
Blood Transcriptome Profiling Links Immunity to Disease Severity in Myotonic Dystrophy Type 1 (DM1)

 , and
, and
Abstract
:1. Introduction
2. Results
2.1. Descriptive Analyses of the OPTIMISTIC and DMBDI Samples
2.2. Data Analysis of the Transcriptome of DM1 Patients Cohorts
RNA Sequencing and Exploratory Analyses in OPTIMISTIC Cohort
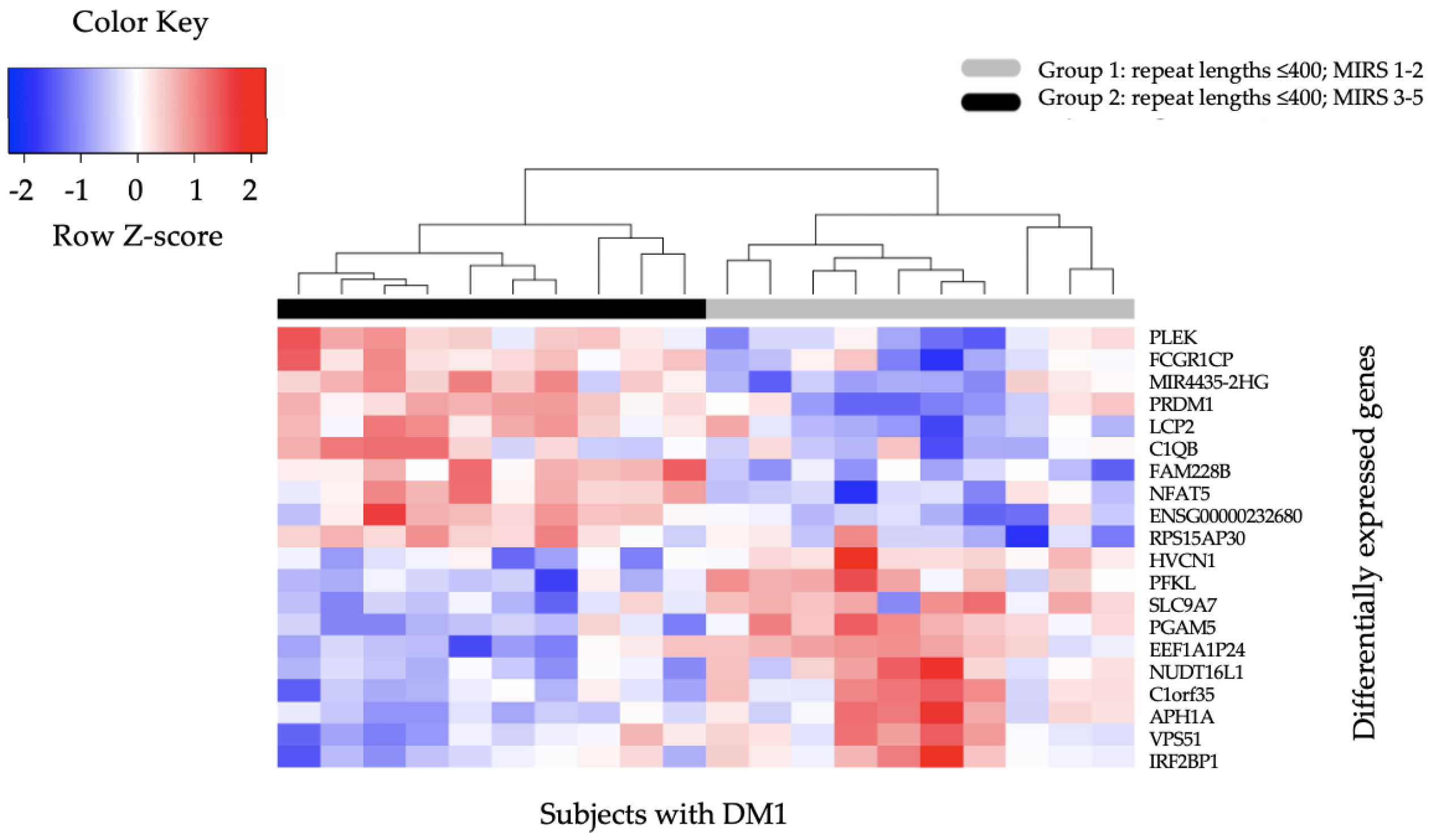
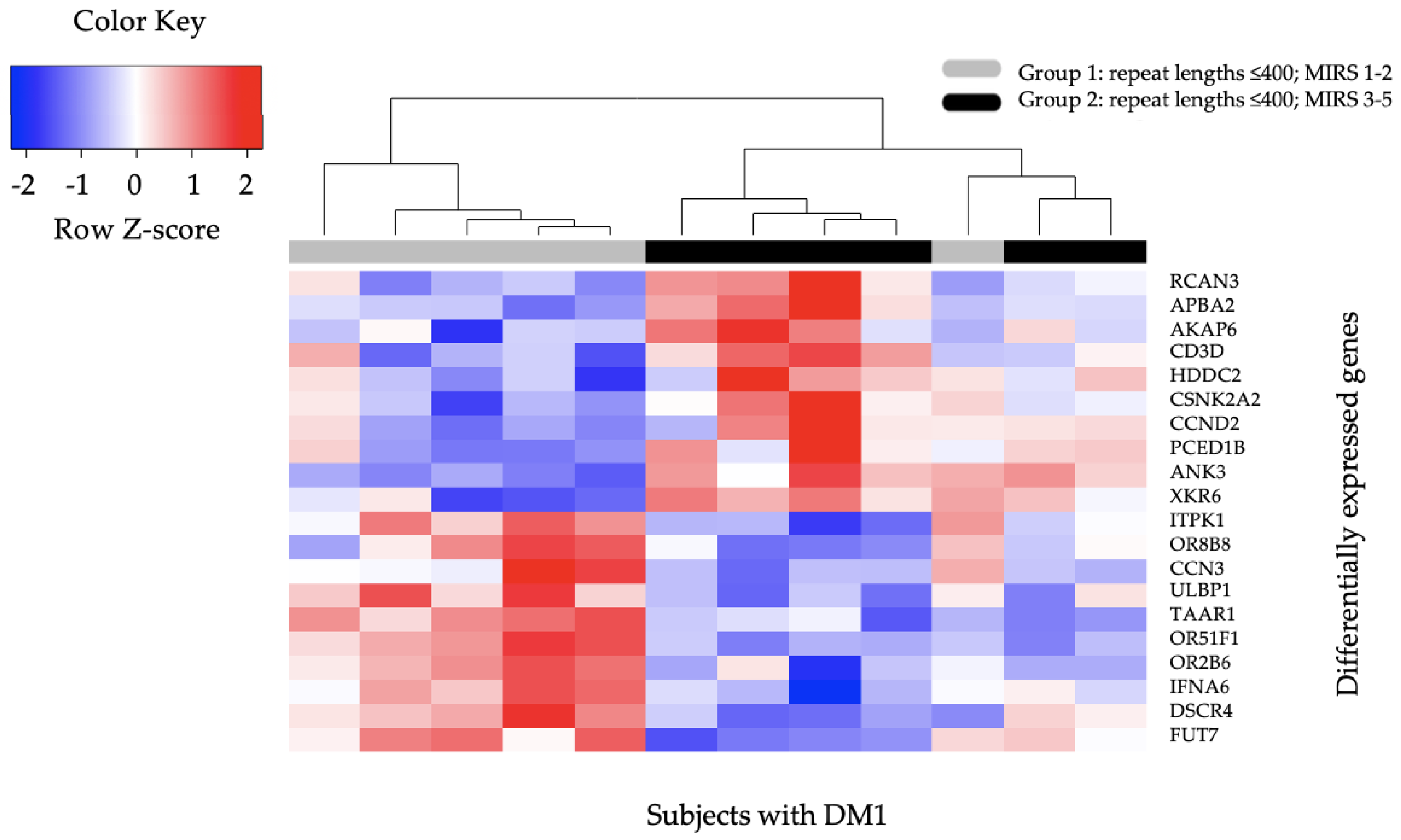
2.3. Differential Gene Expression
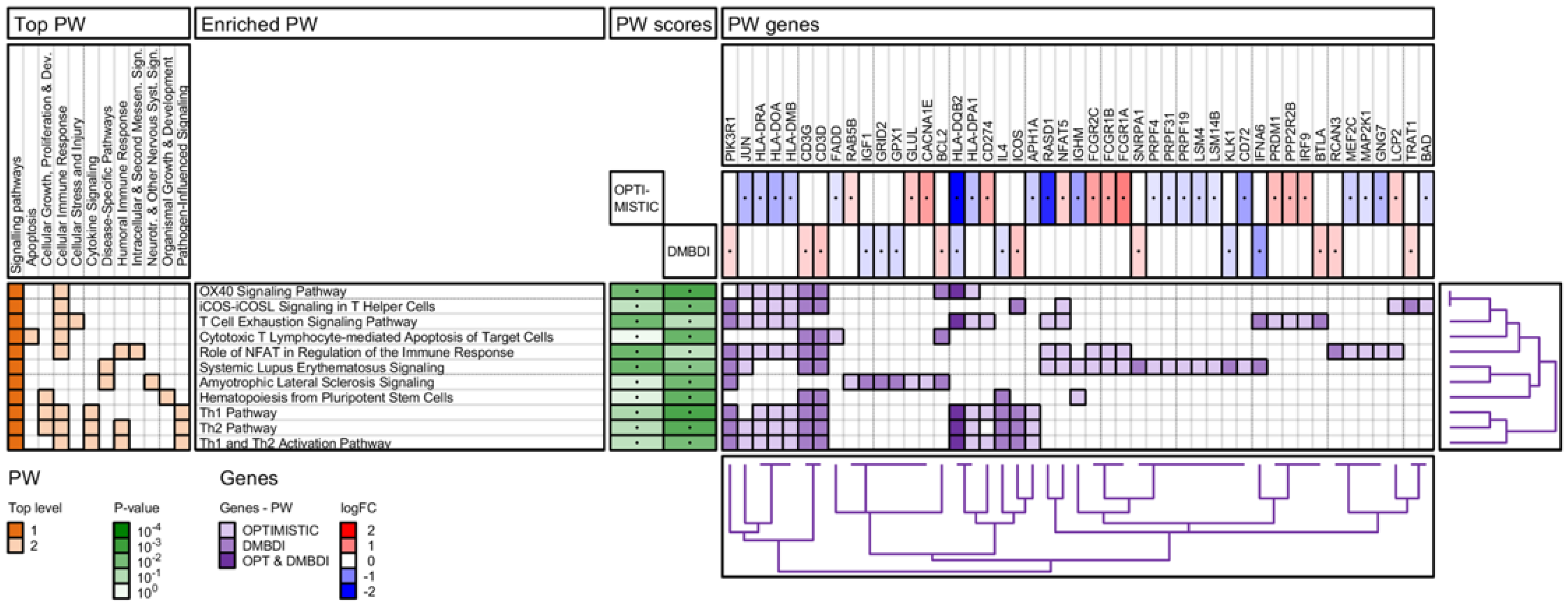
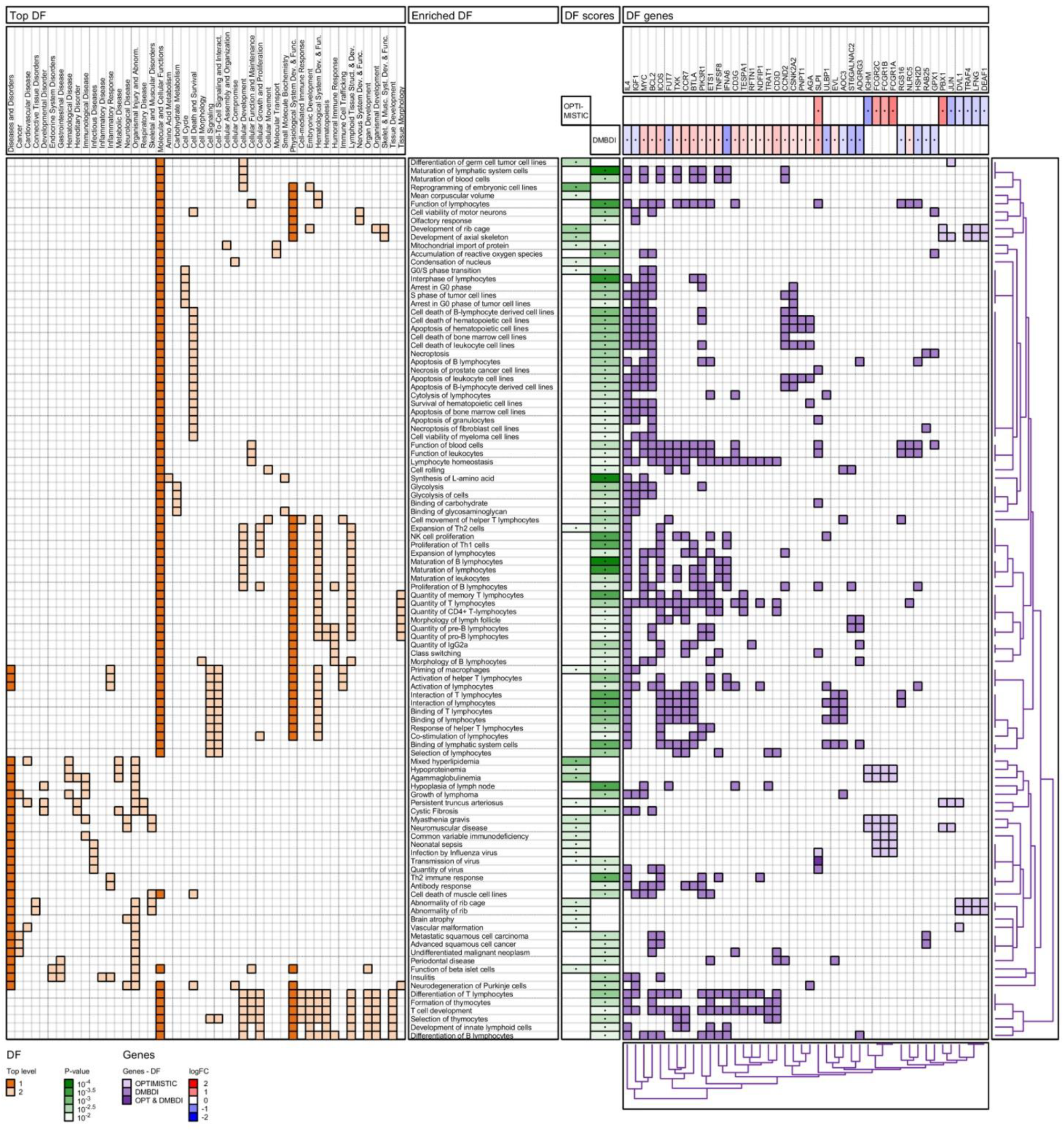
2.4. Enrichment Analysis
2.4.1. IPA Pathway Analysis
2.4.2. IPA Diseases and Functions Analysis
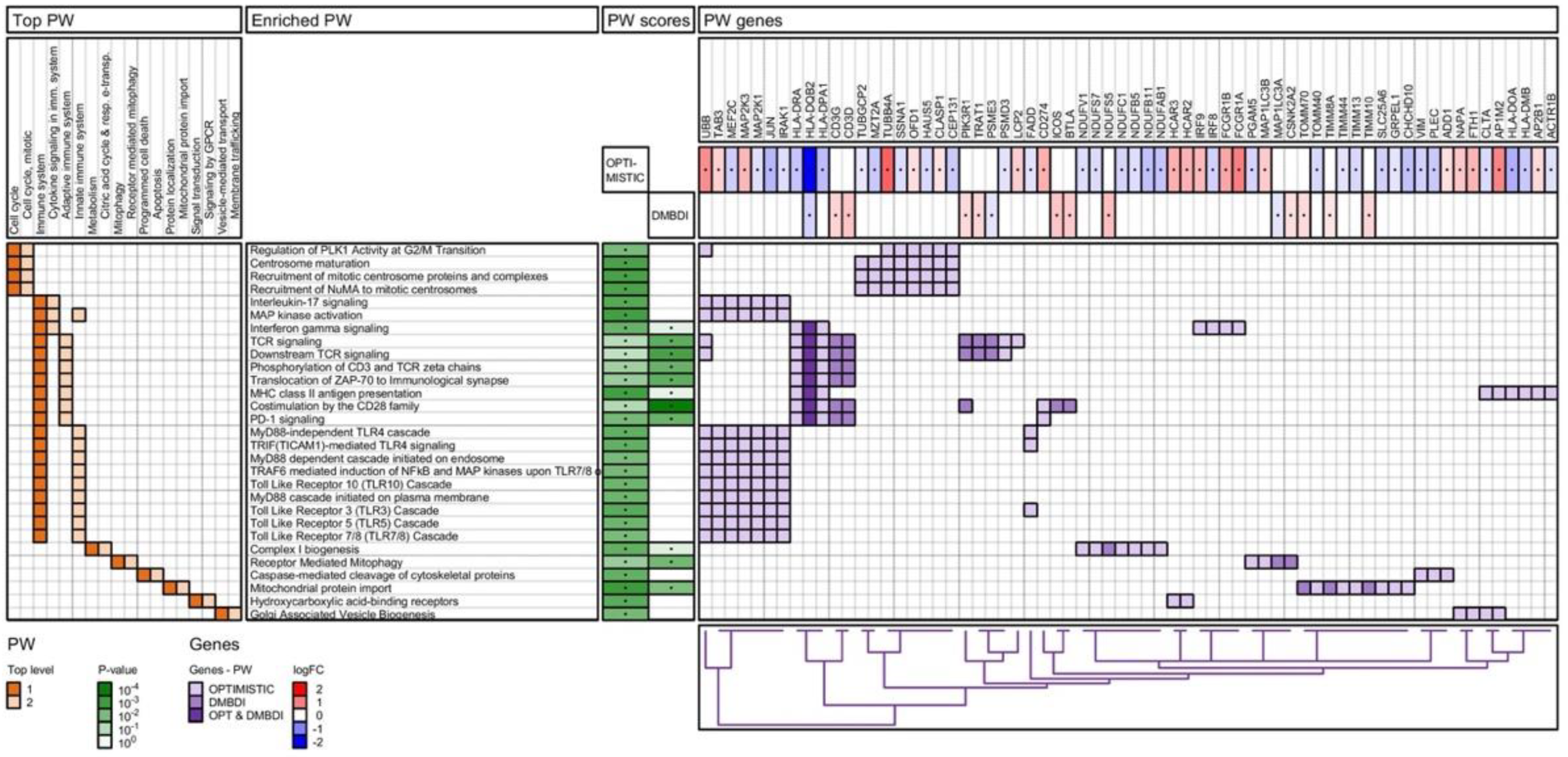
2.4.3. Reactome Pathway Analysis
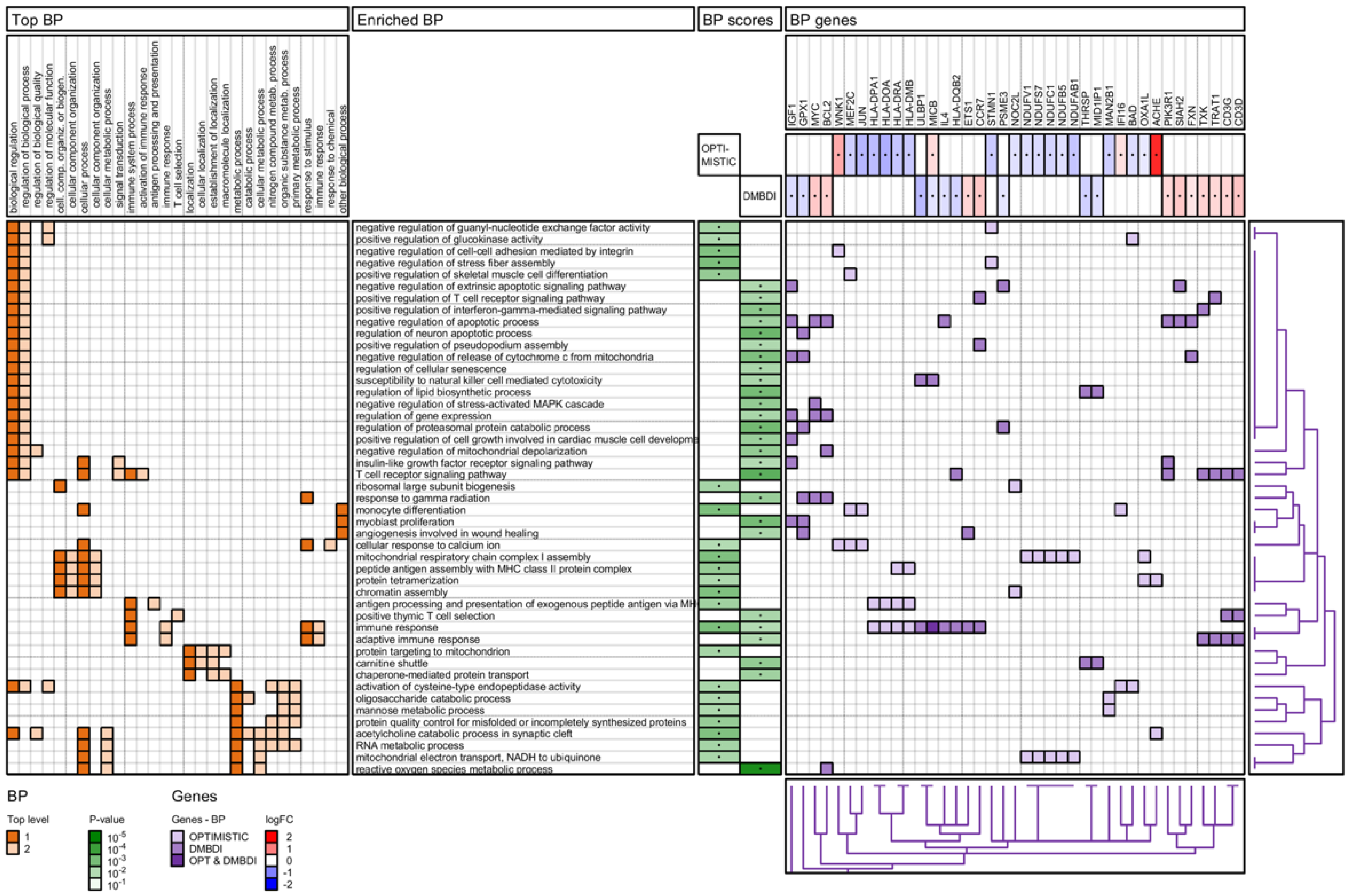
2.4.4. Gene Ontology (GO) Biological Process Analysis
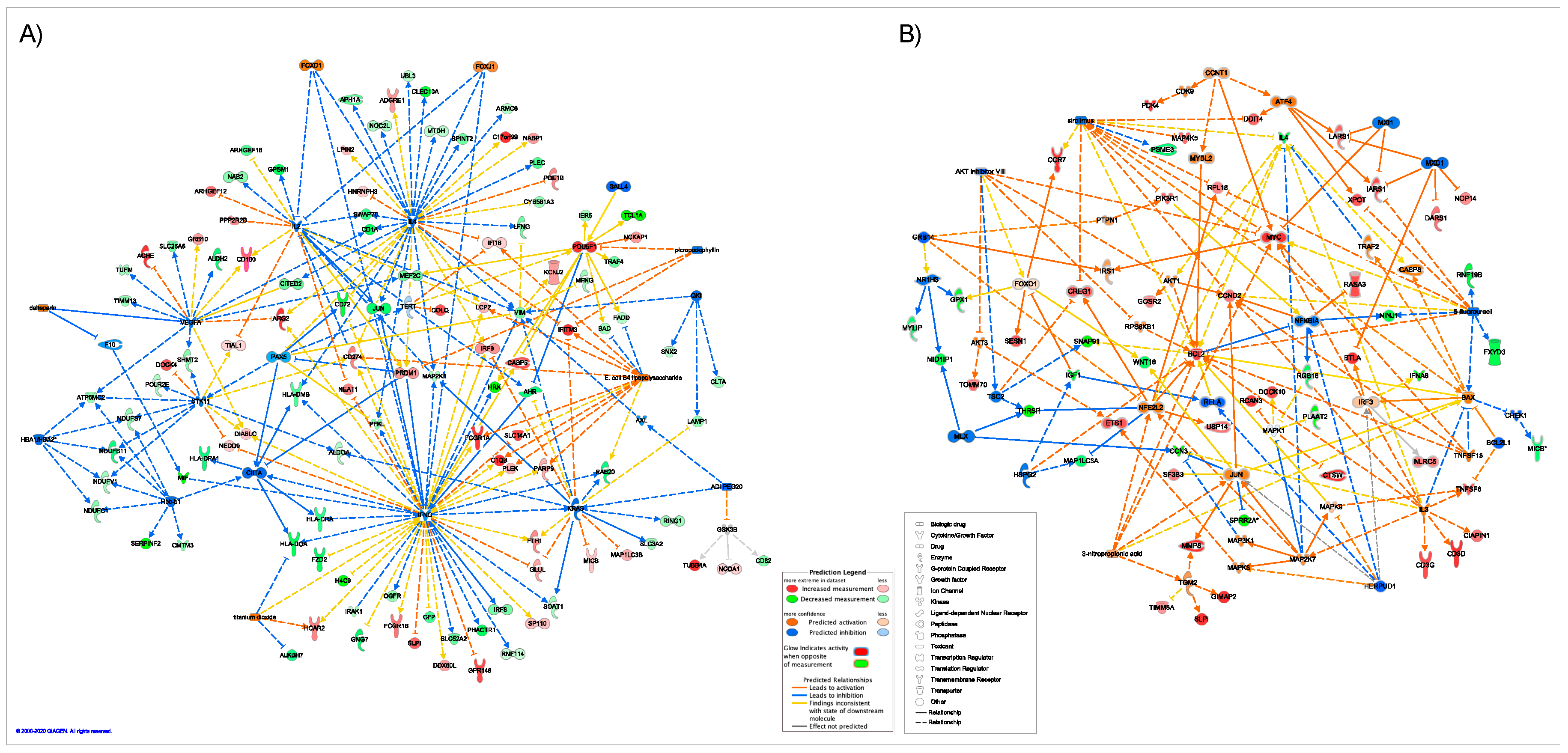
2.5. Master Regulators of Gene Expression
2.6. Splice Variant Analysis
3. Discussion
3.1. Limitations of the Current Study
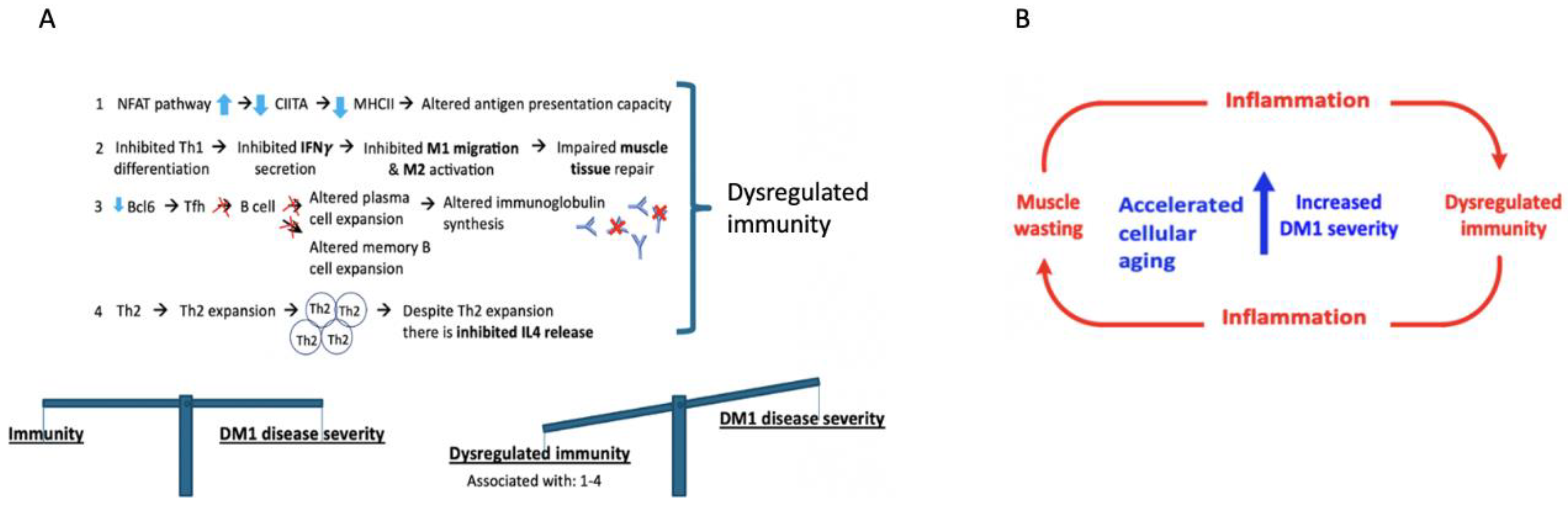
3.2. A Role for Dysregulated Immunity in DM1
3.3. Pathway Analysis—Shared Pathways
3.4. Pathway Analysis—Non-Shared Pathways
3.4.1. NFAT Regulation of the Immune Response
3.4.2. T-Cell Exhaustion
3.5. Biological Processes and Functions
3.5.1. Mitochondrial Import of Proteins
3.5.2. Macrophage Priming
3.5.3. Th2 Cell Expansion
3.5.4. Additional Evidence for Immune-Related Changes in the Blood of DM1 Patients
3.5.5. Immune System Involvement in Other Muscular Dystrophies
3.6. Master Regulators
3.6.1. Immune System Related Master Regulators
3.6.2. Muscle Related Master Regulators
3.7. Increased DM1 Severity in Relation to Aging as Result of Dysregulated Immunity and Muscle-Wasting
4. Materials and Methods
4.1. RNA Isolation, Sequencing, and Differential Gene Expression (DGE) Analysis
4.1.1. RNA Isolation and Sequencing
4.1.2. Data Analysis and Differential Expression Analysis
4.2. Functional Analysis
4.2.1. Gene Enrichment Analysis in IPA
4.2.2. Causal Network Analysis in IPA (Master Regulators)
4.2.3. Gene Enrichment Analysis in Reactome
4.3. Splice Variant Analysis
4.4. Study Participants and Clinical Information
4.5. Ethical Approval
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kornblum, C.; Lutterbey, G.; Bogdanow, M.; Kesper, K.; Schild, H.; Schroder, R.; Wattjes, M.P. Distinct neuromuscular phenotypes in myotonic dystrophy types 1 and 2: A whole body highfield MRI study. J. Neurol. 2006, 253, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Bhakta, D.; Shen, C.; Kron, J.; Epstein, A.E.; Pascuzzi, R.M.; Groh, W.J. Pacemaker and implantable cardioverter-defibrillator use in a US myotonic dystrophy type 1 population. J. Cardiovasc. Electrophysiol. 2011, 22, 1369–1375. [Google Scholar] [CrossRef]
- Bellini, M.; Biagi, S.; Stasi, C.; Costa, F.; Mumolo, M.G.; Ricchiuti, A.; Marchi, S. Gastrointestinal manifestations in myotonic muscular dystrophy. World J. Gastroenterol. 2006, 12, 1821–1828. [Google Scholar] [CrossRef]
- Bird, T.D. Myotonic Dystrophy Type 1. In GeneReviews(R); Pagon, R.A., Adam, M.P., Ardinger, H.H., Wallace, S.E., Amemiya, A., Bean, L.J.H., Bird, T.D., Ledbetter, N., Mefford, H.C., Smith, R.J.H., et al., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Meola, G.; Cardani, R. Myotonic dystrophies: An update on clinical aspects, genetic, pathology, and molecular pathomechanisms. Biochim. Biophys. Acta 2015, 1852, 594–606. [Google Scholar] [CrossRef] [Green Version]
- Delaporte, C. Personality patterns in patients with myotonic dystrophy. Arch. Neurol. 1998, 55, 635–640. [Google Scholar] [CrossRef] [Green Version]
- Winblad, S.; Lindberg, C.; Hansen, S. Temperament and character in patients with classical myotonic dystrophy type 1 (DM-1). Neuromuscul. Disord. 2005, 15, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Peric, S.; Mandic-Stojmenovic, G.; Markovic, I.; Stefanova, E.; Ilic, V.; Parojcic, A.; Misirlic-Dencic, S.; Ostojic, M.; Rakocevic-Stojanovic, V.; Kostic, V. Cerebrospinal fluid biomarkers of neurodegeneration in patients with juvenile and classic myotonic dystrophy type 1. Eur. J. Neurol. 2014, 21, 231–237. [Google Scholar] [CrossRef]
- Mathieu, J.; Boivin, H.; Meunier, D.; Gaudreault, M.; Begin, P. Assessment of a disease-specific muscular impairment rating scale in myotonic dystrophy. Neurology 2001, 56, 336–340. [Google Scholar] [CrossRef]
- Heatwole, C.; Bode, R.; Johnson, N.; Dekdebrun, J.; Dilek, N.; Heatwole, M.; Hilbert, J.E.; Luebbe, E.; Martens, W.; McDermott, M.P.; et al. Myotonic Dystrophy Health Index: Initial evaluation of a disease-specific outcome measure. Muscle Nerve 2014, 49, 906–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermans, M.C.; Faber, C.G.; De Baets, M.H.; de Die-Smulders, C.E.; Merkies, I.S. Rasch-built myotonic dystrophy type 1 activity and participation scale (DM1-Activ). Neuromuscul. Disord. 2010, 20, 310–318. [Google Scholar] [CrossRef]
- Koch, K.S.; Leffert, H.L. Giant hairpins formed by CUG repeats in myotonic dystrophy messenger RNAs might sterically block RNA export through nuclear pores. J. Theor. Biol. 1998, 192, 505–514. [Google Scholar] [CrossRef]
- Meola, G.; Moxley, R.T., 3rd. Myotonic dystrophy type 2 and related myotonic disorders. J. Neurol. 2004, 251, 1173–1182. [Google Scholar] [CrossRef] [PubMed]
- Perseghin, G.; Comola, M.; Scifo, P.; Benedini, S.; De Cobelli, F.; Lanzi, R.; Costantino, F.; Lattuada, G.; Battezzati, A.; Del Maschio, A.; et al. Postabsorptive and insulin-stimulated energy and protein metabolism in patients with myotonic dystrophy type 1. Am. J. Clin. Nutr. 2004, 80, 357–364. [Google Scholar] [CrossRef] [Green Version]
- Napierała, M.; Krzyzosiak, W.J. CUG repeats present in myotonin kinase RNA form metastable “slippery” hairpins. J. Biol. Chem. 1997, 272, 31079–31085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, P.S.; Han, J.; Reddy, S. In situ hybridization analysis of Dmpk mRNA in adult mouse tissues. Neuromuscul. Disord. 2004, 14, 497–506. [Google Scholar] [CrossRef]
- de Leon, M.B.; Cisneros, B. Myotonic dystrophy 1 in the nervous system: From the clinic to molecular mechanisms. J. Neurosci. Res. 2008, 86, 18–26. [Google Scholar] [CrossRef]
- Pantic, B.; Trevisan, E.; Citta, A.; Rigobello, M.P.; Marin, O.; Bernardi, P.; Salvatori, S.; Rasola, A. Myotonic dystrophy protein kinase (DMPK) prevents ROS-induced cell death by assembling a hexokinase II-Src complex on the mitochondrial surface. Cell Death Dis. 2013, 4, e858. [Google Scholar] [CrossRef]
- Nakamori, M.; Thornton, C. Epigenetic changes and non-coding expanded repeats. Neurobiol. Dis. 2010, 39, 21–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wenninger, S.; Cumming, S.A.; Gutschmidt, K.; Okkersen, K.; Jimenez-Moreno, A.C.; Daidj, F.; Lochmüller, H.; Hogarth, F.; Knoop, H.; Bassez, G.; et al. Associations Between Variant Repeat Interruptions and Clinical Outcomes in Myotonic Dystrophy Type 1. Neurol. Genet. 2021, 7, e572. [Google Scholar] [CrossRef] [PubMed]
- Heskamp, L.; van Nimwegen, M.; Ploegmakers, M.J.; Bassez, G.; Deux, J.F.; Cumming, S.A.; Monckton, D.G.; van Engelen, B.G.M.; Heerschap, A. Lower extremity muscle pathology in myotonic dystrophy type 1 assessed by quantitative MRI. Neurology 2019, 92, e2803–e2814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antoury, L.; Hu, N.; Balaj, L.; Das, S.; Georghiou, S.; Darras, B.; Clark, T.; Breakefield, X.O.; Wheeler, T.M. Analysis of extracellular mRNA in human urine reveals splice variant biomarkers of muscular dystrophies. Nat. Commun. 2018, 9, 3906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gosho, M.; Nagashima, K.; Sato, Y. Study designs and statistical analyses for biomarker research. Sensors 2012, 12, 8966–8986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perfetti, A.; Greco, S.; Cardani, R.; Fossati, B.; Cuomo, G.; Valaperta, R.; Ambrogi, F.; Cortese, A.; Botta, A.; Mignarri, A.; et al. Validation of plasma microRNAs as biomarkers for myotonic dystrophy type 1. Sci. Rep. 2016, 6, 38174. [Google Scholar] [CrossRef] [Green Version]
- Thornton, C.A.; Wang, E.; Carrell, E.M. Myotonic dystrophy: Approach to therapy. Curr. Opin. Genet. Dev. 2017, 44, 135–140. [Google Scholar] [CrossRef]
- van Engelen, B. Cognitive behaviour therapy plus aerobic exercise training to increase activity in patients with myotonic dystrophy type 1 (DM1) compared to usual care (OPTIMISTIC): Study protocol for randomised controlled trial. Trials 2015, 16, 224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurkiewicz, A.; Cooper, A.; McIlwaine, E.; Cumming, S.A.; Adam, B.; Krahe, R.; Puymirat, J.; Schoser, B.; Timchenko, L.; Ashizawa, T.; et al. Towards development of a statistical framework to evaluate myotonic dystrophy type 1 mRNA biomarkers in the context of a clinical trial. PLoS ONE 2020, 15, e0231000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dogan, C.; De Antonio, M.; Hamroun, D.; Varet, H.; Fabbro, M.; Rougier, F.; Amarof, K.; Arne Bes, M.C.; Bedat-Millet, A.L.; Behin, A.; et al. Gender as a Modifying Factor Influencing Myotonic Dystrophy Type 1 Phenotype Severity and Mortality: A Nationwide Multiple Databases Cross-Sectional Observational Study. PLoS ONE 2016, 11, e0148264. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.T.; Treacy, D.; Eichinger, K.; Struck, A.; Estabrook, J.; Olafson, H.; Wang, T.T.; Bhatt, K.; Westbrook, T.; Sedehizadeh, S.; et al. Transcriptome alterations in myotonic dystrophy skeletal muscle and heart. Hum. Mol. Genet. 2019, 28, 1312–1321. [Google Scholar] [CrossRef]
- Goodwin, M.; Mohan, A.; Batra, R.; Lee, K.Y.; Charizanis, K.; Fernandez Gomez, F.J.; Eddarkaoui, S.; Sergeant, N.; Buee, L.; Kimura, T.; et al. MBNL Sequestration by Toxic RNAs and RNA Misprocessing in the Myotonic Dystrophy Brain. Cell Rep. 2015, 12, 1159–1168. [Google Scholar] [CrossRef] [Green Version]
- Bachinski, L.L.; Baggerly, K.A.; Neubauer, V.L.; Nixon, T.J.; Raheem, O.; Sirito, M.; Unruh, A.K.; Zhang, J.; Nagarajan, L.; Timchenko, L.T.; et al. Most expression and splicing changes in myotonic dystrophy type 1 and type 2 skeletal muscle are shared with other muscular dystrophies. Neuromuscul. Disord. 2014, 24, 227–240. [Google Scholar] [CrossRef] [Green Version]
- Todorow, V.; Hintze, S.; Kerr, A.R.W.; Hehr, A.; Schoser, B.; Meinke, P. Transcriptome Analysis in a Primary Human Muscle Cell Differentiation Model for Myotonic Dystrophy Type 1. Int. J. Mol. Sci. 2021, 22, 8607. [Google Scholar] [CrossRef] [PubMed]
- Azotla-Vilchis, C.N.; Sanchez-Celis, D.; Agonizantes-Juárez, L.E.; Suárez-Sánchez, R.; Hernández-Hernández, J.M.; Peña, J.; Vázquez-Santillán, K.; Leyva-García, N.; Ortega, A.; Maldonado, V.; et al. Transcriptome Analysis Reveals Altered Inflammatory Pathway in an Inducible Glial Cell Model of Myotonic Dystrophy Type 1. Biomolecules 2021, 11, 159. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, J.D.; Lott, M.C.; Russell, S.L.; Moulton, V.; Sanderson, J.; Wormstone, I.M.; Broadway, D.C. Activation of the innate immune response and interferon signalling in myotonic dystrophy type 1 and type 2 cataracts. Hum. Mol. Genet. 2012, 21, 852–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voellenkle, C.; Perfetti, A.; Carrara, M.; Fuschi, P.; Renna, L.V.; Longo, M.; Sain, S.B.; Cardani, R.; Valaperta, R.; Silvestri, G.; et al. Dysregulation of Circular RNAs in Myotonic Dystrophy Type 1. Int. J. Mol. Sci. 2019, 20, 1938. [Google Scholar] [CrossRef] [Green Version]
- Koutsoulidou, A.; Kyriakides, T.C.; Papadimas, G.K.; Christou, Y.; Kararizou, E.; Papanicolaou, E.Z.; Phylactou, L.A. Elevated Muscle-Specific miRNAs in Serum of Myotonic Dystrophy Patients Relate to Muscle Disease Progress. PLoS ONE 2015, 10, e0125341. [Google Scholar] [CrossRef]
- Tidball, J.G.; Villalta, S.A. Regulatory interactions between muscle and the immune system during muscle regeneration. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R1173–R1187. [Google Scholar] [CrossRef] [Green Version]
- Johnston, R.J.; Poholek, A.C.; DiToro, D.; Yusuf, I.; Eto, D.; Barnett, B.; Dent, A.L.; Craft, J.; Crotty, S. Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science 2009, 325, 1006–1010. [Google Scholar] [CrossRef] [Green Version]
- Jones, E.Y.; Fugger, L.; Strominger, J.L.; Siebold, C. MHC class II proteins and disease: A structural perspective. Nat. Rev. Immunol. 2006, 6, 271–282. [Google Scholar] [CrossRef]
- Afzali, A.M.; Müntefering, T.; Wiendl, H.; Meuth, S.G.; Ruck, T. Skeletal muscle cells actively shape (auto)immune responses. Autoimmun. Rev. 2018, 17, 518–529. [Google Scholar] [CrossRef]
- Ding, M.; Huang, T.; Zhu, R.; Gu, R.; Shi, D.; Xiao, J.; Guo, M.; Li, J.; Hu, J.; Liao, H. Immunological Behavior Analysis of Muscle Cells under IFN-γ Stimulation in Vitro and in Vivo. Anat. Rec. 2018, 301, 1551–1563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, B.; Bruder, J.; Kovesdi, I.; Huard, J. Muscle stem cells can act as antigen-presenting cells: Implication for gene therapy. Gene Ther. 2004, 11, 1321–1330. [Google Scholar] [CrossRef] [Green Version]
- Giroux, M.; Schmidt, M.; Descoteaux, A. IFN-gamma-induced MHC class II expression: Transactivation of class II transactivator promoter IV by IFN regulatory factor-1 is regulated by protein kinase C-alpha. J. Immunol. 2003, 171, 4187–4194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaminsky, P.; Lesesve, J.F.; Jonveaux, P.; Pruna, L. IgG deficiency and expansion of CTG repeats in myotonic dystrophy. Clin. Neurol. Neurosurg. 2011, 113, 464–468. [Google Scholar] [CrossRef]
- Pan-Hammarstrom, Q.; Wen, S.; Ghanaat-Pour, H.; Solders, G.; Forsberg, H.; Hammarstrom, L. Lack of correlation between the reduction of serum immunoglobulin concentration and the CTG repeat expansion in patients with type 1 dystrophia [correction of Dystrofia] myotonica. J. Neuroimmunol. 2003, 144, 100–104. [Google Scholar] [CrossRef]
- Suzumura, A.; Yamada, H.; Matsuoka, Y.; Sobue, I. Immunoglobulin abnormalities in patients with myotonic dystrophy. Acta Neurol. Scand. 1986, 74, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Madaro, L.; Bouché, M. From innate to adaptive immune response in muscular dystrophies and skeletal muscle regeneration: The role of lymphocytes. BioMed Res. Int. 2014, 2014, 438675. [Google Scholar] [CrossRef]
- Sakkas, L.I.; Boulbou, M.; Kyriakou, D.; Makri, I.; Sinani, C.; Germenis, A.; Stathakis, N. Immunological features of visceral leishmaniasis may mimic systemic lupus erythematosus. Clin. Biochem. 2008, 41, 65–68. [Google Scholar] [CrossRef] [PubMed]
- Prockop, L.D. Myotonia, procaine amide, and lupus-like syndrome. Arch. Neurol. 1966, 14, 326–330. [Google Scholar] [CrossRef]
- Andrews, J.S.; Trupin, L.; Schmajuk, G.; Barton, J.; Margaretten, M.; Yazdany, J.; Yelin, E.H.; Katz, P.P. Muscle strength, muscle mass, and physical disability in women with systemic lupus erythematosus. Arthritis Care Res. 2015, 67, 120–127. [Google Scholar] [CrossRef] [Green Version]
- Vaeth, M.; Feske, S. NFAT control of immune function: New Frontiers for an Abiding Trooper. F1000Research 2018, 7, 260. [Google Scholar] [CrossRef] [Green Version]
- Yi, J.S.; Cox, M.A.; Zajac, A.J. T-cell exhaustion: Characteristics, causes and conversion. Immunology 2010, 129, 474–481. [Google Scholar] [CrossRef] [PubMed]
- McCullagh, K.J.; Calabria, E.; Pallafacchina, G.; Ciciliot, S.; Serrano, A.L.; Argentini, C.; Kalhovde, J.M.; Lømo, T.; Schiaffino, S. NFAT is a nerve activity sensor in skeletal muscle and controls activity-dependent myosin switching. Proc. Natl. Acad. Sci. USA 2004, 101, 10590–10595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- John, S.A.; Garrett-Sinha, L.A. Blimp1: A conserved transcriptional repressor critical for differentiation of many tissues. Exp. Cell Res. 2009, 315, 1077–1084. [Google Scholar] [CrossRef]
- André, L.M.; Ausems, C.R.M.; Wansink, D.G.; Wieringa, B. Abnormalities in Skeletal Muscle Myogenesis, Growth, and Regeneration in Myotonic Dystrophy. Front. Neurol. 2018, 9, 368. [Google Scholar] [CrossRef] [PubMed]
- Ivashkiv, L.B. IFNγ: Signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat. Reviews. Immunol. 2018, 18, 545–558. [Google Scholar] [CrossRef]
- Teixeira, L.K.; Fonseca, B.P.; Vieira-de-Abreu, A.; Barboza, B.A.; Robbs, B.K.; Bozza, P.T.; Viola, J.P. IFN-gamma production by CD8+ T cells depends on NFAT1 transcription factor and regulates Th differentiation. J. Immunol. 2005, 175, 5931–5939. [Google Scholar] [CrossRef] [Green Version]
- Shin, H.; Blackburn, S.D.; Intlekofer, A.M.; Kao, C.; Angelosanto, J.M.; Reiner, S.L.; Wherry, E.J. A role for the transcriptional repressor Blimp-1 in CD8(+) T cell exhaustion during chronic viral infection. Immunity 2009, 31, 309–320. [Google Scholar] [CrossRef] [Green Version]
- Kallies, A.; Xin, A.; Belz, G.T.; Nutt, S.L. Blimp-1 transcription factor is required for the differentiation of effector CD8(+) T cells and memory responses. Immunity 2009, 31, 283–295. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Geng, J.; Wen, X.; Bi, E.; Kossenkov, A.V.; Wolf, A.I.; Tas, J.; Choi, Y.S.; Takata, H.; Day, T.J.; et al. The transcription factor Foxp1 is a critical negative regulator of the differentiation of follicular helper T cells. Nat. Immunol. 2014, 15, 667–675. [Google Scholar] [CrossRef]
- Joseph, A.M.; Adhihetty, P.J.; Buford, T.W.; Wohlgemuth, S.E.; Lees, H.A.; Nguyen, L.M.; Aranda, J.M.; Sandesara, B.D.; Pahor, M.; Manini, T.M.; et al. The impact of aging on mitochondrial function and biogenesis pathways in skeletal muscle of sedentary high- and low-functioning elderly individuals. Aging Cell 2012, 11, 801–809. [Google Scholar] [CrossRef] [Green Version]
- Doki, T.; Yamashita, S.; Wei, F.Y.; Hara, K.; Yamamoto, T.; Zhang, Z.; Zhang, X.; Tawara, N.; Hino, H.; Uyama, E.; et al. Mitochondrial localization of PABPN1 in oculopharyngeal muscular dystrophy. Lab. Investig. 2019, 99, 1728–1740. [Google Scholar] [CrossRef] [PubMed]
- Gramegna, L.L.; Giannoccaro, M.P.; Manners, D.N.; Testa, C.; Zanigni, S.; Evangelisti, S.; Bianchini, C.; Oppi, F.; Poda, R.; Avoni, P.; et al. Mitochondrial dysfunction in myotonic dystrophy type 1. Neuromuscul. Disord. 2018, 28, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Loro, E.; Rinaldi, F.; Malena, A.; Masiero, E.; Novelli, G.; Angelini, C.; Romeo, V.; Sandri, M.; Botta, A.; Vergani, L. Normal myogenesis and increased apoptosis in myotonic dystrophy type-1 muscle cells. Cell Death Differ. 2010, 17, 1315–1324. [Google Scholar] [CrossRef] [Green Version]
- Deng, S.; Wu, Q.; Yu, K.; Zhang, Y.; Yao, Y.; Li, W.; Deng, Z.; Liu, G.; Li, W.; Lian, Z. Changes in the relative inflammatory responses in sheep cells overexpressing of toll-like receptor 4 when stimulated with LPS. PLoS ONE 2012, 7, e47118. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, D.W.; Kornbluth, J. E3 ubiquitin ligase NKLAM is a macrophage phagosome protein and plays a role in bacterial killing. Cell. Immunol. 2012, 279, 46–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Chen, S.; Chen, J.; Xie, X.; Gao, S.; Zhang, C.; Zhou, S.; Wang, J.; Mai, R.; Lin, Q.; et al. Germline genetic patterns underlying familial rheumatoid arthritis, systemic lupus erythematosus and primary Sjögren’s syndrome highlight T cell-initiated autoimmunity. Ann. Rheum. Dis. 2020, 79, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Maldonado, M.A. The ubiquitin-proteasome system and its role in inflammatory and autoimmune diseases. Cell. Mol. Immunol. 2006, 3, 255–261. [Google Scholar]
- Egerer, K.; Kuckelkorn, U.; Rudolph, P.E.; Ruckert, J.C.; Dorner, T.; Burmester, G.R.; Kloetzel, P.M.; Feist, E. Circulating proteasomes are markers of cell damage and immunologic activity in autoimmune diseases. J. Rheumatol. 2002, 29, 2045–2052. [Google Scholar]
- Yamashita, M.; Katsumata, M.; Iwashima, M.; Kimura, M.; Shimizu, C.; Kamata, T.; Shin, T.; Seki, N.; Suzuki, S.; Taniguchi, M.; et al. T cell receptor-induced calcineurin activation regulates T helper type 2 cell development by modifying the interleukin 4 receptor signaling complex. J. Exp. Med. 2000, 191, 1869–1879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaier, M.; Gottschalk, C.; Kälble, F.; Uhlmann, L.; Eckstein, V.; Müller-Tidow, C.; Meuer, S.; Mahnke, K.; Lorenz, H.M.; Zeier, M.; et al. The onset of active disease in systemic lupus erythematosus patients is characterised by excessive regulatory CD4+-T-cell differentiation. Clin. Exp. Rheumatol. 2020, 39, 279–288. [Google Scholar]
- Heatwole, C.R.; Miller, J.; Martens, B.; Moxley, R.T., 3rd. Laboratory abnormalities in ambulatory patients with myotonic dystrophy type 1. Arch. Neurol. 2006, 63, 1149–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikolic-Kokic, A.; Marinkovic, D.; Peric, S.; Stevic, Z.; Spasic, M.B.; Blagojevic, D.; Rakoc evic-Stojanovic, V. Redox imbalance in peripheral blood of type 1 myotonic dystrophy patients. Redox Rep. Commun. Free. Radic. Res. 2016, 21, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Nakamori, M.; Hamanaka, K.; Thomas, J.D.; Wang, E.T.; Hayashi, Y.K.; Takahashi, M.P.; Swanson, M.S.; Nishino, I.; Mochizuki, H. Aberrant Myokine Signaling in Congenital Myotonic Dystrophy. Cell Rep. 2017, 21, 1240–1252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tidball, J.G.; Welc, S.S.; Wehling-Henricks, M. Immunobiology of Inherited Muscular Dystrophies. Compr. Physiol. 2018, 8, 1313–1356. [Google Scholar] [CrossRef] [PubMed]
- Salucci, S.; Bartoletti Stella, A.; Battistelli, M.; Burattini, S.; Bavelloni, A.; Cocco, L.I.; Gobbi, P.; Faenza, I. How Inflammation Pathways Contribute to Cell Death in Neuro-Muscular Disorders. Biomolecules 2021, 11, 1109. [Google Scholar] [CrossRef] [PubMed]
- Villalta, S.A.; Nguyen, H.X.; Deng, B.; Gotoh, T.; Tidball, J.G. Shifts in macrophage phenotypes and macrophage competition for arginine metabolism affect the severity of muscle pathology in muscular dystrophy. Hum. Mol. Genet. 2009, 18, 482–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darras, B.T.; Urion, D.K.; Ghosh, P.S. Dystrophinopathies. In GeneReviews(®); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Mirzaa, G.M., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993; revised 2022. [Google Scholar]
- Long, K.; Danoff, S.K. Interstitial Lung Disease in Polymyositis and Dermatomyositis. Clin. Chest Med. 2019, 40, 561–572. [Google Scholar] [CrossRef]
- Arahata, K.; Ishihara, T.; Fukunaga, H.; Orimo, S.; Lee, J.H.; Goto, K.; Nonaka, I. Inflammatory response in facioscapulohumeral muscular dystrophy (FSHD): Immunocytochemical and genetic analyses. Muscle Nerve Suppl. 1995, 18, S56–S66. [Google Scholar] [CrossRef]
- McKinney, E.F.; Lee, J.C.; Jayne, D.R.; Lyons, P.A.; Smith, K.G. T-cell exhaustion, co-stimulation and clinical outcome in autoimmunity and infection. Nature 2015, 523, 612–616. [Google Scholar] [CrossRef] [Green Version]
- Carney, E.F. Immunology: T-cell exhaustion limits immune reactivity and is associated with good prognosis in autoimmune disease. Nat. Rev. Rheumatol. 2015, 11, 501. [Google Scholar] [CrossRef]
- Huang, Y.P.; Perrin, L.H.; Miescher, P.A.; Zubler, R.H. Correlation of T and B cell activities in vitro and serum IL-2 levels in systemic lupus erythematosus. J. Immunol. 1988, 141, 827–833. [Google Scholar] [PubMed]
- Lin, L.; Peng, S.L. Coordination of NF-kappaB and NFAT antagonism by the forkhead transcription factor Foxd1. J. Immunol. 2006, 176, 4793–4803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, J.; Kirou, K.; Lee, C.; Crow, M.K. Functional assay of type I interferon in systemic lupus erythematosus plasma and association with anti-RNA binding protein autoantibodies. Arthritis Rheum. 2006, 54, 1906–1916. [Google Scholar] [CrossRef]
- Sandhu, S.K.; Volinia, S.; Costinean, S.; Galasso, M.; Neinast, R.; Santhanam, R.; Parthun, M.R.; Perrotti, D.; Marcucci, G.; Garzon, R.; et al. miR-155 targets histone deacetylase 4 (HDAC4) and impairs transcriptional activity of B-cell lymphoma 6 (BCL6) in the Eμ-miR-155 transgenic mouse model. Proc. Natl. Acad. Sci. USA 2012, 109, 20047–20052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Vanpouille-Box, C. BAX and BAK at the Gates of Innate Immunity. Trends Cell Biol. 2018, 28, 343–345. [Google Scholar] [CrossRef]
- de Las Heras-Saldana, S.; Chung, K.Y.; Lee, S.H.; Gondro, C. Gene expression of Hanwoo satellite cell differentiation in longissimus dorsi and semimembranosus. BMC Genom. 2019, 20, 156. [Google Scholar] [CrossRef] [Green Version]
- Rajan, S.; Chu Pham Dang, H.; Djambazian, H.; Zuzan, H.; Fedyshyn, Y.; Ketela, T.; Moffat, J.; Hudson, T.J.; Sladek, R. Analysis of early C2C12 myogenesis identifies stably and differentially expressed transcriptional regulators whose knock-down inhibits myoblast differentiation. Physiol. Genom. 2012, 44, 183–197. [Google Scholar] [CrossRef] [Green Version]
- Londhe, P.; Davie, J.K. Interferon-γ resets muscle cell fate by stimulating the sequential recruitment of JARID2 and PRC2 to promoters to repress myogenesis. Sci. Signal. 2013, 6, ra107. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Takakura, N.; Oike, Y.; Imanaka, T.; Araki, K.; Suda, T.; Kaname, T.; Kondo, T.; Abe, K.; Yamamura, K. Defective smooth muscle development in qkI-deficient mice. Dev. Growth Differ. 2003, 45, 449–462. [Google Scholar] [CrossRef]
- van der Veer, E.P.; de Bruin, R.G.; Kraaijeveld, A.O.; de Vries, M.R.; Bot, I.; Pera, T.; Segers, F.M.; Trompet, S.; van Gils, J.M.; Roeten, M.K.; et al. Quaking, an RNA-binding protein, is a critical regulator of vascular smooth muscle cell phenotype. Circ. Res. 2013, 113, 1065–1075. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Wang, C.; Miao, X.; Wu, Y.; Yuan, J.; Ding, M.; Li, J.; Shi, Z. Age-Related Insulin-Like Growth Factor Binding Protein-4 Overexpression Inhibits Osteogenic Differentiation of Rat Mesenchymal Stem Cells. Cell. Physiol. Biochem. 2017, 42, 640–650. [Google Scholar] [CrossRef] [PubMed]
- Messina, S.; Mazzeo, A.; Bitto, A.; Aguennouz, M.; Migliorato, A.; De Pasquale, M.G.; Minutoli, L.; Altavilla, D.; Zentilin, L.; Giacca, M.; et al. VEGF overexpression via adeno-associated virus gene transfer promotes skeletal muscle regeneration and enhances muscle function in mdx mice. FASEB J. 2007, 21, 3737–3746. [Google Scholar] [CrossRef] [Green Version]
- Fröhlich, T.; Kemter, E.; Flenkenthaler, F.; Klymiuk, N.; Otte, K.A.; Blutke, A.; Krause, S.; Walter, M.C.; Wanke, R.; Wolf, E.; et al. Progressive muscle proteome changes in a clinically relevant pig model of Duchenne muscular dystrophy. Sci. Rep. 2016, 6, 33362. [Google Scholar] [CrossRef]
- Heredia, J.E.; Mukundan, L.; Chen, F.M.; Mueller, A.A.; Deo, R.C.; Locksley, R.M.; Rando, T.A.; Chawla, A. Type 2 innate signals stimulate fibro/adipogenic progenitors to facilitate muscle regeneration. Cell 2013, 153, 376–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunt, L.C.; Xu, B.; Finkelstein, D.; Fan, Y.; Carroll, P.A.; Cheng, P.F.; Eisenman, R.N.; Demontis, F. The glucose-sensing transcription factor MLX promotes myogenesis via myokine signaling. Genes Dev. 2015, 29, 2475–2489. [Google Scholar] [CrossRef] [Green Version]
- Bulaklak, K.; Xiao, B.; Qiao, C.; Li, J.; Patel, T.; Jin, Q.; Li, J.; Xiao, X. MicroRNA-206 Downregulation Improves Therapeutic Gene Expression and Motor Function in mdx Mice. Mol. Ther. Nucleic Acids 2018, 12, 283–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.D.; Guo, Q.; Quan, A.; Lopez, J.; Alonso-Escalante, J.C.; Lough, D.M.; Lee, W.P.A.; Brandacher, G.; Kumar, A.R. Vascular Endothelial Growth Factor Induction of Muscle-Derived Stem Cells Enhances Vascular Phenotype While Preserving Myogenic Potential. Ann. Plast. Surg. 2017, 79, 404–409. [Google Scholar] [CrossRef]
- Frost, R.A.; Nystrom, G.J.; Lang, C.H. Lipopolysaccharide regulates proinflammatory cytokine expression in mouse myoblasts and skeletal muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2002, 283, R698–R709. [Google Scholar] [CrossRef]
- Eghtesad, S.; Jhunjhunwala, S.; Little, S.R.; Clemens, P.R. Rapamycin ameliorates dystrophic phenotype in mdx mouse skeletal muscle. Mol. Med. 2011, 17, 917–924. [Google Scholar] [CrossRef]
- Foltz, S.J.; Luan, J.; Call, J.A.; Patel, A.; Peissig, K.B.; Fortunato, M.J.; Beedle, A.M. Four-week rapamycin treatment improves muscular dystrophy in a fukutin-deficient mouse model of dystroglycanopathy. Skelet. Muscle 2016, 6, 20. [Google Scholar] [CrossRef] [Green Version]
- Kawakami, Y.; Hambright, W.S.; Takayama, K.; Mu, X.; Lu, A.; Cummins, J.H.; Matsumoto, T.; Yurube, T.; Kuroda, R.; Kurosaka, M.; et al. Rapamycin Rescues Age-Related Changes in Muscle-Derived Stem/Progenitor Cells from Progeroid Mice. Mol. Ther. Methods Clin. Dev. 2019, 14, 64–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meinke, P.; Hintze, S.; Limmer, S.; Schoser, B. Myotonic Dystrophy-A Progeroid Disease? Front. Neurol. 2018, 9, 601. [Google Scholar] [CrossRef] [PubMed]
- Vainshtein, A.; Sandri, M. Signaling Pathways That Control Muscle Mass. Int. J. Mol. Sci. 2020, 21, 4759. [Google Scholar] [CrossRef]
- Blau, H.M.; Cosgrove, B.D.; Ho, A.T. The central role of muscle stem cells in regenerative failure with aging. Nat. Med. 2015, 21, 854–862. [Google Scholar] [CrossRef] [Green Version]
- Sousa-Victor, P.; García-Prat, L.; Serrano, A.L.; Perdiguero, E.; Muñoz-Cánoves, P. Muscle stem cell aging: Regulation and rejuvenation. Trends Endocrinol. Metab. TEM 2015, 26, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Merritt, E.K.; Stec, M.J.; Thalacker-Mercer, A.; Windham, S.T.; Cross, J.M.; Shelley, D.P.; Craig Tuggle, S.; Kosek, D.J.; Kim, J.S.; Bamman, M.M. Heightened muscle inflammation susceptibility may impair regenerative capacity in aging humans. J. Appl. Physiol. 2013, 115, 937–948. [Google Scholar] [CrossRef] [PubMed]
- Nieuwenhuis, S.; Okkersen, K.; Widomska, J.; Blom, P.; t Hoen, P.A.C.; van Engelen, B.; Glennon, J.C. Insulin Signaling as a Key Moderator in Myotonic Dystrophy Type 1. Front. Neurol. 2019, 10, 1229. [Google Scholar] [CrossRef]
- Mudali, S.; Dobs, A.S. Effects of testosterone on body composition of the aging male. Mech. Ageing Dev. 2004, 125, 297–304. [Google Scholar] [CrossRef]
- Seidman, S.N. Androgens and the aging male. Psychopharmacol. Bull. 2007, 40, 205–218. [Google Scholar]
- Antonini, G.; Clemenzi, A.; Bucci, E.; De Marco, E.; Morino, S.; Di Pasquale, A.; Latino, P.; Ruga, G.; Lenzi, A.; Vanacore, N.; et al. Hypogonadism in DM1 and its relationship to erectile dysfunction. J. Neurol. 2011, 258, 1247–1253. [Google Scholar] [CrossRef]
- Mateos-Aierdi, A.J.; Goicoechea, M.; Aiastui, A.; Fernandez-Torron, R.; Garcia-Puga, M.; Matheu, A.; Lopez de Munain, A. Muscle-wasting in myotonic dystrophies: A model of premature aging. Front. Aging Neurosci. 2015, 7, 125. [Google Scholar] [CrossRef] [PubMed]
- Campione, E.; Botta, A.; Di Prete, M.; Rastelli, E.; Gibellini, M.; Petrucci, A.; Bernardini, S.; Novelli, G.; Bianchi, L.; Orlandi, A.; et al. Cutaneous features of myotonic dystrophy types 1 and 2: Implication of premature aging and vitamin D homeostasis. Neuromuscul. Disord. 2017, 27, 163–169. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Available online: http://broadinstitute.github.io/picard/ (accessed on 14 May 2021).
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, W.; Carey, V.J.; Gentleman, R.; Anders, S.; Carlson, M.; Carvalho, B.S.; Bravo, H.C.; Davis, S.; Gatto, L.; Girke, T.; et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods 2015, 12, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Carvalho, B.S.; Irizarry, R.A. A framework for oligonucleotide microarray preprocessing. Bioinformatics 2010, 26, 2363–2367. [Google Scholar] [CrossRef]
- Bolstad, B.M.; Irizarry, R.A.; Astrand, M.; Speed, T.P. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 2003, 19, 185–193. [Google Scholar] [CrossRef] [Green Version]
- Krämer, A.; Green, J.; Pollard, J., Jr.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef]
- Newman, M.E. Modularity and community structure in networks. Proc. Natl. Acad. Sci. USA 2006, 103, 8577–8582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okkersen, K.; Jimenez-Moreno, C.; Wenninger, S.; Daidj, F.; Glennon, J.; Cumming, S.; Littleford, R.; Monckton, D.G.; Lochmuller, H.; Catt, M.; et al. Cognitive behavioural therapy with optional graded exercise therapy in patients with severe fatigue with myotonic dystrophy type 1: A multicentre, single-blind, randomised trial. Lancet Neurol. 2018, 17, 671–680. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group Size (n) | Age (Year) | Age at Disease Onset (Year) | Male | Female | MIRS (1–5) | Modal CTG Repeat Length | |
|---|---|---|---|---|---|---|---|
| OPTIMISTIC gr. 1 | 10 | 48.5 (8.3) | 40.2 (12.1) | 4 (40%) | 6 (60%) | 1.9 (1–2) | 179.8 (114.6) |
| OPTIMISTIC gr. 2 | 10 | 48.3 (2.1) | 34.1 (8.1) | 6 (60%) | 4 (40%) | 3.8 (3–5) | 291.0 (71.2) |
| OPTIMISTIC gr. 3 | 10 | 49.0 (1.9) | 27.6 (10.0) 1 | 4 (40%) | 6 (60%) | 3.9 (3–5) | 728.5 (228.4) |
| DMBDI group 1 | 6 | 45.5 (14.0) | 41.5 (13.2) 2 | 3 (50%) | 3 (50%) | 1.5 (1–2) | 209.3 (252.8) |
| DMBDI group 2 | 6 | 46.3 (9.5) | 22.7 (13.6) | 2 (33.3) | 4 (66.7%) | 3.2 (3–5) | 252.8 (77.1) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nieuwenhuis, S.; Widomska, J.; Blom, P.; ‘t Hoen, P.-B.A.C.; van Engelen, B.G.M.; Glennon, J.C.; on behalf of the OPTIMISTIC Consortium. Blood Transcriptome Profiling Links Immunity to Disease Severity in Myotonic Dystrophy Type 1 (DM1). Int. J. Mol. Sci. 2022, 23, 3081. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23063081
Nieuwenhuis S, Widomska J, Blom P, ‘t Hoen P-BAC, van Engelen BGM, Glennon JC, on behalf of the OPTIMISTIC Consortium. Blood Transcriptome Profiling Links Immunity to Disease Severity in Myotonic Dystrophy Type 1 (DM1). International Journal of Molecular Sciences. 2022; 23(6):3081. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23063081
Chicago/Turabian StyleNieuwenhuis, Sylvia, Joanna Widomska, Paul Blom, Peter-Bram A. C. ‘t Hoen, Baziel G. M. van Engelen, Jeffrey C. Glennon, and on behalf of the OPTIMISTIC Consortium. 2022. "Blood Transcriptome Profiling Links Immunity to Disease Severity in Myotonic Dystrophy Type 1 (DM1)" International Journal of Molecular Sciences 23, no. 6: 3081. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23063081