutic Avenue. Int. J. Mol. Sci. 2022, 23, 2101. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23042101
AMA Style
Korkutata M, Agrawal L, Lazarus M. Allosteric Modulation of Adenosine A2A Receptors as a New Therapeutic Avenue. International Journal of Molecular Sciences. 2022; 23(4):2101. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23042101
Chicago/Turabian StyleKorkutata, Mustafa, Lokesh Agrawal, and Michael Lazarus. 2022. "Allosteric Modulation of Adenosine A2A Receptors as a New Therapeutic Avenue" International Journal of Molecular Sciences 23, no. 4: 2101. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23042101
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.