
PTK7, a Catalytically Inactive Receptor Tyrosine Kinase, Increases Oncogenic Phenotypes in Xenograft Tumors of Esophageal Squamous Cell Carcinoma KYSE-30 Cells

 , and
, and 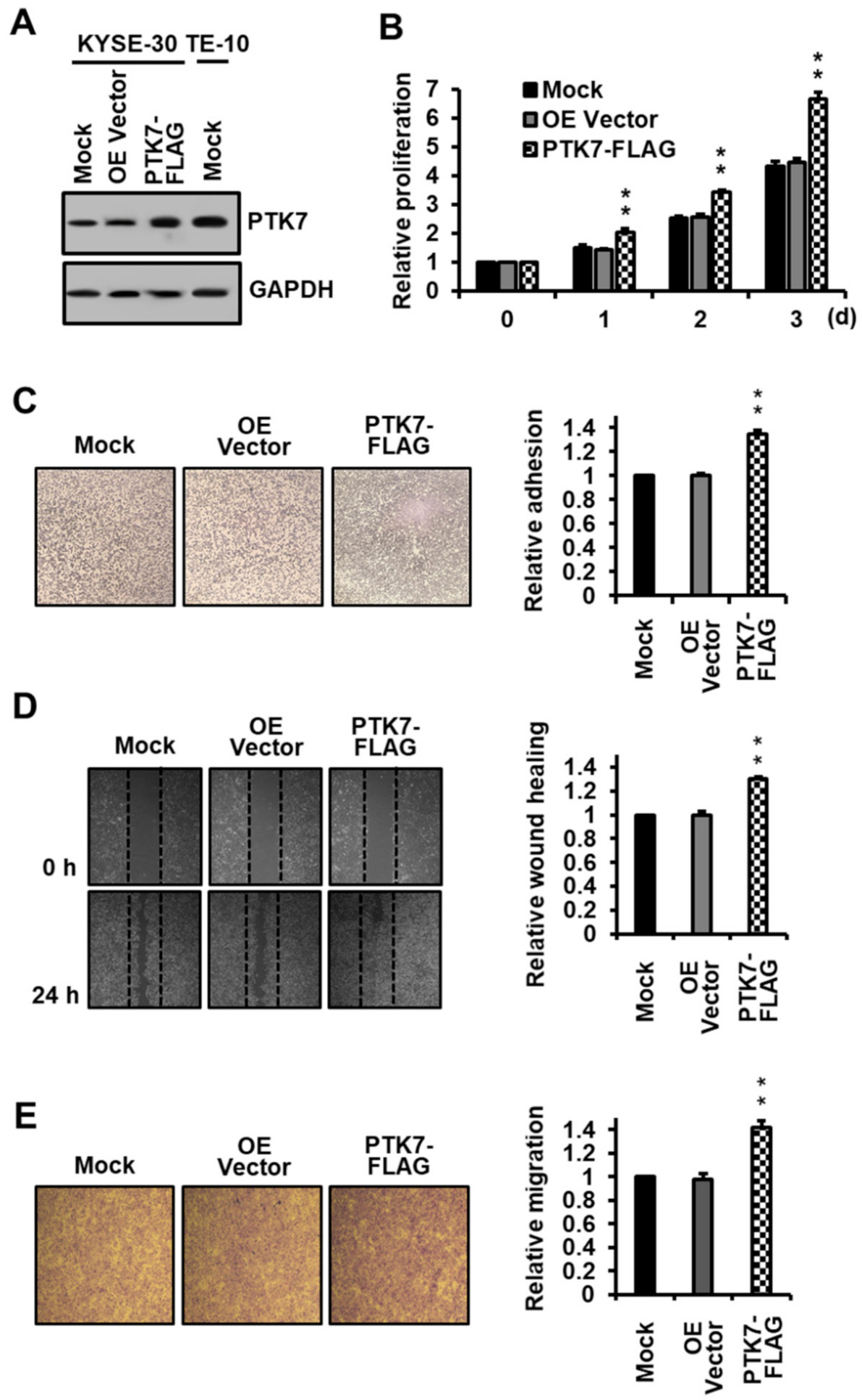{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. PTK7 Overexpression Increases Proliferation, Adhesion, and Migration in ESCC KYSE-30 Cells
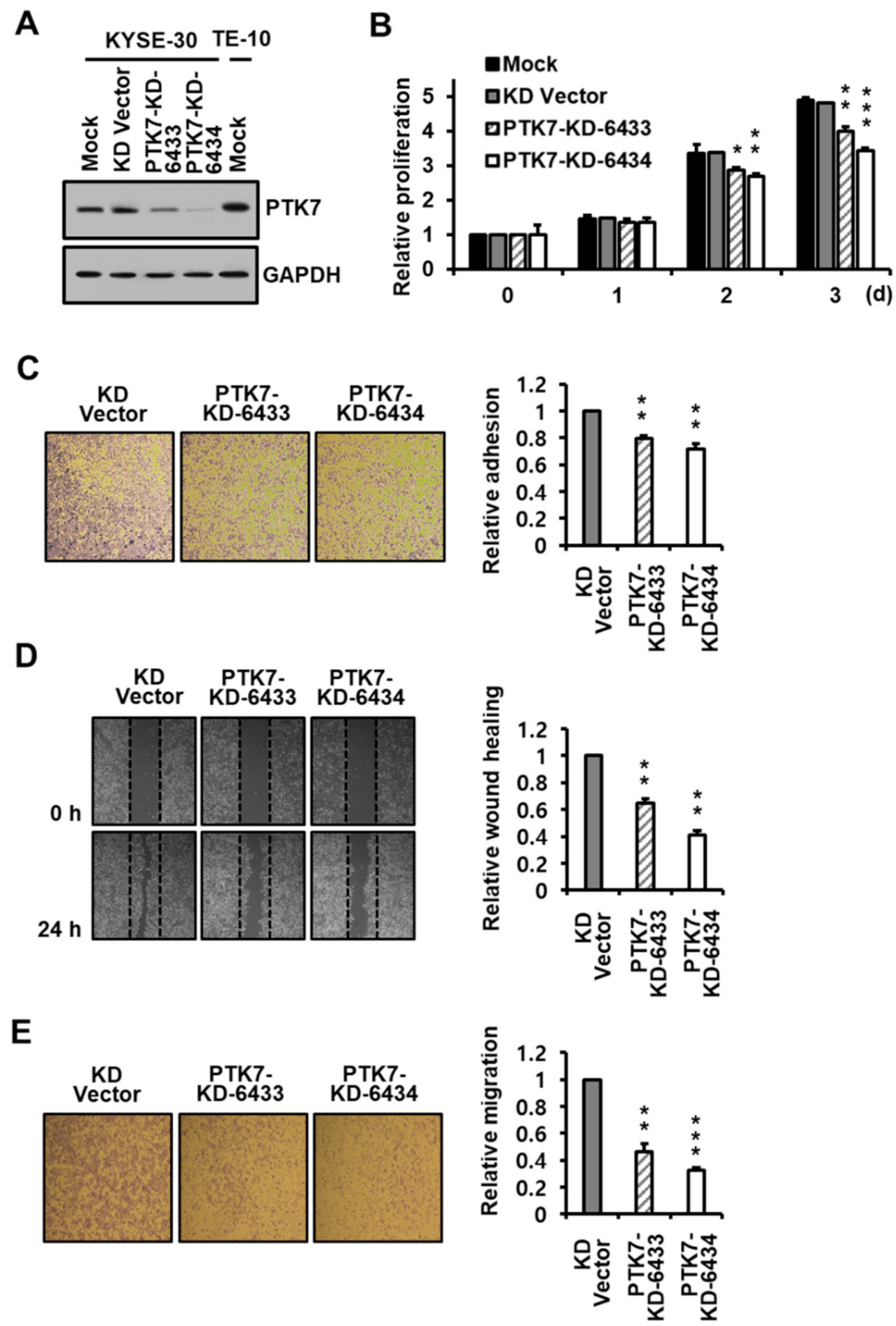
2.2. PTK7 Knockdown Reduces Proliferation, Adhesion, and Migration in ESCC KYSE-30 Cells
2.3. PTK7 Increases the Activation of Oncogenic Signaling Proteins
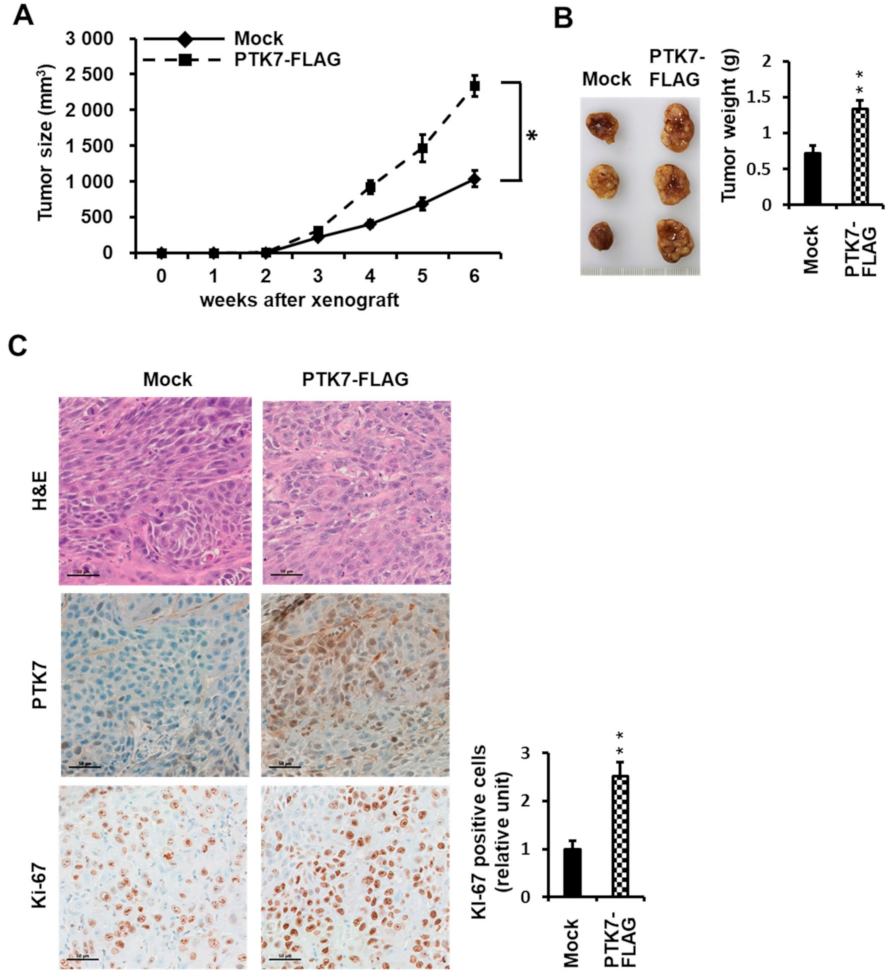
2.4. PTK7 Expression Correlates with the Tumorigenic Effect of ESCC KYSE-30 Cells In Vivo
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Generation of PTK7 Overexpression and Knockdown Lentiviruses and Infection of KYSE-30 Cells
4.3. Antibodies
4.4. Western Blot Analysis
4.5. Cell Proliferation Assay
4.6. Cell Adhesion Assay
4.7. Wound Healing and Chemotactic Migration Assay
4.8. Xenograft Mouse Model
4.9. Ethics Statement
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DMEM | Dulbecco’s modified Eagle medium |
| EAC | esophageal adenocarcinoma |
| EC | esophageal cancer |
| EGFR | epidermal growth factor receptor |
| ESCC | esophageal squamous cell carcinoma |
| FBS | fetal bovine serum |
| FGFR | fibroblast growth factor receptor |
| HEK293 | human embryonic kidney 293 |
| PBS | phosphate-buffered saline |
| PCP | planar cell polarity |
| PCR | polymerase chain reaction |
| PTK7 | protein tyrosine kinase 7 |
| RPTK | receptor protein tyrosine kinase |
References
- Park, S.-K.; Lee, H.-S.; Lee, S.-T. Characterization of the human full-length PTK7 cDNA encoding a receptor protein tyrosine kinase-like molecule closely related to chick KLG. J. Biochem. 1996, 119, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Borchers, A.G.; Jolicoeur, C.; Rayburn, H.; Baker, J.C.; Tessier-Lavigne, M. PTK7/CCK-4 is a novel regulator of planar cell polarity in vertebrates. Nature 2004, 430, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Puppo, F.; Thome, V.; Lhoumeau, A.C.; Cibois, M.; Gangar, A.; Lembo, F.; Belotti, E.; Marchetto, S.; Lecine, P.; Prebet, T.; et al. Protein tyrosine kinase 7 has a conserved role in Wnt/beta-catenin canonical signalling. EMBO Rep. 2011, 12, 43–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peradziryi, H.; Kaplan, N.A.; Podleschny, M.; Liu, X.; Wehner, P.; Borchers, A.; Tolwinski, N.S. PTK7/Otk interacts with Wnts and inhibits canonical Wnt signalling. EMBO J. 2011, 30, 3729–3740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, S.; Scerbo, P.; Giordano, M.; Daulat, A.M.; Lhoumeau, A.-C.; Thomé, V.; Kodjabachian, L.; Borg, J.-P. The PTK7 and ROR2 protein receptors interact in the vertebrate WNT/planar cell polarity (PCP) pathway. J. Biol. Chem. 2015, 290, 30562–30572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, W.-S.; Kwon, J.; Lee, H.W.; Kang, M.C.; Na, H.-W.; Lee, S.-T.; Park, J.H. Oncogenic role of protein tyrosine kinase 7 in esophageal squamous cell carcinoma. Cancer Sci. 2013, 104, 1120–1126. [Google Scholar] [CrossRef]
- Kashyap, M.K.; Abdel-Rahman, O. Expression, regulation and targeting of receptor tyrosine kinases in esophageal squamous cell carcinoma. Mol. Cancer 2018, 17, 1–11. [Google Scholar] [CrossRef]
- Mossie, K.; Jallal, B.; Alves, F.; Sures, I.; Plowman, G.D.; Ullrich, A. Colon carcinoma kinase-4 defines a new subclass of the receptor tyrosine kinase family. Oncogene 1995, 11, 2179–2184. [Google Scholar]
- Lhoumeau, A.-C.; Martinez, S.; Boher, J.-M.; Monges, G.; Castellano, R.; Goubard, A.; Doremus, M.; Poizat, F.; Lelong, B.; De Chaisemartin, C.; et al. Overexpression of the promigratory and prometastatic PTK7 receptor is associated with an adverse clinical outcome in colorectal cancer. PLoS ONE 2015, 10, e0123768. [Google Scholar] [CrossRef]
- Jin, X.; Huang, T.; Ma, C.; Duan, J.; Li, R.; Zhang, W.; Tian, W. Protein tyrosine kinase 7-knockdown inhibits oral squamous cell carcinoma cell viability, proliferation, migration and invasion via downregulating dishevelled segment polarity protein 3 expression. Exp. Ther. Med. 2021, 22, 1–9. [Google Scholar] [CrossRef]
- Duan, F.; Tang, J.; Kong, F.-L.; Zou, H.-W.; Ni, B.-L.; Yu, J.-C. Identification of PTK7 as a promising therapeutic target for thyroid cancer. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 6809–6817. [Google Scholar] [PubMed]
- Zou, R.-C.; Liang, Y.; Li, L.-L.; Tang, J.-Z.; Yang, Y.-P.; Geng, Y.-C.; He, J.; Luo, L.-Y.; Li, W.X.; Sun, Z.-W.; et al. Bioinformatics analysis identifies protein tyrosine kinase 7 (PTK7) as a potential prognostic and therapeutic biomarker in stages I to IV hepatocellular carcinoma. Med. Sci. Monit. 2019, 25, 8618–8627. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Sefah, K.; O’Donoghue, M.B.; Zhu, G.; Shangguan, D.; Noorali, A.; Chen, Y.; Zhou, L.; Tan, W. Silencing of PTK7 in colon cancer cells: Caspase-10-dependent apoptosis via mitochondrial pathway. PLoS ONE 2010, 5, e14018. [Google Scholar] [CrossRef] [PubMed]
- Prebet, T.; Lhoumeau, A.-C.; Arnoulet, C.; Aulas, A.; Marchetto, S.; Audebert, S.; Puppo, F.; Chabannon, C.; Sainty, D.; Santoni, M.-J.; et al. The cell polarity PTK7 receptor acts as a modulator of the chemotherapeutic response in acute myeloid leukemia and impairs clinical outcome. Blood 2010, 116, 2315–2323. [Google Scholar] [CrossRef] [Green Version]
- Shin, W.S.; Hong, Y.; Lee, H.W.; Lee, S.T. Catalytically defective receptor protein tyrosine kinase PTK7 enhances invasive phenotype by inducing MMP-9 through activation of AP-1 and NF-kappaB in esophageal squamous cell carcinoma cells. Oncotarget 2016, 7, 73242–73256. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Song, G.; Zhang, X.; Li, Q.; Zhao, Y.; Zhou, Y.; Xiong, R.; Hu, X.; Tang, Z.; Feng, G. PTK7 is a novel oncogenic target for esophageal squamous cell carcinoma. World J. Surg. Oncol. 2017, 15, 105. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Li, G.; Yin, Y.; Wang, J.; Wang, H.; Wei, W.; Guo, Q.; Ma, H.; Shi, Q.; Zhou, X.; et al. PTK7 protein is decreased in epithelial ovarian carcinomas with poor prognosis. Int. J. Clin. Exp. Pathol. 2014, 7, 7881–7889. [Google Scholar]
- Kim, J.-H.; Kwon, J.; Lee, H.W.; Kang, M.C.; Yoon, H.-J.; Lee, S.-T.; Park, J.H. Protein tyrosine kinase 7 plays a tumor suppressor role by inhibiting ERK and AKT phosphorylation in lung cancer. Oncol. Rep. 2014, 31, 2708–2712. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424, Erratum in CA Cancer J. Clin. 2020, 70, 313. [Google Scholar] [CrossRef] [Green Version]
- Lagergren, J.; Bergström, R.; Lindgren, A.; Nyrén, O. Symptomatic gastroesophageal reflux as a risk factor for esophageal adenocarcinoma. N. Engl. J. Med. 1999, 340, 825–831. [Google Scholar] [CrossRef]
- Kamangar, F.; Chow, W.-H.; Abnet, C.C.; Dawsey, S.M. Environmental causes of esophageal cancer. Gastroenterol. Clin. N. Am. 2009, 38, 27–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Islami, F.; Fedirko, V.; Tramacere, I.; Bagnardi, V.; Jenab, M.; Scotti, L.; Rota, M.; Corrao, G.; Garavello, W.; Schüz, J.; et al. Alcohol drinking and esophageal squamous cell carcinoma with focus on light-drinkers and never-smokers: A systematic review and meta-analysis. Int. J. Cancer 2010, 129, 2473–2484. [Google Scholar] [CrossRef] [PubMed]
- Shin, W.; Lee, H.W.; Lee, S. Catalytically inactive receptor tyrosine kinase PTK7 activates FGFR1 independent of FGF. FASEB J. 2019, 33, 12960–12971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnas, C.; Martel-Planche, G.; Furukawa, Y.; Hollstein, M.; Montesano, R.; Hainaut, P. Inactivation of the p53 protein in cell lines derived from human esophageal cancers. Int. J. Cancer 1997, 71, 79–87. [Google Scholar] [CrossRef]
- Zhao, L.; He, L.-R.; Xi, M.; Cai, M.-Y.; Shen, J.-X.; Li, Q.-Q.; Liao, Y.-J.; Qian, D.; Feng, Z.-Z.; Zeng, Y.-X.; et al. Nimotuzumab promotes radiosensitivity of EGFR-overexpression esophageal squamous cell carcinoma cells by upregulating IGFBP-3. J. Transl. Med. 2012, 10, 249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimada, Y.; Imamura, M.; Wagata, T.; Yamaguchi, N.; Tobe, T. Characterization of 21 newly established esophageal cancer cell lines. Cancer 1992, 69, 277–284. [Google Scholar] [CrossRef]
- Tanaka, H.; Shibagaki, I.; Shimada, Y.; Wagata, T.; Imamura, M.; Ishizaki, K. Characterization of p53 gene mutations in esophageal squamous cell carcinoma cell lines: Increased frequency and different spectrum of mutations from primary tumors. Int. J. Cancer 1996, 65, 372–376. [Google Scholar] [CrossRef]
- Song, S.; Chang, D.; Cui, Y.; Hu, J.; Gong, M.; Ma, K.; Ding, F.; Liu, Z.-H.; Wang, T.-Y. New orthotopic implantation model of human esophageal squamous cell carcinoma in athymic nude mice. Thorac. Cancer 2014, 5, 417–424. [Google Scholar] [CrossRef]
- Luo, L.-L.; Zhao, L.; Xi, M.; He, L.-R.; Shen, J.-X.; Li, Q.-Q.; Liu, S.-L.; Zhang, P.; Xie, D.; Liu, M.-Z. Association of insulin-like growth factor-binding protein-3 with radiotherapy response and prognosis of esophageal squamous cell carcinoma. Chin. J. Cancer 2015, 34, 514–521. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.J.; Pan, W.W.; Liu, S.B.; Shen, Z.F.; Xu, Y.; Hu, L.L. ERK/MAPK signalling pathway and tumorigenesis. Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in cancer: Mechanisms and advances in clinical trials. Mol. Cancer 2019, 18, 1–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanteti, R.; Batra, S.K.; Lennon, F.E.; Salgia, R. FAK and paxillin, two potential targets in pancreatic cancer. Oncotarget 2016, 7, 31586–31601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, D.-C.; Hao, J.-J.; Nagata, Y.; Xu, L.; Shang, L.; Meng, X.; Sato, Y.; Okuno, Y.; Varela, A.M.; Ding, L.-W.; et al. Genomic and molecular characterization of esophageal squamous cell carcinoma. Nat. Genet. 2014, 46, 467–473. [Google Scholar] [CrossRef]
- Testa, U.; Castelli, G.; Pelosi, E. Esophageal cancer: Genomic and molecular characterization, stem cell compartment and clonal evolution. Medicines 2017, 4, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.; Li, P.; Xing, W.; Qiu, H. Heterogeneous genomic aberrations in esophageal squamous cell carcinoma: A review. Am. J. Transl. Res. 2020, 12, 1553–1568. [Google Scholar]
- Hanawa, M.; Suzuki, S.; Dobashi, Y.; Yamane, T.; Kono, K.; Enomoto, N.; Ooi, A. EGFR protein overexpression and gene amplification in squamous cell carcinomas of the esophagus. Int. J. Cancer 2006, 118, 1173–1180. [Google Scholar] [CrossRef]
- Shigaki, H.; Baba, Y.; Watanabe, M.; Murata, A.; Ishimoto, T.; Iwatsuki, M.; Iwagami, S.; Nosho, K.; Baba, H. PIK3CA mutation is associated with a favorable prognosis among patients with curatively resected esophageal squamous cell carcinoma. Clin. Cancer Res. 2013, 19, 2451–2459. [Google Scholar] [CrossRef] [Green Version]
- Chang, D.; Wang, T.-Y.; Li, H.-C.; Wei, J.-C.; Song, J.-X. Prognostic significance of PTEN expression in esophageal squamous cell carcinoma from Linzhou City, a high incidence area of northern China. Dis. Esophagus 2007, 20, 491–496. [Google Scholar] [CrossRef]
- Gao, Y.-B.; Chen, Z.-L.; Li, J.-G.; Hu, X.-D.; Shi, X.-J.; Sun, Z.-M.; Zhang, F.; Zhao, Z.-R.; Li, Z.-T.; Liu, Z.-Y.; et al. Genetic landscape of esophageal squamous cell carcinoma. Nat. Genet. 2014, 46, 1097–1102. [Google Scholar] [CrossRef]
- Sawada, G.; Niida, A.; Uchi, R.; Hirata, H.; Shimamura, T.; Suzuki, Y.; Shiraishi, Y.; Chiba, K.; Imoto, S.; Takahashi, Y.; et al. Genomic landscape of esophageal squamous cell carcinoma in a Japanese population. Gastroenterology 2016, 150, 1171–1182. [Google Scholar] [CrossRef] [Green Version]
- Abedi-Ardekani, B.; Dar, N.A.; Mir, M.M.; Zargar, S.A.; Lone, M.M.; Martel-Planche, G.; Villar, S.; Mounawar, M.; Saidi, F.; Malekzadeh, R.; et al. Epidermal growth factor receptor (EGFR) mutations and expression in squamous cell carcinoma of the esophagus in central Asia. BMC Cancer 2012, 12, 602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, H.; Wang, C.; Wang, J.; Chen, D.; Deng, J.; Deng, J.; Fan, J.; Badakhshi, H.; Huang, X.; Zhang, L.; et al. A subset of esophageal squamous cell carcinoma patient-derived xenografts respond to cetuximab, which is predicted by high EGFR expression and amplification. J. Thorac. Dis. 2018, 10, 5328–5338. [Google Scholar] [CrossRef] [PubMed]
- Dutton, S.J.; Ferry, D.R.; Blazeby, J.; Abbas, H.; Dahle-Smith, A.; Mansoor, W.; Thompson, J.; Harrison, M.; Chatterjee, A.; Falk, S.; et al. Gefitinib for oesophageal cancer progressing after chemotherapy (COG): A phase 3, multicentre, double-blind, placebo-controlled randomised trial. Lancet Oncol. 2014, 15, 894–904. [Google Scholar] [CrossRef]
- Wang, X.; Niu, H.; Fan, Q.; Lu, P.; Ma, C.; Liu, W.; Liu, Y.; Li, W.; Hu, S.; Ling, Y.; et al. Predictive value of EGFR overexpression and gene amplification on icotinib efficacy in patients with advanced esophageal squamous cell car-cinoma. Oncotarget 2016, 7, 24744–24751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshioka, M.; Ohashi, S.; Ida, T.; Nakai, Y.; Kikuchi, O.; Amanuma, Y.; Matsubara, J.; Yamada, A.; Miyamoto, S.; Natsuizaka, M.; et al. Distinct effects of EGFR inhibitors on epithelial- and mesenchymal-like esophageal squamous cell carcinoma cells. J. Exp. Clin. Cancer Res. 2017, 36, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Nishihira, T.; Hashimoto, Y.; Katayama, M.; Mori, S.; Kuroki, T. Molecular and cellular features of esophageal cancer cells. J. Cancer Res. Clin. Oncol. 1993, 119, 441–449. [Google Scholar] [CrossRef]
- Yang, Y.; Li, W.; Wei, B.; Wu, K.; Liu, D.; Zhu, D.; Zhang, C.; Wen, F.; Fan, Y.; Zhao, S. MicroRNA let-7i inhibits histone lysine demethylase KDM5B to halt esophageal cancer progression. Mol. Ther. Nucleic Acids 2020, 22, 846–861. [Google Scholar] [CrossRef]
- Liu, M.; Hu, Y.; Zhang, M.-F.; Luo, K.-J.; Xie, X.-Y.; Wen, J.; Fu, J.-H.; Yang, H. MMP1 promotes tumor growth and metastasis in esophageal squamous cell carcinoma. Cancer Lett. 2016, 377, 97–104. [Google Scholar] [CrossRef]
- Lee, N.P.; Chan, C.M.; Tung, L.N.; Wang, H.K.; Law, S. Tumor xenograft animal models for esophageal squamous cell car-cinoma. J. Biomed. Sci. 2018, 25, 66. [Google Scholar] [CrossRef]
- Shin, W.-S.; Gim, J.; Won, S.; Lee, S.-T. Biphasic regulation of tumorigenesis by PTK7 expression level in esophageal squamous cell carcinoma. Sci. Rep. 2018, 8, 8519. [Google Scholar] [CrossRef]
- Kodama, T.; Koma, Y.-I.; Arai, N.; Kido, A.; Urakawa, N.; Nishio, M.; Shigeoka, M.; Yokozaki, H. CCL3–CCR5 axis contributes to progression of esophageal squamous cell carcinoma by promoting cell migration and invasion via Akt and ERK pathways. Lab. Investig. 2020, 100, 1140–1157. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Zhang, Y.; Da, J.; Jia, Z.; Wu, H.; Gu, K. Downregulation of SPARC expression decreases cell migration and invasion involving epithelial-mesenchymal transition through the p-FAK/p-ERK pathway in esophageal squamous cell carcinoma. J. Cancer 2020, 11, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhao, D.; Wang, Y.; Zhang, W.; Zhang, J.; Fan, J.; Zhan, Q.; Chen, J. Focal adhesion kinase (FAK) inhibitor-defactinib suppresses the malignant progression of human esophageal squamous cell carcinoma (ESCC) cells via effective blockade of PI3K/AKT axis and downstream molecular network. Mol. Carcinog. 2021, 60, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Quan, J.; Xiao, H.; Luo, J.; Zhang, Q.; Pi, G.; Ye, Y.; He, R.; Liu, Y.; Su, X.; et al. FGFR inhibitor AZD4547 can enhance sensitivity of esophageal squamous cell carcinoma cells with epithelial-mesenchymal transition to gefitinib. Oncol. Rep. 2018, 39, 2270–2278. [Google Scholar] [CrossRef] [PubMed]
- Cui, N.-P.; Qiao, S.; Jiang, S.; Hu, J.-L.; Wang, T.-T.; Liu, W.-W.; Qin, Y.; Wang, Y.-N.; Zheng, L.-S.; Zhang, J.-C.; et al. Protein tyrosine kinase 7 regulates EGFR/Akt signaling pathway and correlates with malignant progression in triple-negative breast cancer. Front. Oncol. 2021, 11, 699889. [Google Scholar] [CrossRef]
- Na, H.W.; Shin, W.S.; Ludwig, A.; Lee, S.T. The cytosolic domain of PTK7, generated from sequential cleavage by ADAM17 and gamma-secretase, enhances cell proliferation and migration in colon cancer cells. J. Biol. Chem. 2012, 287, 25001–25009. [Google Scholar] [CrossRef] [Green Version]
- Shin, W.-S.; Maeng, Y.-S.; Jung, J.-W.; Min, J.-K.; Kwon, Y.-G.; Lee, S.-T. Soluble PTK7 inhibits tube formation, migration, and invasion of endothelial cells and angiogenesis. Biochem. Biophys. Res. Commun. 2008, 371, 793–798. [Google Scholar] [CrossRef]
- Lee, Y.H.; Seo, E.K.; Lee, S.-T. Skullcapflavone II inhibits degradation of type I collagen by suppressing MMP-1 transcription in human skin fibroblasts. Int. J. Mol. Sci. 2019, 20, 2734. [Google Scholar] [CrossRef] [Green Version]
- Shin, W.-S.; Shim, H.J.; Lee, Y.H.; Pyo, M.; Park, J.S.; Ahn, S.Y.; Lee, S.-T. PTK6 localized at the plasma membrane promotes cell proliferation and migration through phosphorylation of Eps8. J. Cell. Biochem. 2017, 118, 2887–2895. [Google Scholar] [CrossRef]
- Shin, W.; Park, M.K.; Lee, Y.H.; Kim, K.W.; Lee, H.; Lee, S. The catalytically defective receptor protein tyrosine kinase EphA10 promotes tumorigenesis in pancreatic cancer cells. Cancer Sci. 2020, 111, 3292–3302. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shin, W.-S.; Park, M.-K.; Kim, J.H.; Oh, S.W.; Jang, J.-Y.; Lee, H.; Lee, S.-T. PTK7, a Catalytically Inactive Receptor Tyrosine Kinase, Increases Oncogenic Phenotypes in Xenograft Tumors of Esophageal Squamous Cell Carcinoma KYSE-30 Cells. Int. J. Mol. Sci. 2022, 23, 2391. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23042391
Shin W-S, Park M-K, Kim JH, Oh SW, Jang J-Y, Lee H, Lee S-T. PTK7, a Catalytically Inactive Receptor Tyrosine Kinase, Increases Oncogenic Phenotypes in Xenograft Tumors of Esophageal Squamous Cell Carcinoma KYSE-30 Cells. International Journal of Molecular Sciences. 2022; 23(4):2391. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23042391
Chicago/Turabian StyleShin, Won-Sik, Mi-Kyung Park, Jae Hoon Kim, Si Won Oh, Ji-Yun Jang, Ho Lee, and Seung-Taek Lee. 2022. "PTK7, a Catalytically Inactive Receptor Tyrosine Kinase, Increases Oncogenic Phenotypes in Xenograft Tumors of Esophageal Squamous Cell Carcinoma KYSE-30 Cells" International Journal of Molecular Sciences 23, no. 4: 2391. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23042391