
Identification of D Modification Sites Using a Random Forest Model Based on Nucleotide Chemical Properties



Abstract
:1. Introduction
2. Results
2.1. Sequence Encoding Scheme and Partition Rate Analysis
2.2. Oversampling and Comparison to Other Algorithms
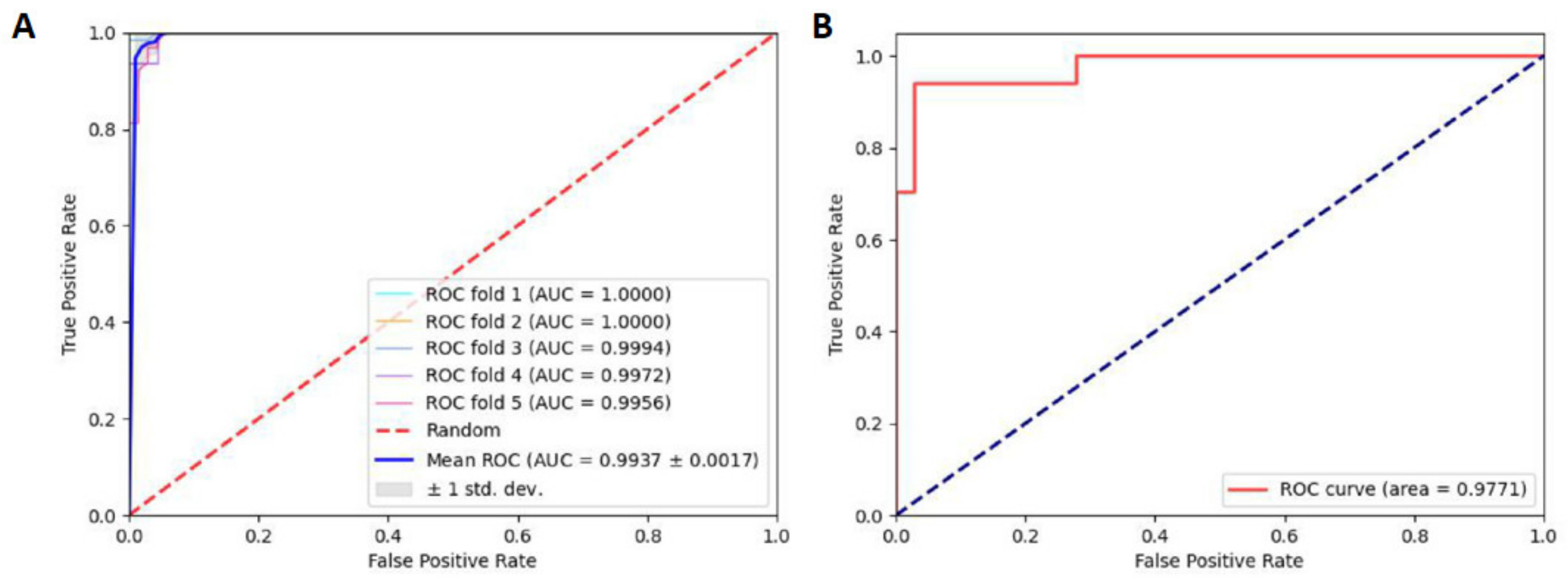
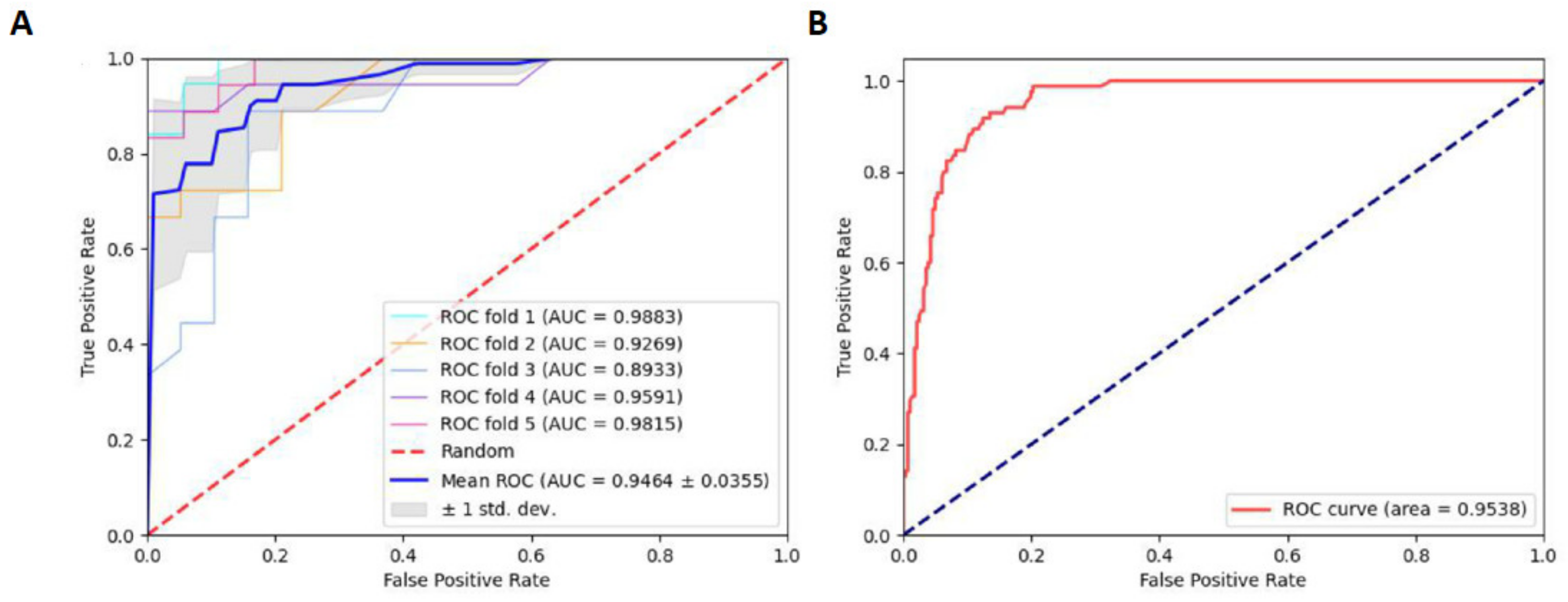
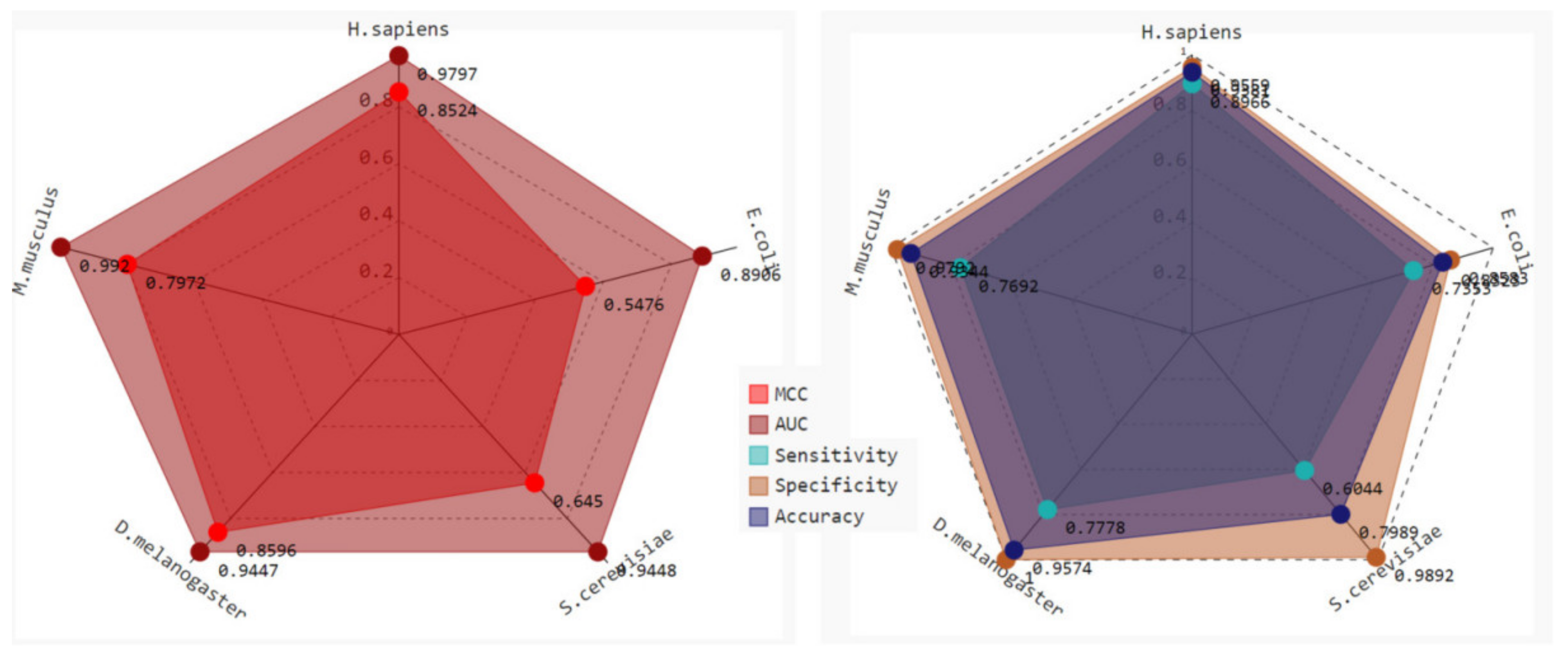
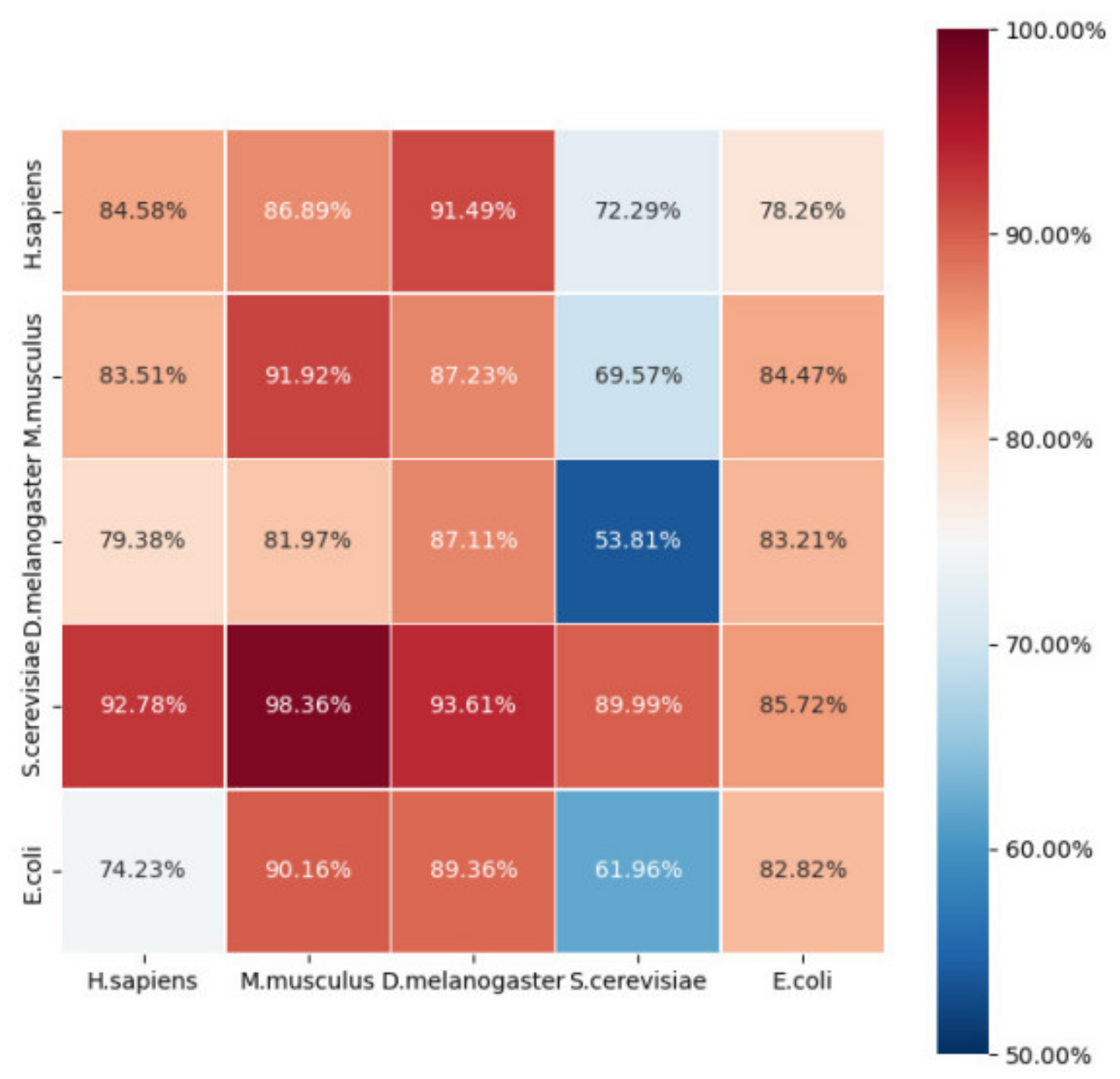
2.3. Robustness and Reliability Analysis
2.4. Comparisons with Other Tools
3. Materials and Methods
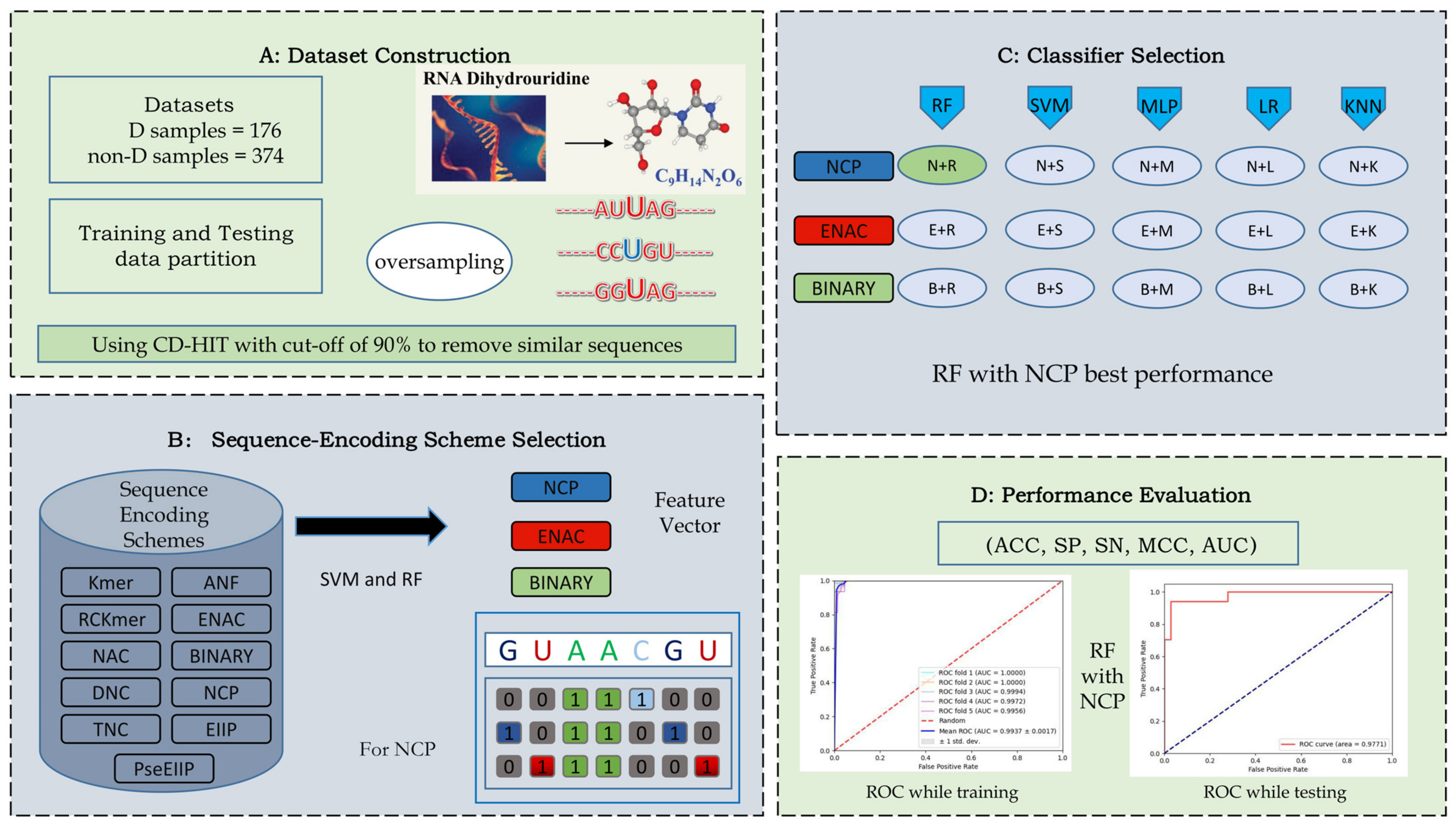
3.1. Benchmark Datasets
3.2. Sequence Encoding Scheme
3.2.1. Nucleotide Chemical Property
3.2.2. Binary
3.2.3. Enhanced Nucleic Acid Composition
3.3. Classifiers
3.4. Performance Evaluation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kirchner, S.; Ignatova, Z. Emerging roles of tRNA in adaptive translation, signalling dynamics and disease. Nat. Rev. Genet. 2014, 16, 98–112. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Mason, C. The pivotal regulatory landscape of RNA modifications. Annual Review of Genomics and Human Genetics. Ann. Rev. Genom. Hum. Gen. 2014, 15, 127–150. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.; Jaffrey, S. The dynamic epitranscriptome: N6-methyladenosine and gene expression control. Nat. Rev. Mol. Cell Biol. 2014, 15, 313–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roundtree, I.; Evans, M.; Pan, T. Dynamic RNA modifications in gene expression regulation. Cells 2017, 169, 1187–1200. [Google Scholar] [CrossRef] [Green Version]
- Boccaletto, P.; Machnicka, M.A.; Purta, E.; Piatkowski, P.; Baginski, B.; Wirecki, T.K.; de Crecy-Lagard, V.; Ross, R.; Limbach, P.A.; Kotter, A.; et al. MODOMICS: A database of RNA modification pathways. Nucleic Acids Res. 2018, 46, D303–D307. [Google Scholar] [CrossRef]
- Guohua, H.; Jincheng, L. Feature extractions for computationally predicting protein post-translational modifications. Curr. Bioinf. 2018, 13, 387–395. [Google Scholar] [CrossRef]
- Xuan, J.J.; Sun, W.J.; Lin, P.H.; Zhou, K.R.; Liu, S.; Zheng, L.L.; Qu, L.H.; Yang, J.H. RMBase v2.0: Deciphering the map of RNA modifications from epitranscriptome sequencing data. Nucleic Acids Res. 2018, 46, D327–D334. [Google Scholar] [CrossRef]
- Lv, H.; Zhang, Z.-M.; Li, S.-H. Evaluation of different computational methods on 5-methylcytosine sites identification. Brief. Bioinform. 2019, 21, 982–995. [Google Scholar] [CrossRef]
- Dao, F.-Y.; Lv, H.; Yang, Y.-H. Computational identification of N6-methyladenosine sites in multiple 396 tissues of mammals. Comput. Struct. Biotechnol. J. 2020, 18, 1084–1091. [Google Scholar] [CrossRef]
- Frye, M.; Jaffrey, S.R.; Pan, T.; Rechavi, G.; Suzuki, T. RNA modifications: What have we learned and where are we 399 headed? Nat. Rev. Genet. 2016, 17, 365–372. [Google Scholar] [CrossRef]
- Madison, J.; Holley, R. The presence of 5,6-dihydrouridylic acid in yeast “soluble” ribonucleic acid. Biochem. Biophys. Res. Commun. 1965, 18, 153–157. [Google Scholar] [CrossRef]
- Edmonds, C.; Crain, R.; Gupta, R. Posttranscriptional modification of tRNA in thermophilic archaea (Archaebacteria). J. Bacteriol. 1991, 173, 3138–3148. [Google Scholar] [CrossRef] [Green Version]
- Sprinzl, M.; Vassilenko, K. Compilation of tRNA sequences and sequences of tRNA genes. Nucleic Acids Res. 2005, 33, D139–D140. [Google Scholar] [CrossRef] [Green Version]
- Yu, F.; Tanaka, Y.; Yamashita, K.; Suzuki, T.; Nakamura, A.; Hirano, N.; Suzuki, T.; Yao, M.; Tanaka, I. Molecular basis of dihydrouridine formation on tRNA. Proc. Natl. Acad. Sci. USA 2011, 108, 19593–19598. [Google Scholar] [CrossRef] [Green Version]
- Dalluge, J.; Hashizume, T.; Sopchik, A. Conformational flexibility in RNA: The role of dihydrouridine. Nucleic Acids Res. 1996, 24, 1073–1079. [Google Scholar] [CrossRef] [Green Version]
- Sundaralingam, M. Molecular conformation of dihydrouridine: Puckered base nucleoside of transfer RNA. Science 1971, 172, 725–727. [Google Scholar] [CrossRef]
- Jones, C.; Spencer, A.; Hsu, J. A counterintuitive Mg2+-dependent and modification-assisted functional folding of mitochondrial tRNAs. J. Mol. Biol. 2006, 362, 771–786. [Google Scholar] [CrossRef] [Green Version]
- Kuchino, Y.; Borek, E. Tumour-specific phenylalanine tRNA contains two supernumerary methylated bases. Nature 1978, 271, 126–129. [Google Scholar] [CrossRef]
- Kato, T.; Daigo, Y.; Hayama, S. A novel human tRNA-dihydrouridine synthase involved in pulmonary carcinogenesis. Cancer Res. 2005, 65, 5638. [Google Scholar] [CrossRef] [Green Version]
- Jacobson, M.; Hedgcoth, C. Levels of 5, 6-dihydrouridine in relaxed and chloramphenicol transfer ribonucleic acid. Biochemistry 1970, 9, 2513–2519. [Google Scholar] [CrossRef]
- Randerath, K. 3H and 32P derivative methods for base composition and sequence analysis of RNA. Methods Enzymol. 1980, 65, 638–680. [Google Scholar] [CrossRef]
- Dalluge, J.; Hamamoto, T.; Horikoshi, K. Quantitative measurement of dihydrouridine in RNA using isotope dilution liquid chromatography-mass spectrometry (LC/MS). Nucleic Acids Res. 1996, 24, 3242–3245. [Google Scholar] [CrossRef] [Green Version]
- Kellner, S.; Ochel, A.; Thüring, K. Absolute and relative quantification of RNA modifications via biosynthetic isotopomers. Nucleic Acids Res. 2014, 42, e142. [Google Scholar] [CrossRef] [Green Version]
- Hiley, S.L.; Jackman, J.; Babak, T.; Trochesset, M.; Morris, Q.D.; Phizicky, E.; Hughes, T.R. Detection and discovery of RNA modifications using microarrays. Nucleic Acids Res. 2005, 33, e2. [Google Scholar] [CrossRef] [Green Version]
- Motorin, Y.; Muller, S.; Behm-Ansmant, I.; Branlant, C. Identification of Modified Residues in RNAs by Reverse Transcription-Based Methods. RNA Modif. 2007, 425, 21–53. [Google Scholar]
- Luvino, D.; Smietana, M.; Vasseur, J.-J. Selective fluorescence-based detection of dihydrouridine with boronic acids. Tetrahedron Lett. 2006, 47, 9253–9256. [Google Scholar] [CrossRef]
- Bishop, A.C.; Xu, J.; Johnson, R.C.; Schimmel, P.; de Crecy-Lagard, V. Identification of the tRNA-dihydrouridine synthase family. J. Biol. Chem. 2002, 277, 25090–25095. [Google Scholar] [CrossRef] [Green Version]
- Feng, P.; Xu, Z.; Yang, H.; Lv, H.; Ding, H.; Liu, L. Identification of D Modification Sites by Integrating Heterogeneous Features in Saccharomyces cerevisiae. Molecules 2019, 24, 380. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.C.; Feng, P.M.; Yang, H.; Qiu, W.R.; Chen, W.; Lin, H. iRNAD: A computational tool for identifying D modification sites in RNA sequence. Bioinformatics 2019, 35, 4922–4929. [Google Scholar] [CrossRef]
- Dou, L.; Zhou, W.; Zhang, L.; Xu, L.; Han, K. Accurate identification of RNA D modification using 438 multiple features. RNA Biol. 2021, 18, 2236–2246. [Google Scholar] [CrossRef]
- Branco, P.; Torgo, L.; Ribeiro, R. A survey of predictive modeling on imbalanced domains. ACM Comput. Surv. 2016, 49, 31. [Google Scholar] [CrossRef]
- Kaur, H.; Pannu, H.; Malhi, A. A systematic review on imbalanced data challenges in machine learning: Applications and solutions. ACM Comput. Surv. 2019, 52, 79. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhao, P.; Li, F.; Wang, Y.; Smith, A.I.; Webb, G.I.; Akutsu, T.; Baggag, A.; Bensmail, H.; Song, J. Comprehensive review and assessment of computational methods for predicting RNA post-transcriptional modification sites from RNA sequences. Brief. Bioinform. 2020, 21, 1676–1696. [Google Scholar] [CrossRef]
- Chen, Z.; Zhao, P.; Li, F.; Andre, L.; Marquez-Lago, T.T.; Wang, Y.; Webb, G.I.; Ian, S.A.; Daly, R.J.; Kuo-Chen, C.J.B. iFeature: A python package and web server for features extraction and selection from protein and peptide sequences. Bioinformatics 2018, 34, 14. [Google Scholar] [CrossRef] [Green Version]
- Feng, P.; Ding, H.; Yang, H.; Chen, W.; Lin, H.; Chou, K.C. iRNA-PseColl: Identifying the Occurrence Sites of Different RNA Modifications by Incorporating Collective Effects of Nucleotides into PseKNC. Mol. Ther. Nucleic Acids 2017, 7, 155–163. [Google Scholar] [CrossRef] [Green Version]
- Li, G.Q.; Liu, Z.; Shen, H.B.; Yu, D.J. TargetM6A: Identifying N(6)-Methyladenosine Sites From RNA Sequences via Position-Specific Nucleotide Propensities and a Support Vector Machine. IEEE Trans. Nanobiosci. 2016, 15, 674–682. [Google Scholar] [CrossRef]
- Chen, W.; Yang, H.; Feng, P.; Ding, H.; Lin, H. iDNA4mC: Identifying DNA N4-methylcytosine sites based on nucleotide chemical properties. Bioinformatics 2017, 33, 3518–3523. [Google Scholar] [CrossRef]
- Xu, Z.C.; Wang, P.; Qiu, W.R.; Xiao, X. iSS-PC: Identifying Splicing Sites via Physical-Chemical Properties Using Deep Sparse Auto-Encoder. Sci. Rep. 2017, 7, 8222. [Google Scholar] [CrossRef]
- Fang, T.; Zhang, Z.; Sun, R.; Zhu, L.; He, J.; Huang, B.; Xiong, Y.; Zhu, X. RNAm5CPred: Prediction of RNA 5-Methylcytosine Sites Based on Three Different Kinds of Nucleotide Composition. Mol. Ther. Nucleic Acids 2019, 18, 739–747. [Google Scholar] [CrossRef] [Green Version]
- Manavalan, B.; Basith, S.; Shin, T.H.; Lee, D.Y.; Wei, L.; Lee, G. 4mCpred-EL: An Ensemble Learning Framework for Identification of DNA N(4)-methylcytosine Sites in the Mouse Genome. Cells 2019, 8, 1332. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Zhao, P.; Li, F. iLearn: An integrated platform and meta-learner for feature engineering, machine-learning analysis and modeling of DNA, RNA and protein sequence data. Brief. Bioinform. 2019, 21, 1047–1057. [Google Scholar] [CrossRef]
- Zhang, J.; Feng, P.; Lin, H.; Chen, W. Identifying RNA N(6)-Methyladenosine Sites in Escherichia coli Genome. Front. Microbiol. 2018, 9, 955. [Google Scholar] [CrossRef]
- Zhang, L.; Qin, X.; Liu, M.; Xu, Z.; Liu, G. DNN-m6A: A Cross-Species Method for Identifying RNA N6-Methyladenosine Sites Based on Deep Neural Network with Multi-Information Fusion. Genes 2021, 12, 354. [Google Scholar] [CrossRef]
- Han, T.; Jiang, D.; Zhao, Q.; Wang, L.; Yin, K. Comparison of random forest, artificial neural networks and support vector machine for intelligent diagnosis of rotating machinery. Trans. Inst. Measur. Control 2017, 40, 2681–2693. [Google Scholar] [CrossRef]
- Huang, Y.; He, N.; Chen, Y.; Chen, Z.; Li, L. BERMP: A cross-species classifier for predicting m(6)A sites by integrating a deep learning algorithm and a random forest approach. Int. J. Biol. Sci. 2018, 14, 1669–1677. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Zheng, W.; Li, W.; Huang, Y. Large group activity security risk assessment and risk 479 early warning based on random forest algorithm. Pattern Recognit. Lett. 2021, 144, 1–5. [Google Scholar] [CrossRef]
- Velo, R.; López, P.; Maseda, F. Wind speed estimation using multilayer perceptron. Energy Convers. Manag. 2014, 81, 1–9. [Google Scholar] [CrossRef]
- Liu, H.; Tian, H.-Q.; Li, Y.-F.; Zhang, L. Comparison of four Adaboost algorithm based artificial neural networks in wind speed predictions. Energy Convers. Manag. 2015, 92, 67–81. [Google Scholar] [CrossRef]
- Zhang, S.; Li, X.; Zong, M.; Zhu, X.; Wang, R. Efficient kNN Classification With Different Numbers of Nearest Neighbors. IEEE Trans. Neural Netw. Learn. Syst. 2018, 29, 1774–1785. [Google Scholar] [CrossRef]
- Kasza, J.; Wolfe, R. Interpretation of commonly used statistical regression models. Respirology 2014, 19, 14–21. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Yang, W.; Dao, F.Y. A comparison and assessment of computational method for identifying recombination hotspots in Saccharomyces cerevisiae. Brief. Bioinform. 2019, 21, 1568–1580. [Google Scholar] [CrossRef]
- Feng, P.M.; Chen, W.; Lin, H.; Chou, K.C. iHSP-PseRAAAC: Identifying the heat shock protein families using pseudo reduced amino acid alphabet composition. Anal. Biochem. 2013, 442, 118–125. [Google Scholar] [CrossRef]
- Tang, H.; Zhao, Y.W.; Zou, P.; Zhang, C.M.; Chen, R.; Huang, P.; Lin, H. HBPred: A tool to identify growth hormone-binding proteins. Int. J. Biol. Sci. 2018, 14, 957–964. [Google Scholar] [CrossRef]
- Yang, H.; Qiu, W.R.; Liu, G.; Guo, F.B.; Chen, W.; Chou, K.C.; Lin, H. iRSpot-Pse6NC: Identifying recombination spots in Saccharomyces cerevisiae by incorporating hexamer composition into general PseKNC. Int. J. Biol. Sci. 2018, 14, 883–891. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Wang, Y.; Li, F.; Akutsu, T.; Rawlings, N.D.; Webb, G.I.; Chou, K.C. iProt-Sub: A comprehensive package for accurately mapping and predicting protease-specific substrates and cleavage sites. Brief. Bioinform. 2019, 20, 638–658. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.-J.; Feng, C.-Q.; Lai, H.-Y.; Chen, W.; Hao, L. Predicting protein structural classes for low-similarity sequences by evaluating different features. Knowl.-Based Syst. 2019, 163, 787–793. [Google Scholar] [CrossRef]
- Sokolova, M.; Lapalme, G. A systematic analysis of performance measures for classification tasks. Inf. Process. Manag. 2009, 45, 427–437. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Performance | SVM | RF | ||||||
|---|---|---|---|---|---|---|---|---|
| Sn | Sp | Acc | MCC | Sn | Sp | Acc | MCC | |
| Kmer | 0.2955 | 0.8772 | 0.7152 | 0.2056 | 0.5161 | 0.6176 | 0.5859 | 0.1255 |
| RCKmer | 0.1364 | 0.9298 | 0.7089 | 0.1044 | 0.5263 | 0.8056 | 0.7091 | 0.3415 |
| NAC | 0.1136 | 0.9912 | 0.7468 | 0.2459 | 0.4063 | 0.7910 | 0.6667 | 0.2072 |
| DNC | 0.2955 | 0.8772 | 0.7152 | 0.2056 | 0.5806 | 0.7794 | 0.7172 | 0.3542 |
| TNC | 0.5682 | 0.8772 | 0.7911 | 0.4630 | 0.7368 | 0.9444 | 0.8727 | 0.7133 |
| ANF | 0.4773 | 0.8947 | 0.7785 | 0.4102 | 0.6316 | 0.8889 | 0.8000 | 0.5449 |
| ENAC | 0.8864 | 0.9737 | 0.9494 | 0.8727 | 0.8947 | 0.9722 | 0.9455 | 0.8786 |
| BINARY | 0.8636 | 0.9474 | 0.9241 | 0.8110 | 0.8065 | 1.0000 | 0.9394 | 0.8609 |
| NCP | 0.8636 | 0.9474 | 0.9241 | 0.8110 | 0.9063 | 0.9851 | 0.9596 | 0.9071 |
| EIIP | 0.6818 | 0.8860 | 0.8291 | 0.5718 | 0.8125 | 1.0000 | 0.9394 | 0.8636 |
| PseEIIP | 0.5682 | 0.6754 | 0.6456 | 0.2236 | 0.7368 | 0.9444 | 0.8727 | 0.7133 |
| Performance | Sn | Sp | Acc | MCC | |
|---|---|---|---|---|---|
| Encoding Scheme | Testing Data Partition Rate | ||||
| ENAC | 30% | 0.2623 | 0.9100 | 0.6646 | 0.2308 |
| 20% | 0.6111 | 0.8136 | 0.7368 | 0.4327 | |
| 10% | 0.8947 | 0.9167 | 0.9091 | 0.8021 | |
| BINARY | 30% | 0.5738 | 0.9600 | 0.8137 | 0.6044 |
| 20% | 0.5556 | 0.9322 | 0.7895 | 0.5446 | |
| 10% | 0.7895 | 0.9722 | 0.9091 | 0.7975 | |
| NCP | 30% | 0.6957 | 0.9818 | 0.8974 | 0.7482 |
| 20% | 0.7857 | 0.9524 | 0.9011 | 0.7632 | |
| 10% | 0.8421 | 0.9722 | 0.9273 | 0.8379 | |
| Performance | Sn | Sp | Acc | MCC | |
|---|---|---|---|---|---|
| Algorithm | Encoding Scheme | ||||
| RF | ENAC | 0.9375 | 0.9706 | 0.9545 | 0.9093 |
| BINARY | 0.9531 | 0.9559 | 0.9545 | 0.9090 | |
| NCP | 0.9688 | 0.9706 | 0.9697 | 0.9393 | |
| SVM | ENAC | 0.9063 | 0.8235 | 0.8333 | 0.6670 |
| BINARY | 0.8438 | 0.8824 | 0.8939 | 0.7882 | |
| NCP | 0.9688 | 0.8529 | 0.9091 | 0.8247 | |
| KNN | ENAC | 0.9688 | 0.8235 | 0.8939 | 0.7978 |
| BINARY | 0.9531 | 0.7059 | 0.8258 | 0.6764 | |
| NCP | 0.9688 | 0.8676 | 0.9167 | 0.8384 | |
| LR | ENAC | 0.8594 | 0.8235 | 0.8409 | 0.6827 |
| BINARY | 0.9063 | 0.8382 | 0.8712 | 0.7449 | |
| NCP | 0.7500 | 0.9552 | 0.8889 | 0.7406 | |
| MLP | ENAC | 0.9219 | 0.7941 | 0.8561 | 0.7197 |
| BINARY | 0.9219 | 0.8971 | 0.9091 | 0.8186 | |
| NCP | 0.9688 | 0.8971 | 0.9318 | 0.8663 | |
| Tools | Sn (%) | Sp (%) | Acc (%) | MCC | AUC | Pre (%) | F1 |
|---|---|---|---|---|---|---|---|
| iRNAD | 86.11 | 96.05 | 92.86 | 0.83 | 0.98 | N/A | N/A |
| iRNAD_XGBoost | 91.67 | 94.74 | 93.75 | 0.86 | 0.87 | 89.19 | 0.90 |
| This work | 96.88 | 97.06 | 96.97 | 0.94 | 0.98 | 96.29 | 0.85 |
| Species | H. sapiens | M. musculus | D. melanogaster | S. cerevisiae | E. coli |
|---|---|---|---|---|---|
| Pos | 29 | 13 | 9 | 91 | 34 |
| Neg | 68 | 48 | 38 | 93 | 127 |
| Chemical Properties | Classes | Nucleotides |
|---|---|---|
| Ring Structure | Pyrimidine | U, C |
| Purine | G, A | |
| Functional Group | Keto | U, G |
| Amino | C, A | |
| Hydrogen Bond | Weak | U, A |
| Strong | G, C |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, H.; Ao, C.-Y.; Ding, Y.-J.; Hao, H.-X.; Yu, L. Identification of D Modification Sites Using a Random Forest Model Based on Nucleotide Chemical Properties. Int. J. Mol. Sci. 2022, 23, 3044. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23063044
Zhu H, Ao C-Y, Ding Y-J, Hao H-X, Yu L. Identification of D Modification Sites Using a Random Forest Model Based on Nucleotide Chemical Properties. International Journal of Molecular Sciences. 2022; 23(6):3044. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23063044
Chicago/Turabian StyleZhu, Huan, Chun-Yan Ao, Yi-Jie Ding, Hong-Xia Hao, and Liang Yu. 2022. "Identification of D Modification Sites Using a Random Forest Model Based on Nucleotide Chemical Properties" International Journal of Molecular Sciences 23, no. 6: 3044. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23063044