
Long Non-Coding RNAs Might Regulate Phenotypic Switch of Vascular Smooth Muscle Cells Acting as ceRNA: Implications for In-Stent Restenosis



Abstract
:1. Introduction
2. Results
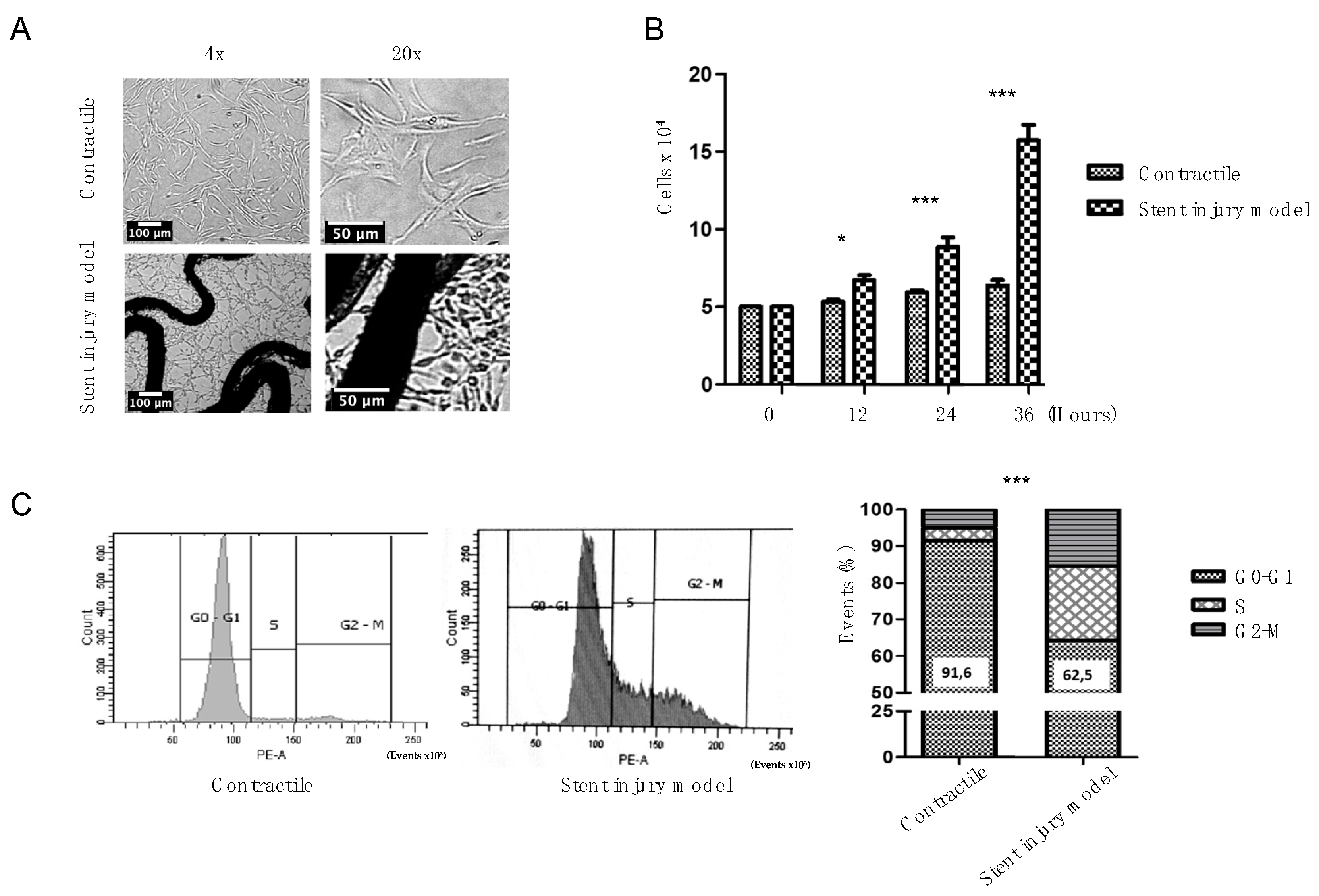
2.1. Phenotypic Change Induction by Stent and PDGF-BB
2.2. Stent Injury Model Promotes Cells Migration and the Expression of Genetic Markers of Phenotypic Switch
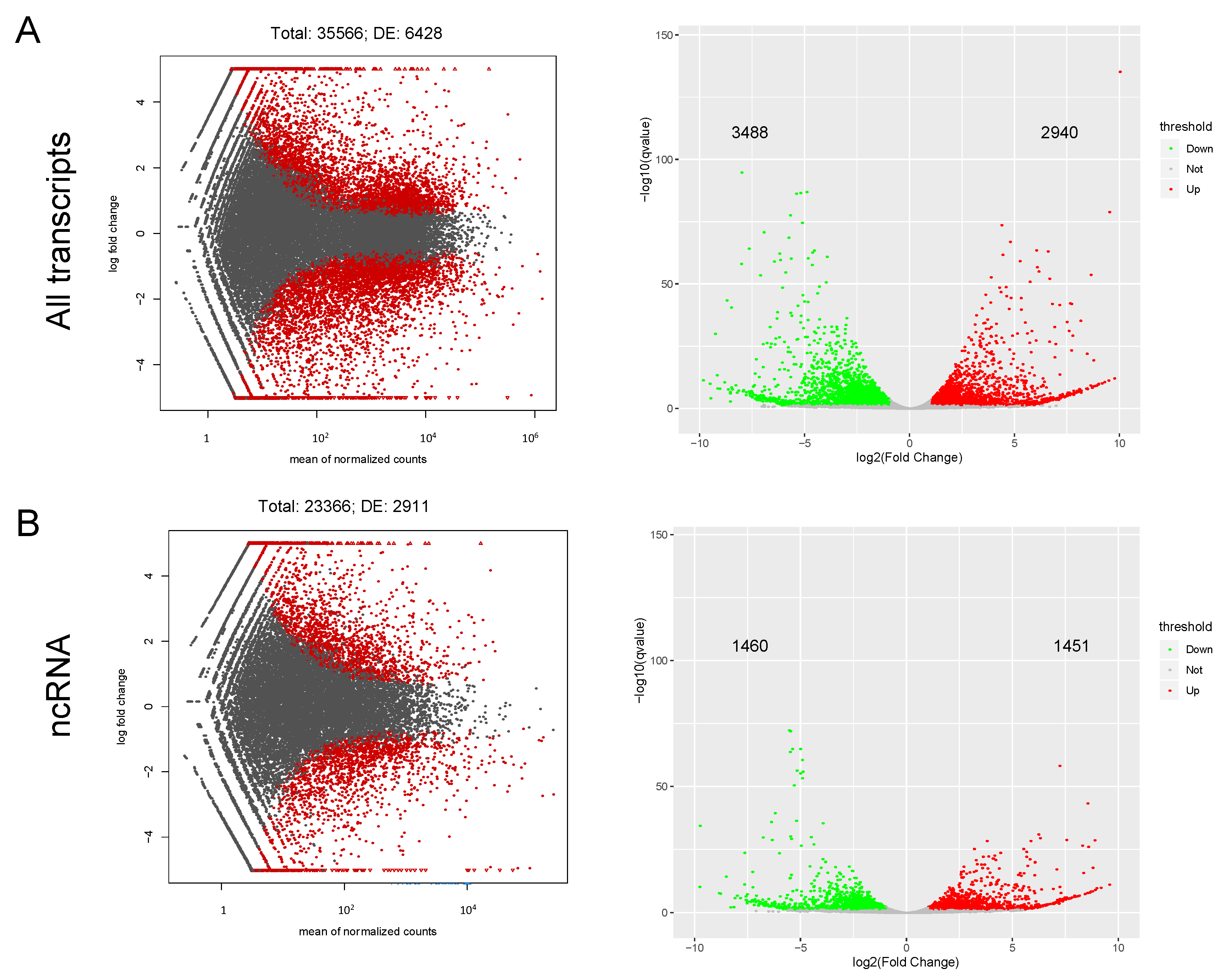
2.3. Stent Injury Model Drives Activation of ncRNA Genes along with mRNA Downregulation
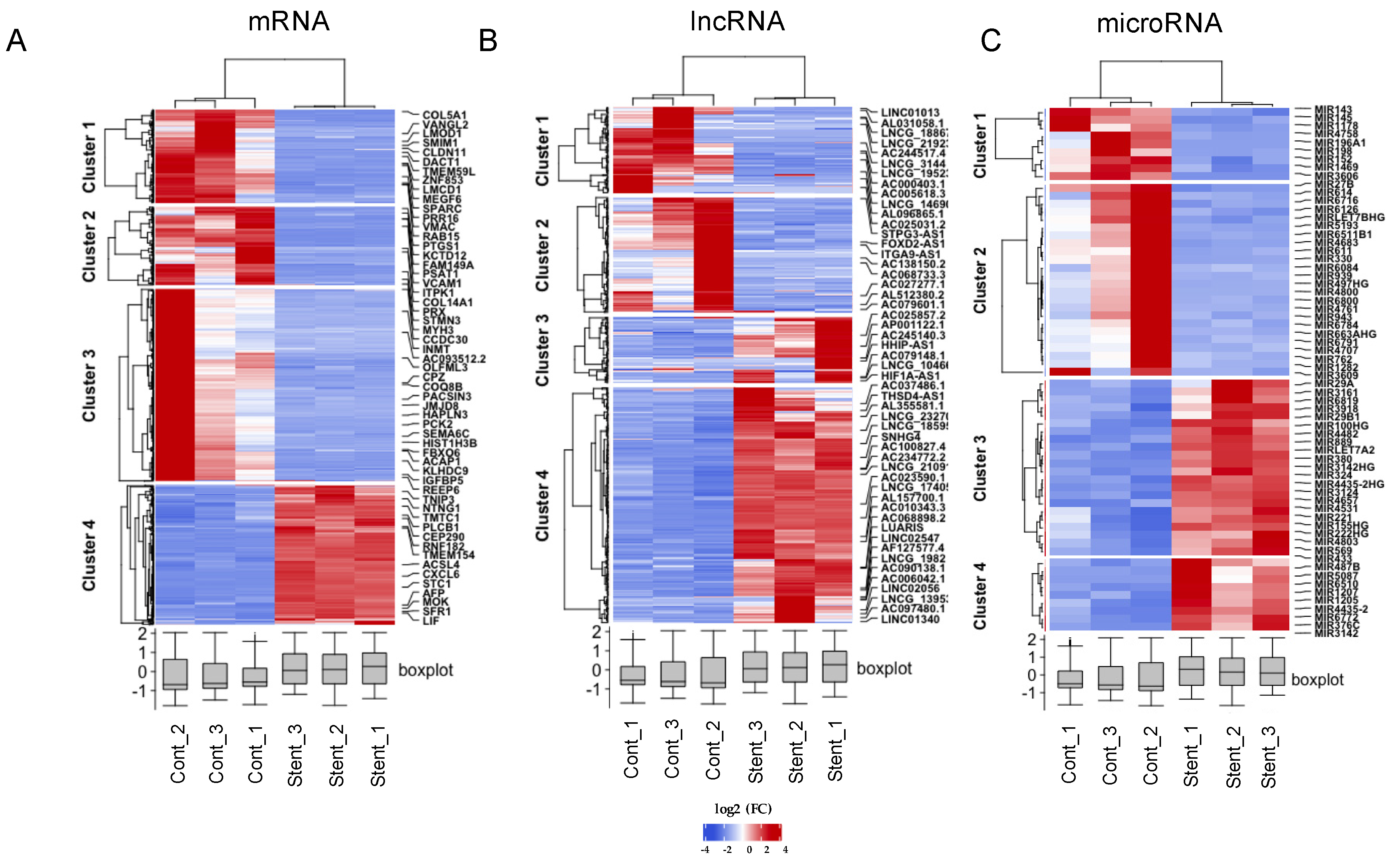
2.4. Identification of Differentially Expressed Genes in HUASMC Exposed to PDGF and Stent
2.5. Gene Enrichment Analysis of DE mRNA and Cis-Target mRNA of lncRNAs
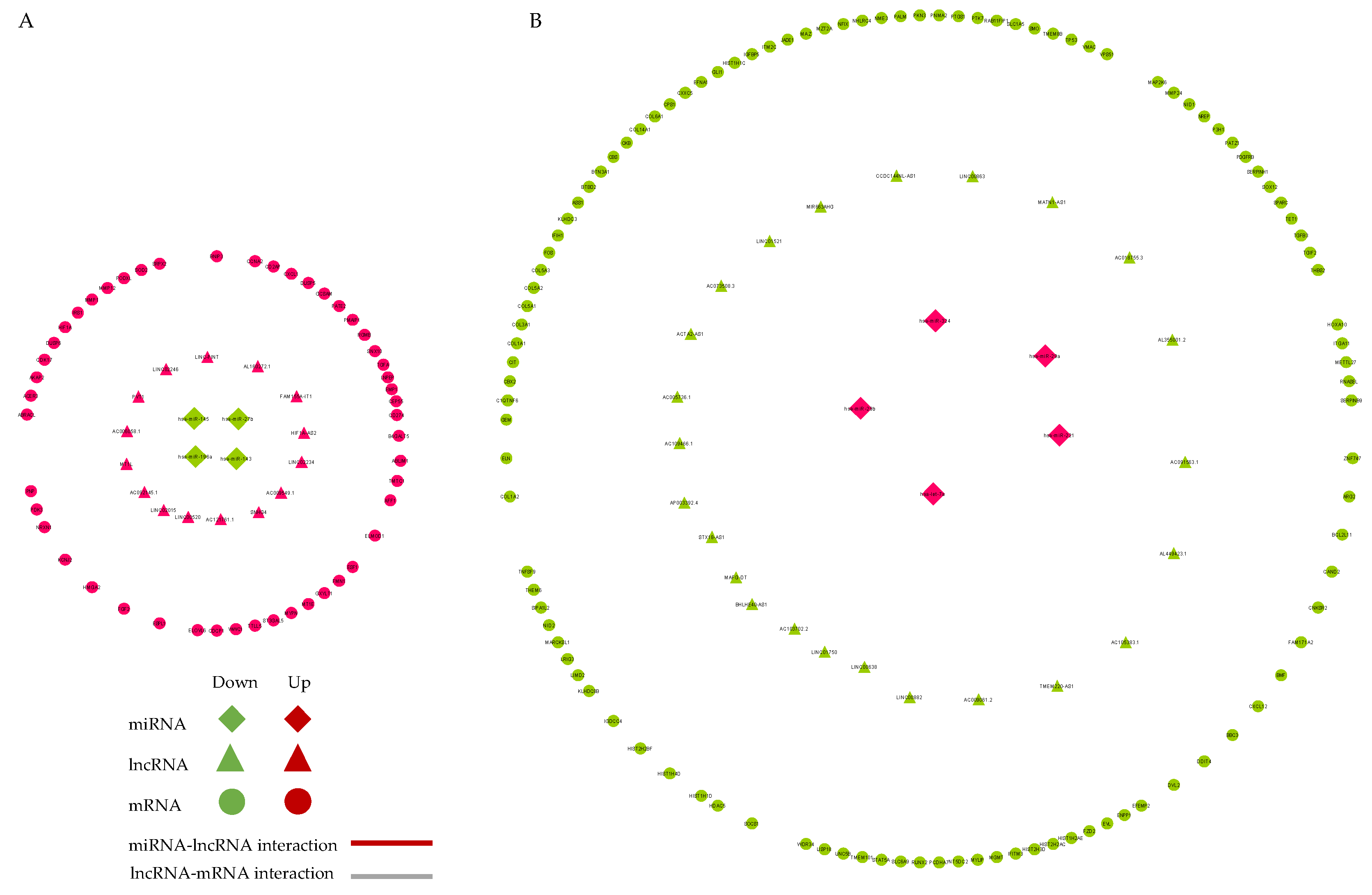
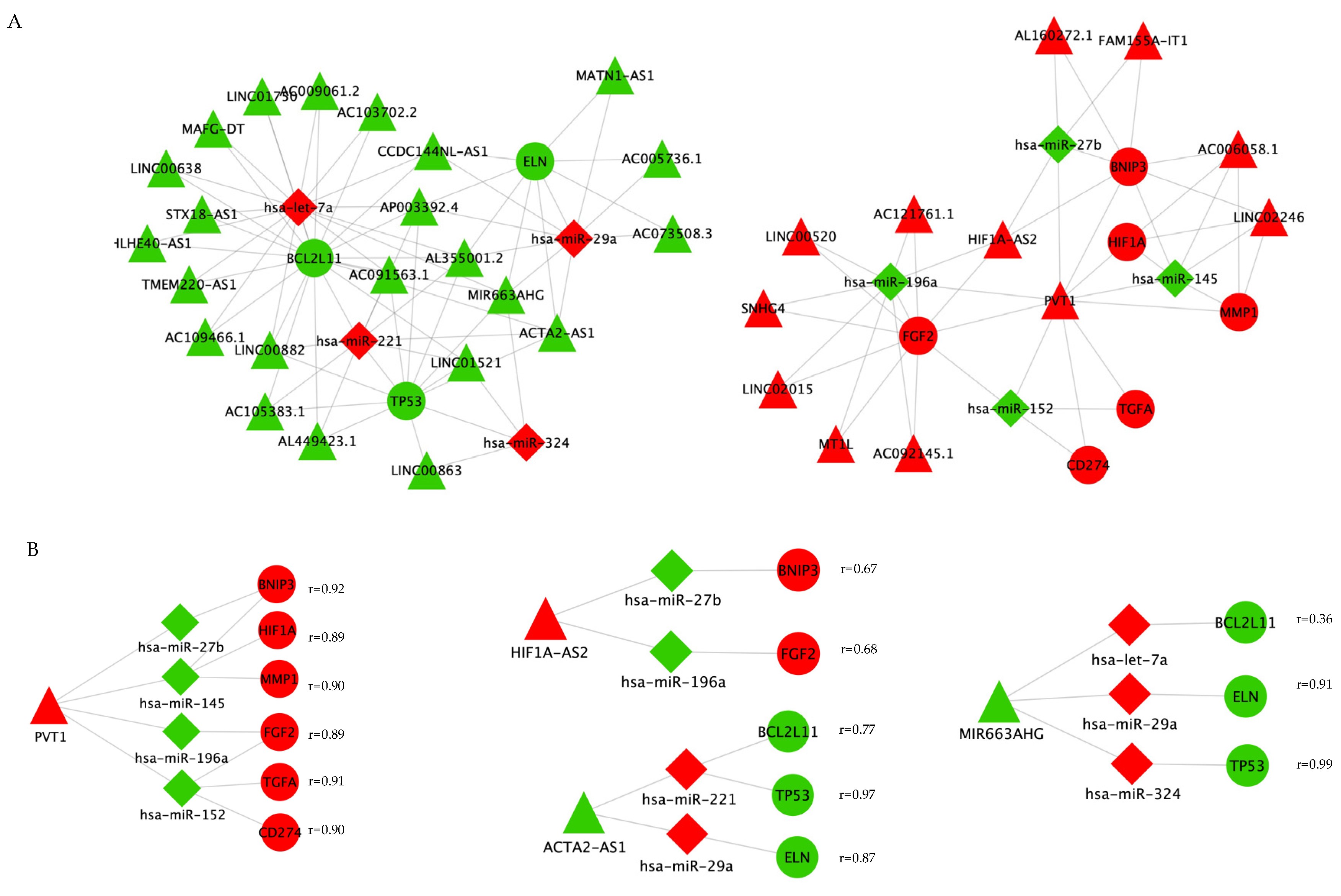
2.6. Competing Endogenous RNA (ceRNA) Network Construction
2.7. PPI Network Analysis
2.8. Key CeRNA Selection
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Stent-Induced Injury Model
4.3. Cell Migration
4.4. Cell Cycle Analysis
4.5. RNA Extraction
4.6. RNAseq Analysis
4.7. Gene Expression Quantitative Analysis
4.8. Filtering of Candidate lncRNAs
- (1)
- Screening exons, filtering low-expression and low-quality single exon transcripts;
- (2)
- Selecting transcripts which were longer than 200 bp and had more than two exons;
- (3)
- Screening transcripts with known annotations using Cuffcompare;
- (4)
- Transcript expression level filtering by calculating the expression level of each transcript and choosing those with FPKM ≥ 0.5;
- (5)
4.9. Prediction of lncRNA Target Genes
4.10. Functional Analysis
4.11. Construction of lncRNA–miRNa–mRNA Competing Endogenous RNA (ceRNA) Network
4.12. Protein–Protein Interaction Network
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jeewandara, T.M.; Wise, S.G.; Ng, M.K.C. Biocompatibility of Coronary Stents. Materials 2014, 7, 769–786. [Google Scholar] [CrossRef]
- Habib, A.; Finn, A.V. Endothelialization of drug eluting stents and its impact on dual anti-platelet therapy duration. Pharmacol. Res. 2015, 93, 22–27. [Google Scholar] [CrossRef] [Green Version]
- Rensen, S.S.; Doevendans, P.A.; van Eys, G.J. Regulation and characteristics of vascular smooth muscle cell phenotypic diversity. Neth. Heart J. 2007, 15, 100–108. [Google Scholar] [CrossRef] [Green Version]
- Beamish, J.A.; He, P.; Kottke-Marchant, K.; Marchant, R.E. Molecular Regulation of Contractile Smooth Muscle Cell Phenotype: Implications for Vascular Tissue Engineering. Tissue Eng. Part B Rev. 2010, 16, 467–491. [Google Scholar] [CrossRef] [Green Version]
- Timraz, S.B.H.; Farhat, I.A.H.; Alhussein, G.; Christoforou, N.; Teo, J.C.M. In-depth evaluation of commercially available human vascular smooth muscle cells phenotype: Implications for vascular tissue engineering. Exp. Cell Res. 2016, 343, 168–176. [Google Scholar] [CrossRef]
- Li, L.; Wang, R.; Shi, H.H.; Xie, L.; Li, J.D.S.; Kong, W.C.; Tang, J.T.; Ke, D.N.; Zhao, L.Y. In vitro study on the feasibility of magnetic stent hyperthermia for the treatment of cardiovascular restenosis. Exp. Ther. Med. 2013, 6, 347–354. [Google Scholar] [CrossRef] [Green Version]
- Guerra, E.; Byrne, R.A.; Kastrati, A. Pharmacological inhibition of coronary restenosis: Systemic and local approaches. Expert Opin. Pharmacother. 2014, 15, 2155–2171. [Google Scholar] [CrossRef]
- Qiu, M.-T.; Hu, J.-W.; Yin, R.; Xu, L. Long noncoding RNA: An emerging paradigm of cancer research. Tumor Biol. 2013, 34, 613–620. [Google Scholar] [CrossRef]
- Verjans, R.; van Bilsen, M.; Schroen, B. Reviewing the Limitations of Adult Mammalian Cardiac Regeneration: Noncoding RNAs as Regulators of Cardiomyogenesis. Biomolecules 2020, 10, 262. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Shah, R.; Dimmeler, S.; Freedman, J.E.; Holley, C.; Lee, J.M.; Moore, K.; Musunuru, K.; Wang, D.Z.; Xiao, J.; et al. Noncoding RNAs in Cardiovascular Disease: Current Knowledge, Tools and Technologies for Investigation, and Future Directions: A Scientific Statement From the American Heart Association. Circ. Genom. Precis. Med. 2020, 13, e000062. [Google Scholar] [CrossRef]
- Pasmant, E.; Sabbagh, A.; Vidaud, M.; Bieche, I. ANRIL, a long, noncoding RNA, is an unexpected major hotspot in GWAS. FASEB J. 2011, 25, 444–448. [Google Scholar] [CrossRef]
- Burd, C.E.; Jeck, W.R.; Liu, Y.; Sanoff, H.K.; Wang, Z.; Sharpless, N.E. Expression of Linear and Novel Circular Forms of an INK4/ARF-Associated Non-Coding RNA Correlates with Atherosclerosis Risk. PLoS Genet. 2010, 6, e0171448. [Google Scholar] [CrossRef]
- Bell, R.D.; Long, X.C.; Lin, M.Y.; Bergmann, J.H.; Nanda, V.; Cowan, S.L.; Zhou, Q.; Han, Y.; Spector, D.L.; Zheng, D.Y.; et al. Identification and Initial Functional Characterization of a Human Vascular Cell-Enriched Long Noncoding RNA. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1249–1259. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.Z.; Cai, J.; Han, Y.; Chen, J.H.; Huang, Z.P.; Chen, C.Y.; Cai, Y.; Huang, H.F.; Yang, Y.J.; Liu, Y.K.; et al. LincRNA-p21 Regulates Neointima Formation, Vascular Smooth Muscle Cell Proliferation, Apoptosis, and Atherosclerosis by Enhancing p53 Activity. Circulation 2014, 130, 1452–1465. [Google Scholar] [CrossRef] [Green Version]
- Leung, A.; Trac, C.; Jin, W.; Lanting, L.; Akbany, A.; Saetrom, P.; Schones, D.E.; Natarajan, R. Novel Long Noncoding RNAs Are Regulated by Angiotensin II in Vascular Smooth Muscle Cells. Circ. Res. 2013, 113, 266–278. [Google Scholar] [CrossRef] [Green Version]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA hypothesis: The Rosetta Stone of a hidden RNA language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Han, P.; Guo, Q.; Hao, Y.; Qi, Y.; Xin, M.; Cui, B.; Wang, P. Oncogenic Landscape of Somatic Mutations Perturbing Pan-Cancer lncRNA-ceRNA Regulation. Front. Cell Dev. Biol. 2021, 9, 658346. [Google Scholar] [CrossRef]
- Liu, B.; Zhou, X.; Wu, D.; Zhang, X.; Shen, X.; Mi, K.; Qu, Z.; Jiang, Y.; Shang, D. Comprehensive characterization of a drug-resistance-related ceRNA network across 15 anti-cancer drug categories. Mol. Ther. Nucleic Acids 2021, 24, 11–24. [Google Scholar] [CrossRef]
- Song, C.; Zhang, J.; Qi, H.; Feng, C.; Chen, Y.; Cao, Y.; Ba, L.; Ai, B.; Wang, Q.; Huang, W.; et al. The global view of mRNA-related ceRNA cross-talks across cardiovascular diseases. Sci. Rep. 2017, 7, 10185. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Liu, C.; Zhang, Z. lncRNA GAS5 acts as a ceRNA for miR-21 in suppressing PDGF-bb-induced proliferation and migration in vascular smooth muscle cells. J. Cell Biochem. 2019, 120, 15233–15240. [Google Scholar] [CrossRef]
- Lin, Z.; Ge, J.; Wang, Z.; Ren, J.; Wang, X.; Xiong, H.; Gao, J.; Zhang, Y.; Zhang, Q. Let-7e modulates the inflammatory response in vascular endothelial cells through ceRNA crosstalk. Sci. Rep. 2017, 7, 42498. [Google Scholar] [CrossRef] [PubMed]
- Sandison, M.E.; Dempster, J.; McCarron, J.G. The transition of smooth muscle cells from a contractile to a migratory, phagocytic phenotype: Direct demonstration of phenotypic modulation. J. Physiol. 2016, 594, 6189–6209. [Google Scholar] [CrossRef] [Green Version]
- Santin, M.; Morris, C.; Harrison, M.; Mikhalovska, L.; Lloyd, A.; Mikhalovsky, S. Factors inducing in-stent restenosis: An in-vitro model. Med. J. Malays. 2004, 59 (Suppl. B), 93–94. [Google Scholar]
- Guildford, A.L.; Stewart, H.J.S.; Morris, C.; Santin, M. Substrate-induced phenotypic switches of human smooth muscle cells: An in vitro study of in-stent restenosis activation pathways. J. R. Soc. Interface 2011, 8, 641–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caglayan, E.; Vantler, M.; Leppanen, O.; Gerhardt, F.; Mustafov, L.; ten Freyhaus, H.; Kappert, K.; Odenthal, M.; Zimmermann, W.H.; Tallquist, M.D.; et al. Disruption of Platelet-Derived Growth Factor-Dependent Phosphatidylinositol 3-Kinase and Phospholipase C gamma 1 Activity Abolishes Vascular Smooth Muscle Cell Proliferation and Migration and Attenuates Neointima Formation In Vivo. J. Am. Coll. Cardiol. 2011, 57, 2527–2538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, X.; Chen, S.Y.; Liu, M.; Liang, T.M.; Liu, C. Evodiamine inhibits PDGF-BB-induced proliferation of rat vascular smooth muscle cells through the suppression of cell cycle progression and oxidative stress. Mol. Med. Rep. 2016, 14, 4551–4558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharyya, A.; Lin, S.; Sandig, M.; Mequanint, K. Regulation of vascular smooth muscle cell phenotype in three-dimensional coculture system by Jagged1-selective Notch3 signaling. Tissue Eng. Part. A 2014, 20, 1175–1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fellows, B.D.; Ghobrial, N.; Mappus, E.; Hargett, A.; Bolding, M.; Dean, D.; Mefford, O.T. In vitro studies of heparin-coated magnetic nanoparticles for use in the treatment of neointimal hyperplasia. Nanomedicine 2018, 14, 1191–1200. [Google Scholar] [CrossRef] [PubMed]
- Chiu, J.J.; Chen, L.J.; Lee, P.L.; Lee, C.I.; Lo, L.W.; Usami, S.; Chien, S. Shear stress inhibits adhesion molecule expression in vascular endothelial cells induced by coculture with smooth muscle cells. Blood 2003, 101, 2667–2674. [Google Scholar] [CrossRef] [Green Version]
- Zeng, Z.; Yao, J.; Li, Y.; Xue, Y.; Zou, Y.; Shu, Z.; Jiao, Z. Anti-apoptosis endothelial cell-secreted microRNA-195-5p promotes pulmonary arterial smooth muscle cell proliferation and migration in pulmonary arterial hypertension. J. Cell Biochem. 2018, 119, 2144–2155. [Google Scholar] [CrossRef] [PubMed]
- Orr, A.W.; Lee, M.Y.; Lemmon, J.A.; Yurdagul, A.; Gomez, M.F.; Bortz, P.D.; Wamhoff, B.R. Molecular mechanisms of collagen isotype-specific modulation of smooth muscle cell phenotype. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 225–231. [Google Scholar] [CrossRef]
- Ma, Z.; Mao, C.; Jia, Y.; Fu, Y.; Kong, W. Extracellular matrix dynamics in vascular remodeling. Am. J. Physiol. Cell Physiol. 2020, 319, C481–C499. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Du, Y.T. LncRNAs: From Basic Research to Medical Application. Int. J. Biol. Sci. 2017, 13, 295–307. [Google Scholar] [CrossRef] [PubMed]
- Frismantiene, A.; Philippova, M.; Erne, P.; Resink, T.J. Smooth muscle cell-driven vascular diseases and molecular mechanisms of VSMC plasticity. Cell Signal 2018, 52, 48–64. [Google Scholar] [CrossRef]
- Statello, L.; Guo, C.-J.; Chen, L.-L.; Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 2021, 22, 96–118. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.; Ulitsky, I.; Alloza, I.; Vandenbroeck, K.; Miscianinov, V.; Mahmoud, A.; Ballantyne, M.; Rodor, J.; Baker, A. Novel Transcript Discovery Expands the Repertoire of Pathologically-Associated, Long Non-Coding RNAs in Vascular Smooth Muscle Cells. Int. J. Mol. Sci. 2021, 22, 1484. [Google Scholar] [CrossRef]
- Traversa, D.; Simonetti, G.; Tolomeo, D.; Visci, G.; Macchia, G.; Ghetti, M.; Martinelli, G.; Kristensen, L.S.; Storlazzi, C.T. Unravelling similarities and differences in the role of circular and linear PVT1 in cancer and human disease. Br. J. Cancer 2021. [Google Scholar] [CrossRef]
- Li, S.; Zhao, X.; Cheng, S.; Li, J.; Bai, X.; Meng, X. Downregulating long non-coding RNA PVT1 expression inhibited the viability, migration and phenotypic switch of PDGF-BB-treated human aortic smooth muscle cells via targeting miR-27b-3p. Hum. Cell 2021, 34, 335–348. [Google Scholar] [CrossRef]
- Zhang, Z.; Zou, G.; Chen, X.; Lu, W.; Liu, J.; Zhai, S.; Qiao, G. Knockdown of lncRNA PVT1 Inhibits Vascular Smooth Muscle Cell Apoptosis and Extracellular Matrix Disruption in a Murine Abdominal Aortic Aneurysm Model. Mol. Cells 2019, 42, 218–227. [Google Scholar] [CrossRef]
- Ghafouri-Fard, S.; Khoshbakht, T.; Taheri, M.; Jamali, E. A Concise Review on the Role of CircPVT1 in Tumorigenesis, Drug Sensitivity, and Cancer Prognosis. Front. Oncol. 2021, 11, 762960. [Google Scholar] [CrossRef]
- Tian, C.; Hu, S.; Yu, J.; Li, W.; Li, P.; Huang, H. CREB1 transcription-activated lncRNA PVT1 promotes cardiac fibrosis via miR-145/HCN1 axis. Int. J. Cardiol. 2022, in press. [CrossRef] [PubMed]
- Wu, Q.; Chen, Q.; Wang, J.; Fan, D.; Zhou, H.; Yuan, Y.; Shen, D. Long non-coding RNA Pvt1 modulates the pathological cardiac hypertrophy via miR-196b-mediated OSMR regulation. Cell Signal 2021, 86, 110077. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Ji, X.; Wang, J.; Ma, X.; Yang, Y.; Zuo, J.; Cui, J. LncRNA PVT1 Enhances Proliferation and Cisplatin Resistance via Regulating miR-194-5p/HIF1a Axis in Oral Squamous Cell Carcinoma. OncoTargets Ther. 2020, 13, 243–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Meng, X.L.; Yang, W.Q. Long Noncoding RNA PVT1 Acts as a "Sponge" to Inhibit microRNA-152 in Gastric Cancer Cells. Dig. Dis. Sci. 2017, 62, 3021–3028. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Zhang, H.; Yang, R.; Qiao, L.; Shao, H.; Zhang, X. Small interfering RNA-induced silencing lncRNA PVT1 inhibits atherosclerosis via inactivating the MAPK/NF-κB pathway. Aging 2021, 13, 24449–24463. [Google Scholar] [CrossRef] [PubMed]
- Quan, W.; Hu, P.F.; Zhao, X.; Lianhua, C.G.; Batu, B.R. Expression level of lncRNA PVT1 in serum of patients with coronary atherosclerosis disease and its clinical significance. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 6333–6337. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.D.; Liu, X.F.; Lu, C.W.; Pant, O.P.; Liu, X.D. Long non-coding RNA PVT1: Emerging biomarker in digestive system cancer. Cell Prolif. 2017, 50, e12398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Zhang, Y.; Chen, C.; Li, Y. lncRNA HIF1A-AS2: A potential oncogene in human cancers (Review). Biomed. Rep. 2021, 15, 85. [Google Scholar] [CrossRef] [PubMed]
- Mu, L.; Wang, Y.; Su, H.; Lin, Y.; Sui, W.; Yu, X.; Lv, Z. HIF1A-AS2 Promotes the Proliferation and Metastasis of Gastric Cancer Cells Through miR-429/PD-L1 Axis. Dig. Dis. Sci. 2021, 66, 4314–4325. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, K.; Pei, Y.; Tan, J.; Ma, J.; Zhao, J. Long Noncoding RNA HIF1A-AS2 Promotes Non-Small Cell Lung Cancer Progression by the miR-153-5p/S100A14 Axis. OncoTargets Ther. 2020, 13, 8715–8722. [Google Scholar] [CrossRef]
- Li, L.; Wang, M.; Mei, Z.; Cao, W.; Yang, Y.; Wang, Y.; Wen, A. lncRNAs HIF1A-AS2 facilitates the up-regulation of HIF-1α by sponging to miR-153-3p, whereby promoting angiogenesis in HUVECs in hypoxia. Biomed. Pharmacother. 2017, 96, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.; Luo, L. LncRNA HIF1A-AS1 contributes to ventricular remodeling after myocardial ischemia/reperfusion injury by adsorption of microRNA-204 to regulating SOCS2 expression. Cell Cycle 2019, 18, 2465–2480. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.J.; Chen, W.; Gong, M.; Xu, X.; Du, M.Y.; Wang, S.F.; Yang, L.Y.; Wang, Y.; Liu, K.X.; Kong, P.; et al. Expression and Functional Analysis of lncRNAs Involved in Platelet-Derived Growth Factor-BB-Induced Proliferation of Human Aortic Smooth Muscle Cells. Front. Cardiovasc. Med. 2021, 8, 702718. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Huang, Y.; Wang, Y.; Li, D.; Lei, C. LncRNA ACTA2-AS1 suppress colon adenocarcinoma progression by sponging miR-4428 upregulation BCL2L11. Cancer Cell Int. 2021, 21, 203. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.J.; Lv, H.Z. Knockdown of ACTA2-AS1 promotes liver cancer cell proliferation, migration and invasion. Mol. Med. Rep. 2019, 19, 2263–2270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, K.; Wang, L.; Long, G.; Lian, C.; Chen, Z.; Lin, W. ACTA2-AS1 suppresses lung adenocarcinoma progression via sequestering miR-378a-3p and miR-4428 to elevate SOX7 expression. Cell Biol. Int. 2020, 44, 2438–2449. [Google Scholar] [CrossRef] [PubMed]
- Rajpathak, S.N.; Vellarikkal, S.K.; Patowary, A.; Scaria, V.; Sivasubbu, S.; Deobagkar, D.D. Human 45,X fibroblast transcriptome reveals distinct differentially expressed genes including long noncoding RNAs potentially associated with the pathophysiology of Turner syndrome. PLoS ONE 2014, 9, e100076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Li, Y.; Ye, Z.; Cheng, Z.; Lu, S.; Su, K.; Liang, Y.; Li, M.; Huang, L. Prediction of a Potential Mechanism of Intervertebral Disc Degeneration Based on a Novel Competitive Endogenous RNA Network. Biomed. Res. Int. 2021, 2021, 6618834. [Google Scholar] [CrossRef] [PubMed]
- Zhi, Y.; Du, J.; Qian, M.; Song, N. Long non-coding RNA RP11-342L8.2, derived from RNA sequencing and validated via RT-qPCR, is upregulated and correlates with disease severity in psoriasis patients. Ir. J. Med. Sci. 2022. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zeng, H.; Yu, Q.; Huang, H.; Fervers, B.; Chen, Z.S.; Lu, L. A Circulating Exosome RNA Signature Is a Potential Diagnostic Marker for Pancreatic Cancer, a Systematic Study. Cancers 2021, 13, 2565. [Google Scholar] [CrossRef]
- Sudo, R.; Sato, F.; Azechi, T.; Wachi, H. MiR-29-mediated elastin down-regulation contributes to inorganic phosphorus-induced osteoblastic differentiation in vascular smooth muscle cells. Genes Cells 2015, 20, 1077–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, K.; Cui, M.; Fu, X.; Ma, J.; Zhang, K.; Zhang, D.; Zhai, S. LncRNA Xist induces arterial smooth muscle cell apoptosis in thoracic aortic aneurysm through miR-29b-3p/Eln pathway. Biomed. Pharmacother. 2021, 137, 111163. [Google Scholar] [CrossRef] [PubMed]
- Yousef, M.; Kumar, A.; Bakir-Gungor, B. Application of Biological Domain Knowledge Based Feature Selection on Gene Expression Data. Entropy 2020, 23, 2. [Google Scholar] [CrossRef] [PubMed]
- Ouranidis, A.; Davidopoulou, C.; Tashi, R.-K.; Kachrimanis, K. Pharma 4.0 Continuous mRNA Drug Products Manufacturing. Pharmaceutics 2021, 13, 1371. [Google Scholar] [CrossRef] [PubMed]
- Che, H.L.; Bae, I.H.; Lim, K.S.; Song, I.T.; Lee, H.; Lee, D.; Kim, W.J.; Jeong, M.H.; Ahn, Y. Therapeutic Effect of Akt1 siRNA Nanoparticle Eluting Coronary Stent on Suppression of Post-Angioplasty Restenosis. J. Biomed. Nanotechnol. 2016, 12, 1211–1222. [Google Scholar] [CrossRef] [PubMed]
- Koenig, O.; Zengerle, D.; Perle, N.; Hossfeld, S.; Neumann, B.; Behring, A.; Avci-Adali, M.; Walker, T.; Schlensak, C.; Wendel, H.; et al. RNA-Eluting Surfaces for the Modulation of Gene Expression as A Novel Stent Concept. Pharmaceuticals 2017, 10, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Che, H.L.; Bae, I.H.; Lim, K.S.; Uthaman, S.; Song, I.T.; Lee, H.; Lee, D.; Kim, W.J.; Ahn, Y.; Park, I.K.; et al. Novel Fabrication of MicroRNA Nanoparticle-Coated Coronary Stent for Prevention of Post-Angioplasty Restenosis. Korean Circ. J. 2016, 46, 23–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, H.; Wang, S.; Song, M. Long non-coding RNA SENCR alleviates the inhibitory effects of rapamycin on human umbilical vein endothelial cells. Mol. Med. Rep. 2018, 18, 1405–1414. [Google Scholar] [CrossRef] [PubMed]
- Ge, D.; Meng, N.; Su, L.; Zhang, Y.; Zhang, S.L.; Miao, J.Y.; Zhao, J. Human vascular endothelial cells reduce sphingosylphosphorylcholine-induced smooth muscle cell contraction in co-culture system through integrin β4 and Fyn. Acta Pharmacol. Sin. 2012, 33, 57–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iqbal, J.; Chamberlain, J.; Francis, S.E.; Gunn, J. Role of Animal Models in Coronary Stenting. Ann. Biomed. Eng. 2016, 44, 453–465. [Google Scholar] [CrossRef] [PubMed]
- Antoine, E.E.; Cornat, F.P.; Barakat, A.I. The stentable in vitro artery: An instrumented platform for endovascular device development and optimization. J. R. Soc. Interface 2016, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smillie, C.L.; Sirey, T.; Ponting, C.P. Complexities of post-transcriptional regulation and the modeling of ceRNA crosstalk. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 231–245. [Google Scholar] [CrossRef] [PubMed]
- Castellot, J.J.; Favreau, L.V.; Karnovsky, M.J.; Rosenberg, R.D. Inhibition of vascular smooth muscle cell growth by endothelial cell-derived heparin. Possible role of a platelet endoglycosidase. J. Biol. Chem. 1982, 257, 11256–11260. [Google Scholar] [CrossRef]
- Available online: https://github.com/MontpellierRessourcesImagerie/imagej_macros_and_scripts/wiki/Wound-Healing-Tool (accessed on 23 September 2018).
- Zhao, F.Y.; Xu, S.L.; Zhang, C.F.; Liu, J.; Zhang, Y.; Yang, J.; Xing, X.Q. PDGF mediates pulmonary arterial smooth muscle cell proliferation and migration by regulating NFATc2. Mol. Med. Rep. 2021, 23, 39. [Google Scholar] [CrossRef] [PubMed]
- Jonkman, J.E.; Cathcart, J.A.; Xu, F.; Bartolini, M.E.; Amon, J.E.; Stevens, K.M.; Colarusso, P. An introduction to the wound healing assay using live-cell microscopy. Cell Adh. Migr. 2014, 8, 440–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crowley, L.C.; Chojnowski, G.; Waterhouse, N.J. Measuring the DNA Content of Cells in Apoptosis and at Different Cell-Cycle Stages by Propidium Iodide Staining and Flow Cytometry. Cold Spring Harb. Protoc. 2016, 2016, pdb-prot087247. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [Green Version]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Zhang, Y.; Ye, Z.Q.; Liu, X.Q.; Zhao, S.Q.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef] [PubMed]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019, 47, D427–D432. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ma, W.; Zeng, P.; Wang, J.; Geng, B.; Yang, J.; Cui, Q. LncTar: A tool for predicting the RNA targets of long noncoding RNAs. Brief. Bioinform. 2015, 16, 806–812. [Google Scholar] [CrossRef]
- Alexa, A.; Rahnenfuhrer, J. topGO: Enrichment Analysis for Gene Ontology; R package Version 2.18. 0. 2010. Available online: https://bioconductor.org/packages/devel/bioc/manuals/topGO/man/topGO.pdf (accessed on 15 January 2020).
- Bu, D.; Luo, H.; Huo, P.; Wang, Z.; Zhang, S.; He, Z.; Wu, Y.; Zhao, L.; Liu, J.; Guo, J.; et al. KOBAS-i: Intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucleic Acids Res. 2021, 49, W317–W325. [Google Scholar] [CrossRef]
- Lin, W.; Liu, H.; Tang, Y.; Wei, Y.; Wei, W.; Zhang, L.; Chen, J. The development and controversy of competitive endogenous RNA hypothesis in non-coding genes. Mol. Cell Biochem. 2021, 476, 109–123. [Google Scholar] [CrossRef]
- Tay, Y.; Rinn, J.; Pandolfi, P.P. The multilayered complexity of ceRNA crosstalk and competition. Nature 2014, 505, 344–352. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.Y.; Lin, Y.C.; Li, J.; Huang, K.Y.; Shrestha, S.; Hong, H.C.; Tang, Y.; Chen, Y.G.; Jin, C.N.; Yu, Y.; et al. miRTarBase 2020: Updates to the experimentally validated microRNA-target interaction database. Nucleic Acids Res. 2020, 48, D148–D154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Wang, X. miRDB: An online database for prediction of functional microRNA targets. Nucleic Acids Res. 2020, 48, D127–D131. [Google Scholar] [CrossRef] [Green Version]
- McGeary, S.E.; Lin, K.S.; Shi, C.Y.; Pham, T.M.; Bisaria, N.; Kelley, G.M.; Bartel, D.P. The biochemical basis of microRNA targeting efficacy. Science 2019, 366, eaav1741. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Expression | mRNA | ncRNA | ||
|---|---|---|---|---|
| N | % | N | % | |
| Downregulated | 2028 | 57.7 | 1460 | 50.1 |
| Upregulated | 1489 | 42.3 | 1451 | 49.9 |
| Total | 3517 | 100 | 2911 | 100 |
| Expression | mRNA | lncRNA | microRNA | |||
|---|---|---|---|---|---|---|
| N | % | N | % | N | % | |
| Down regulated | 793 | 72.1 | 207 | 40.7 | 33 | 51.6 |
| Up regulated | 306 | 27.8 | 302 | 59.3 | 31 | 48.4 |
| Total | 1099 | 100 | 509 | 100 | 64 | 100 |
| lncRNA | Expression | log2FC | Location | Type |
|---|---|---|---|---|
| LNCG_1012 | up | 11.22 | 1:70947327–70966066 (+) | novel |
| AL031429.1 | up | 10.02 | 1:70947379–70951493 (−) | antisense |
| LNCG_20369 | up | 9.56 | 5:172802872–172807207 (+) | novel |
| LNCG_17378 | up | 8.95 | 3:91874302–92117628 (−) | novel |
| AC003092.1 | up | 8.88 | 7:94022833–94064723 (+) | lincRNA |
| LNCG_21417 | up | 8.86 | 6:137692987–137705416 (+) | novel |
| AC079298.1 | up | 8.77 | 4:154235980–154237598 (−) | antisense |
| LNCG_17377 | up | 8.75 | 3:91825033–91847766 (−) | novel |
| AC097480.1 | up | 8.69 | 4:28435449–28600275 (+) | lincRNA |
| LNCG_22261 | up | 8.56 | 7:94022772–94068906 (+) | novel |
| CEBPA-DT | down | −7.19 | 19:33302857–33305054 (+) | lincRNA |
| AC103702.2 | down | −7.24 | 17:48635923–48647023 (+) | sense-intronic |
| AP005131.7 | down | −7.29 | 18:13526078–13526688 (+) | sense-intronic |
| AC027309.1 | down | −7.55 | 5:172690454–72697720 (−) | antisense |
| AL109924.2 | down | −7.62 | 6:169034197–169035642 (+) | lincRNA |
| AC092687.3 | down | −7.62 | 2:10767875–10770058 (−) | antisense |
| AC023669.1 | down | −8.03 | 7:46261064–46294469 (+) | lincRNA |
| AP001180.4 | down | −8.19 | 18:10704297–10709599 (−) | antisense |
| AL445183.1 | down | −8.55 | 1:53069938–53085502 (−) | antisense |
| LNCG_14690 | down | −9.78 | 2:190677035–190813478 (−) | novel |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arencibia, A.; Lanas, F.; Salazar, L.A. Long Non-Coding RNAs Might Regulate Phenotypic Switch of Vascular Smooth Muscle Cells Acting as ceRNA: Implications for In-Stent Restenosis. Int. J. Mol. Sci. 2022, 23, 3074. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23063074
Arencibia A, Lanas F, Salazar LA. Long Non-Coding RNAs Might Regulate Phenotypic Switch of Vascular Smooth Muscle Cells Acting as ceRNA: Implications for In-Stent Restenosis. International Journal of Molecular Sciences. 2022; 23(6):3074. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23063074
Chicago/Turabian StyleArencibia, Alberto, Fernando Lanas, and Luis A. Salazar. 2022. "Long Non-Coding RNAs Might Regulate Phenotypic Switch of Vascular Smooth Muscle Cells Acting as ceRNA: Implications for In-Stent Restenosis" International Journal of Molecular Sciences 23, no. 6: 3074. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23063074