
Synthesis of Multiple Bispecific Antibody Formats with Only One Single Enzyme Based on Enhanced Trypsiligase †

Abstract
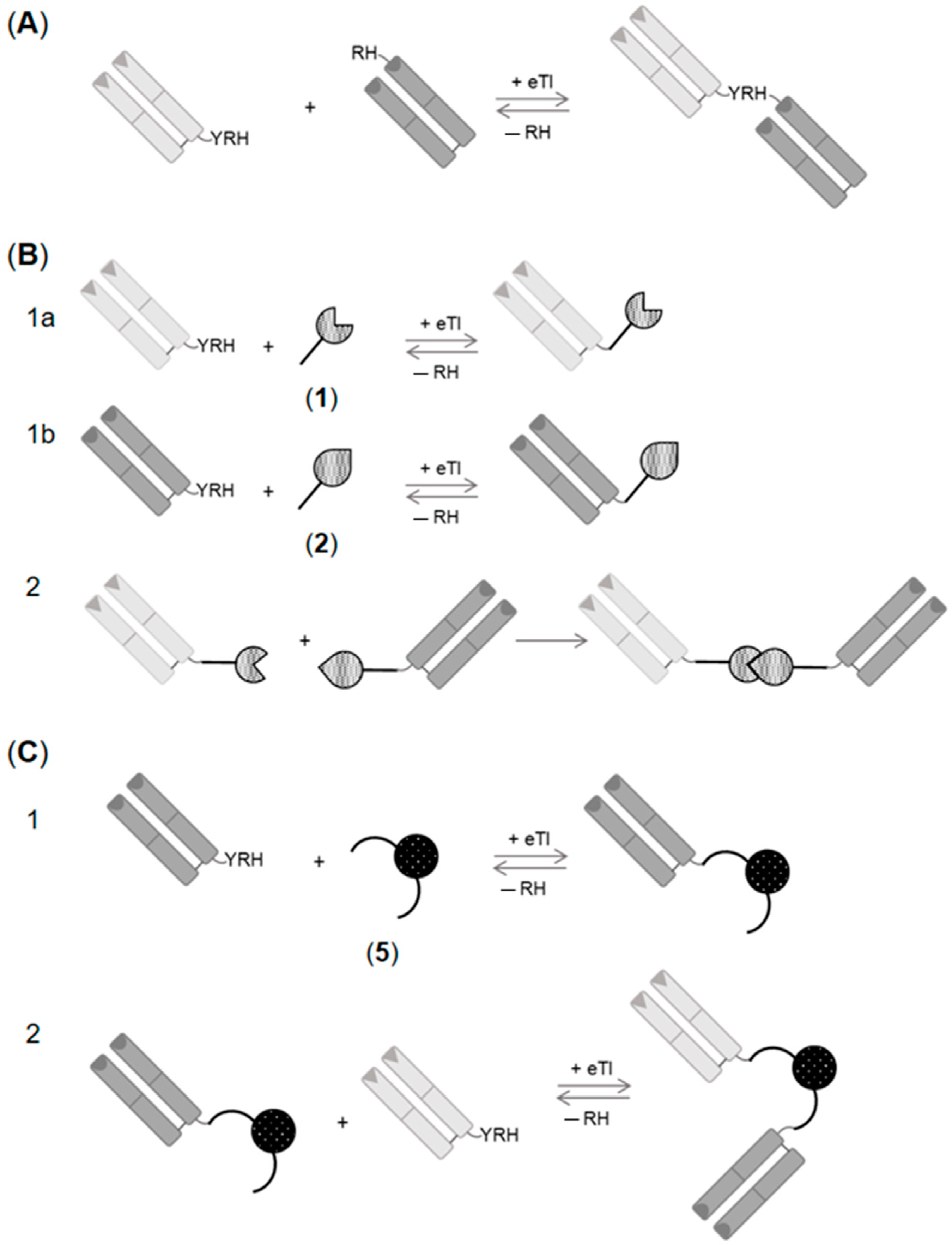
:1. Introduction
2. Results and Discussion
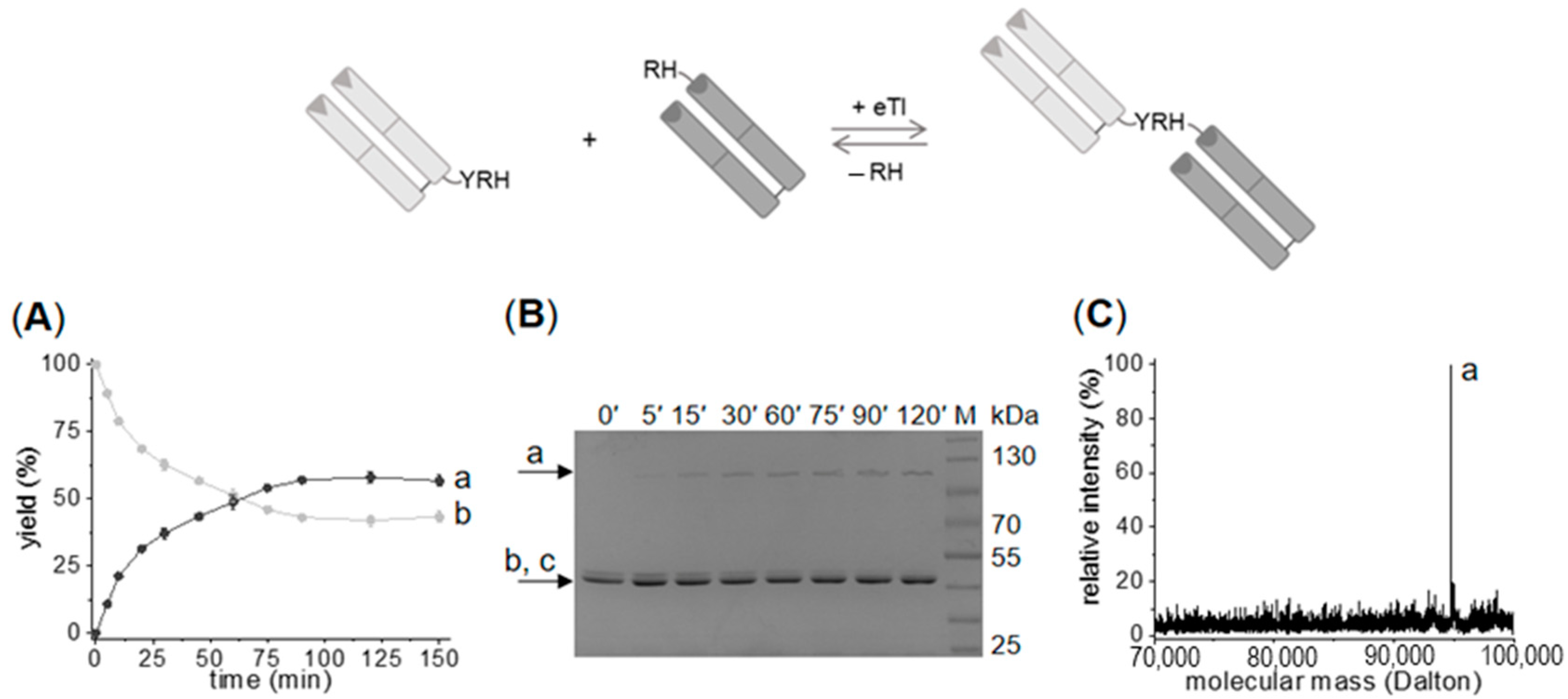
2.1. Enzymatic Synthesis of C- to N-Linked Anti-ErbB3-Anti-ErbB2-bsFab
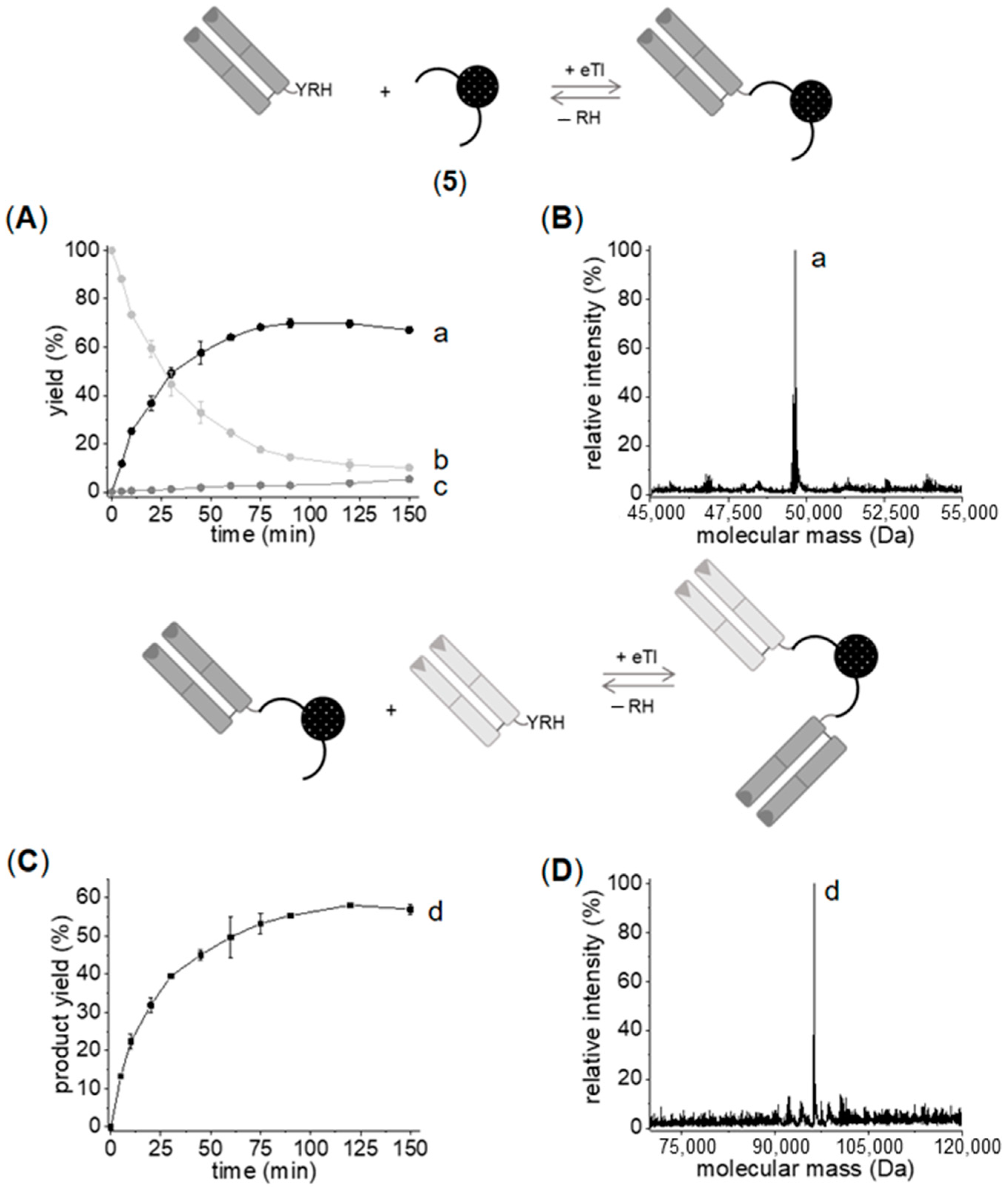
2.2. Generation of C- to C-Linked Anti-ErbB2-Anti-ErbB3-bsFab
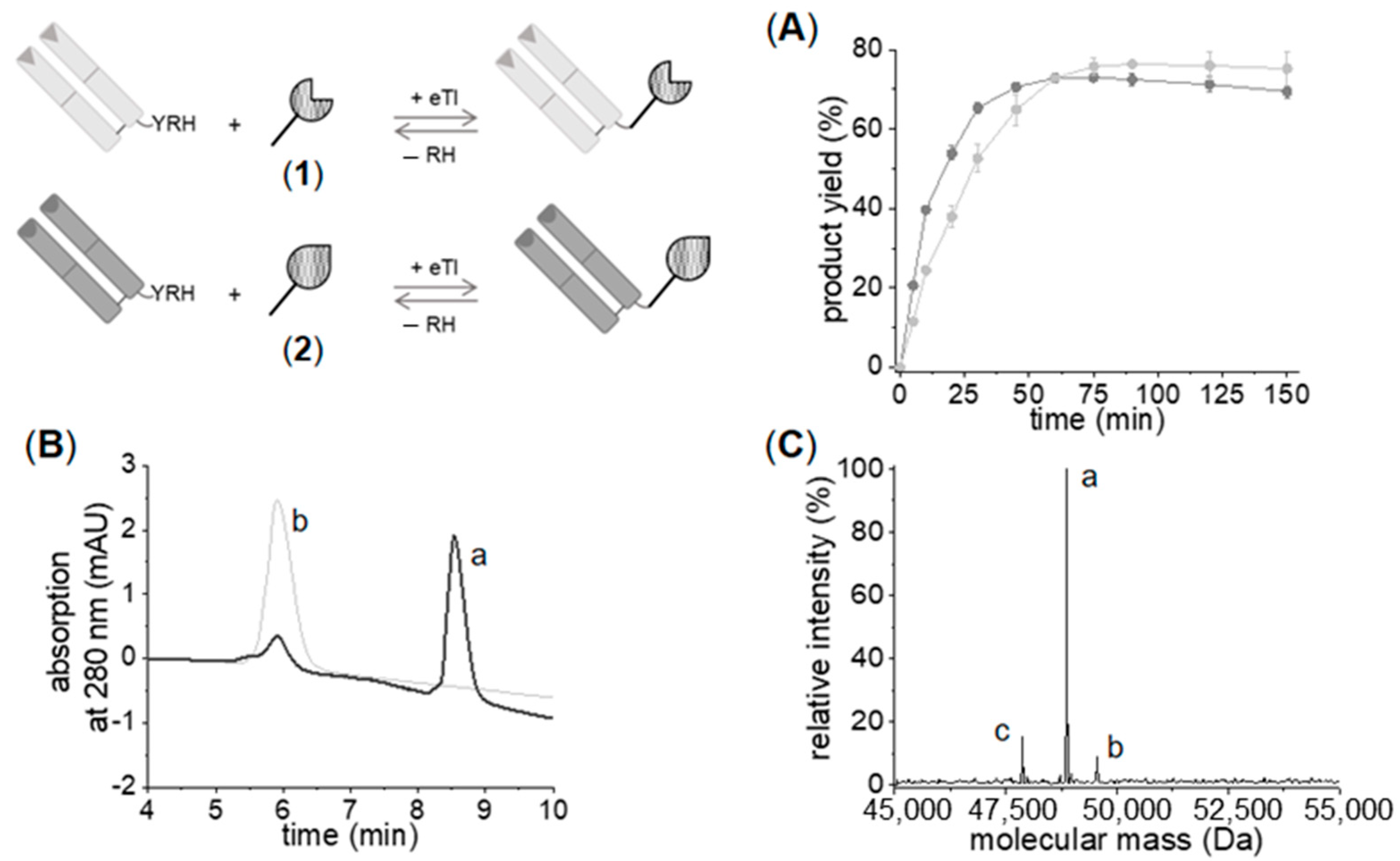
2.2.1. Chemo–Enzymatic Synthesis of C- to C-Linked Anti-ErbB2-Anti-ErbB3-bsFab
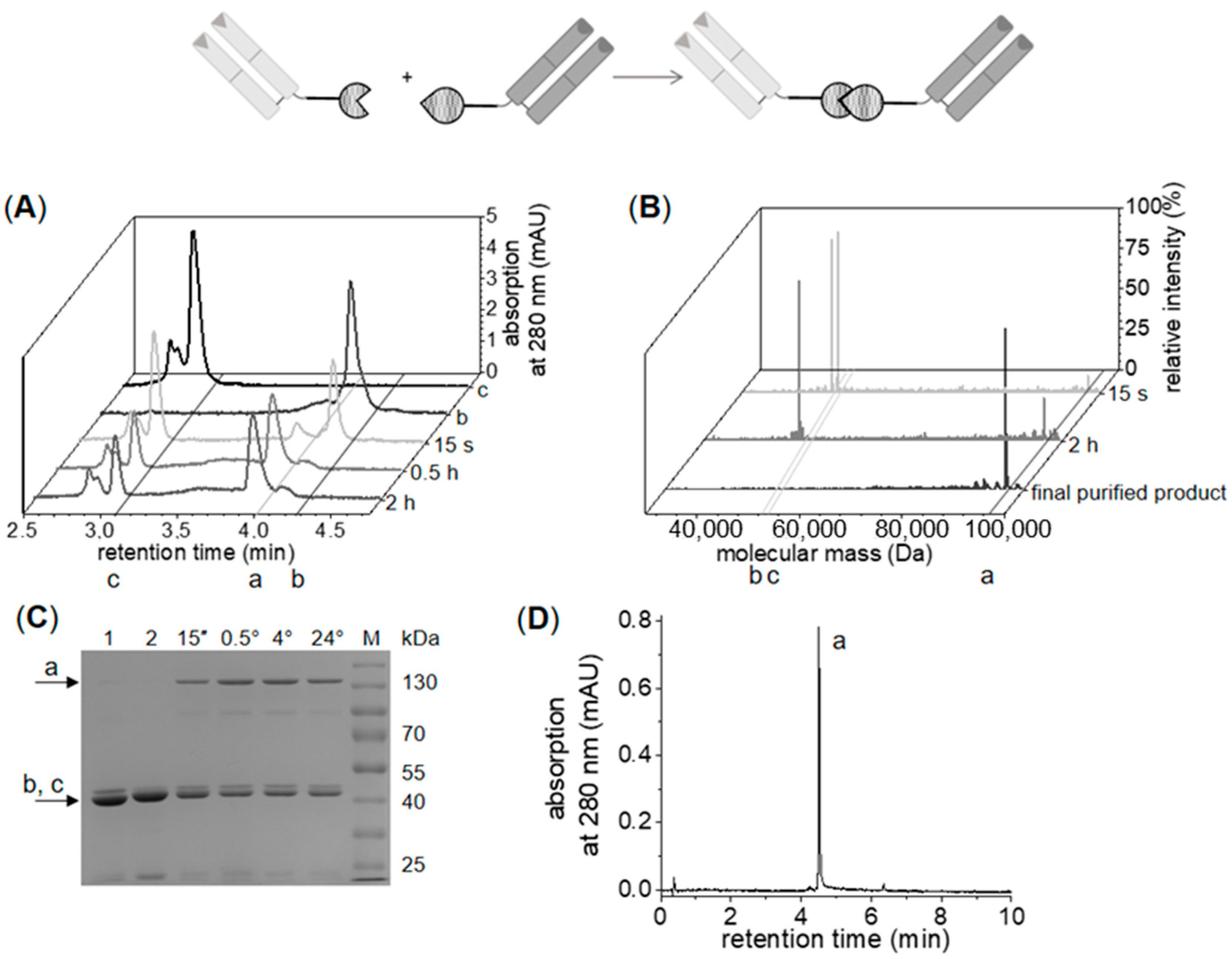
2.2.2. Enzymatic Synthesis of C- to C-Linked Anti-ErbB2-Anti-ErbB3-bsFab
2.3. Analysis of In Vitro Functionality of the Generated bsFabs
2.3.1. Surface Plasmon Resonance (SPR)-Based Activity Assay
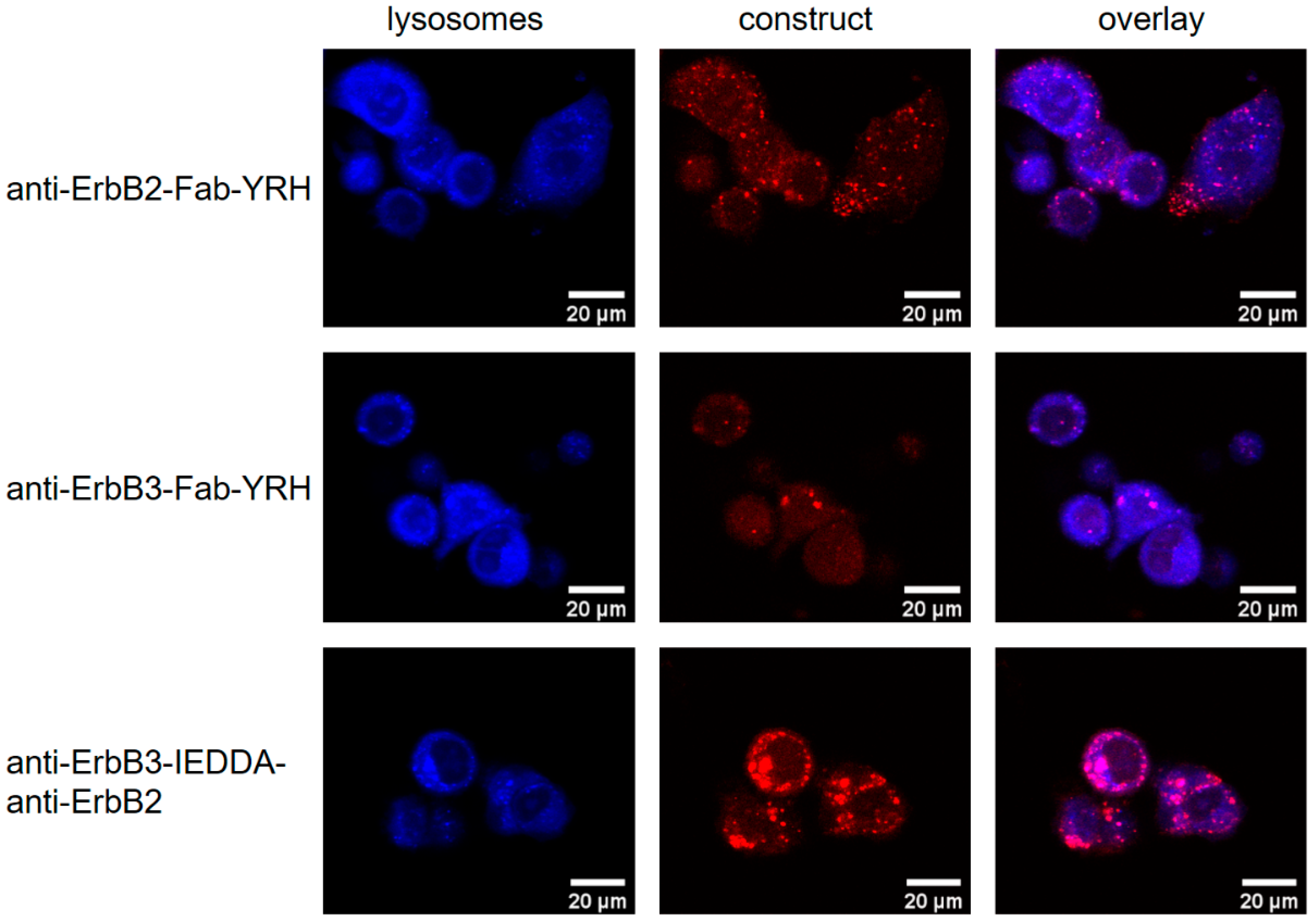
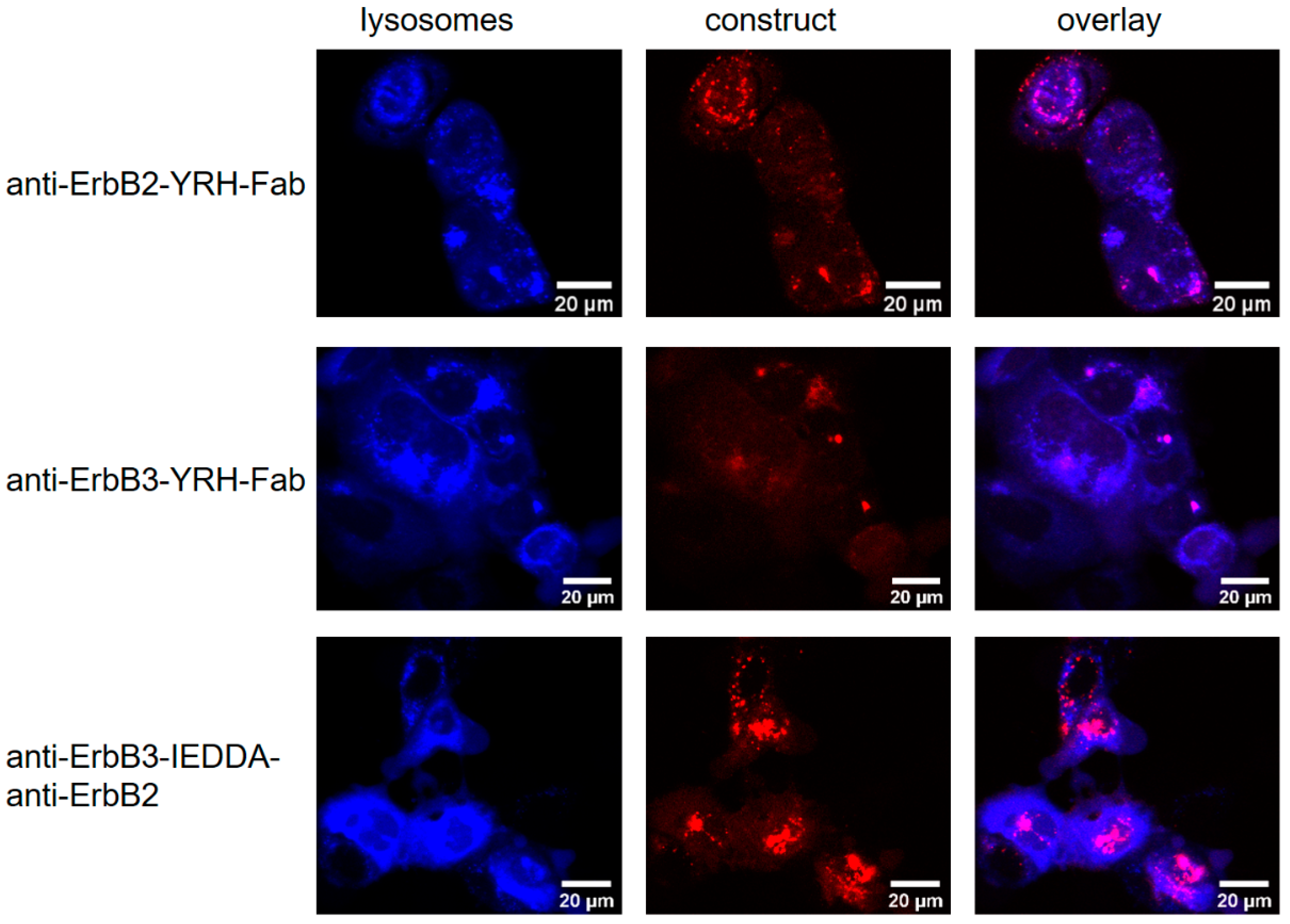
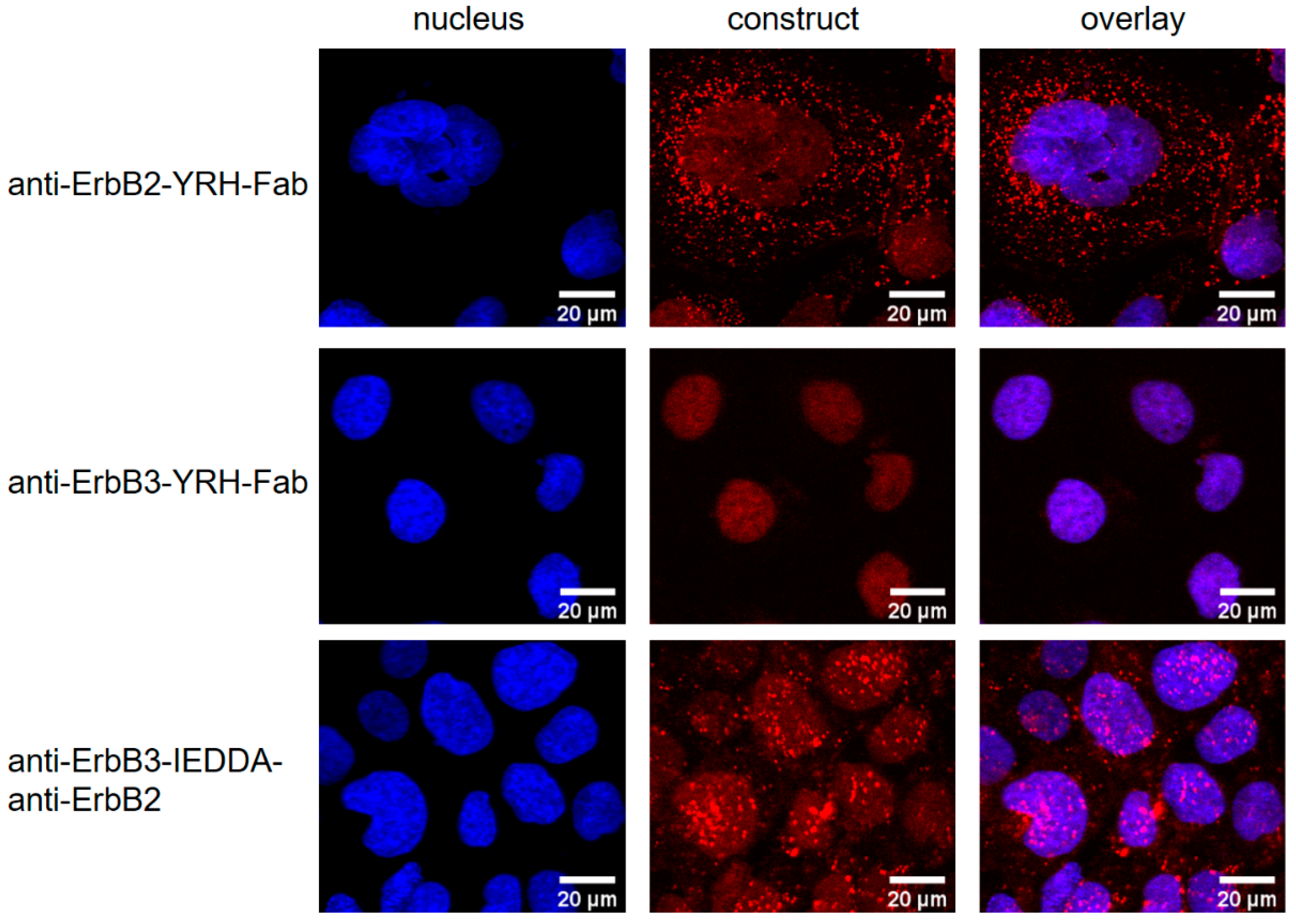
2.3.2. Receptor Internalization Assay
3. Materials and Methods
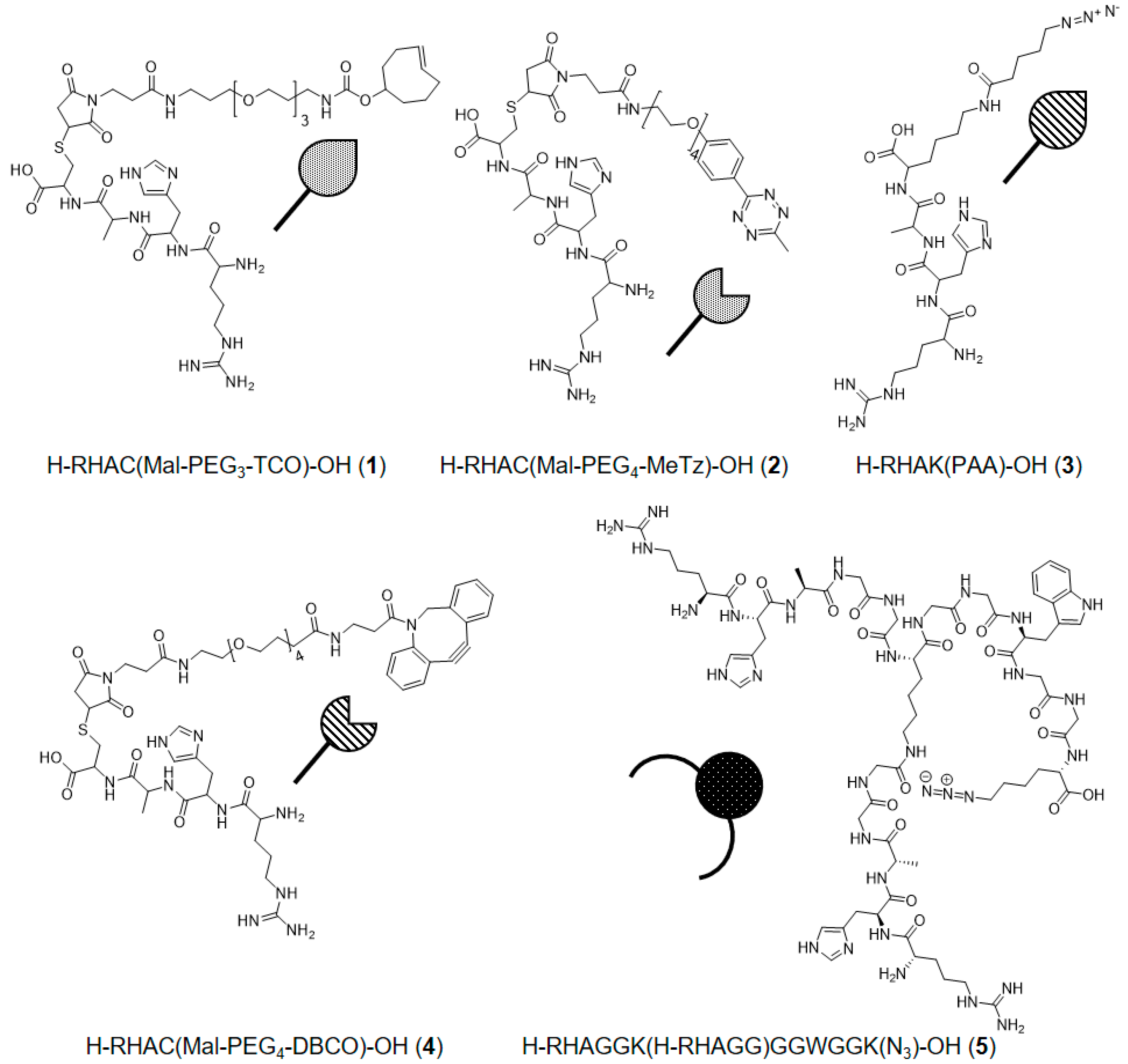
3.1. Chemicals and Peptide Synthesis
3.2. Production of eTl
3.3. Production of Fabs
3.4. C-Terminal Modification of Fabs by eTl
3.5. Chemo–Enzymatic Production of bsFabs
3.6. Enzymatic Production of C- to C- and C- to N-Terminal bsFabs
3.7. SPR-Based Activity Assay
3.8. Receptor Internalization Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Labrijn, A.F.; Janmaat, M.L.; Reichert, J.M.; Parren, P.W.H.I. Bispecific antibodies: A mechanistic review of the pipeline. Nat. Rev. Drug Discov. 2019, 18, 585–608. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, U.; Kontermann, R.E. The making of bispecific antibodies. mAbs 2017, 9, 182–212. [Google Scholar] [CrossRef]
- Ma, J.; Mo, Y.; Tang, M.; Shen, J.; Qi, Y.; Zhao, W.; Huang, Y.; Xu, Y.; Qian, C. Bispecific Antibodies: From Research to Clinical Application. Front. Immunol. 2021, 12, 626616. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.M.; Pyo, K.H.; Soo, R.A.; Cho, B.C. The promise of bispecific antibodies: Clinical applications and challenges. Cancer Treat. Rev. 2021, 99, 102240. [Google Scholar] [CrossRef] [PubMed]
- Milstein, C.; Cuello, A. Hybrid hybridomas and their use in immunohistochemistry. Nature 1983, 305, 537–540. [Google Scholar] [CrossRef] [PubMed]
- Milstein, C.; Cuello, A. Hybrid hybridomas and the production of bi-specific monoclonal antibodies. Immunol. Today 1984, 5, 299–304. [Google Scholar] [CrossRef]
- Suresh, M.R.; Cuello, A.C.; Milstein, C. Advantages of bispecific hybridomas in one-step immunocytochemistry and immunoassays. Proc. Natl. Acad. Sci. USA 1986, 83, 7989–7993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mack, M.; Riethmüller, G.; Kufer, P. A small bispecific antibody construct expressed as a functional single-chain molecule with high tumor cell cytotoxicity. Proc. Natl. Acad. Sci. USA 1995, 92, 7021–7025. [Google Scholar] [CrossRef] [Green Version]
- Ridgway, J.B.; Presta, L.G.; Carter, P. ‘Knobs-into-holes’ engineering of antibody CH3 domains for heavy chain heterodimerization. Protein Eng. Des. Sel. 1996, 9, 617–621. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; Ying, H.; Grinnell, C.; Bryant, S.; Miller, R.; Clabbers, A.; Bose, S.; McCarthy, D.; Zhu, R.-R.; Santora, L.; et al. Simultaneous targeting of multiple disease mediators by a dual-variable-domain immunoglobulin. Nat. Biotechnol. 2007, 25, 1290–1297. [Google Scholar] [CrossRef]
- Wu, P.; Shui, W.; Carlson, B.L.; Hu, N.; Rabuka, D.; Lee, J.; Bertozzi, C.R. Site-specific chemical modification of recombinant proteins produced in mammalian cells by using the genetically encoded aldehyde tag. Proc. Natl. Acad. Sci. USA 2009, 106, 3000–3005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiGiammarino, E.L.; Harlan, J.E.; Walter, K.A.; Ladror, U.S.; Edalji, R.P.; Hutchins, C.W.; Lake, M.R.; Greischar, A.J.; Liu, J.; Ghayur, T.; et al. Ligand association rates to the inner-variable-domain of a dual-variable-domain immunoglobulin are significantly impacted by linker design. mAbs 2011, 3, 487–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinmetz, A.; Vallée, F.O.; Beil, C.; Lange, C.; Baurin, N.; Beninga, J.; Capdevila, C.; Corvey, C.; Dupuy, A.; Ferrari, P.; et al. CODV-Ig, a universal bispecific tetravalent and multifunctional immunoglobulin format for medical applications. mAbs 2016, 8, 867–878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falck, G.; Müller, K.M. Enzyme-Based Labeling Strategies for Antibody–Drug Conjugates and Antibody Mimetics. Antibodies 2018, 7, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Lee, S.; Kim, Y.; Yoo, T.H. Methods to generate site-specific conjugates of antibody and protein. Bioorg. Med. Chem. 2021, 30, 115946. [Google Scholar] [CrossRef] [PubMed]
- Zang, B.; Ren, J.; Li, D.; Huang, C.; Ma, H.; Peng, Q.; Ji, F.; Han, L.; Jia, L. Freezing-assisted synthesis of covalent C-C linked bivalent and bispecific nanobodies. Org. Biomol. Chem. 2019, 17, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Harmand, T.J.; Bousbaine, D.; Chan, A.; Zhang, X.; Liu, D.R.; Tam, J.P.; Ploegh, H.L. One-Pot Dual Labeling of IgG 1 and Preparation of C-to-C Fusion Proteins Through a Combination of Sortase A and Butelase 1. Bioconjug. Chem. 2019, 29, 3245–3249. [Google Scholar] [CrossRef] [PubMed]
- Plagmann, I.; Chalaris, A.; Kruglov, A.A.; Nedospasov, S.; Rosenstiel, P.; Rose-John, S.; Scheller, J. Transglutaminase-catalyzed covalent multimerization of camelidea anti-human TNF single domain antibodies improves neutralizing activity. J. Biotechnol. 2009, 142, 170–178. [Google Scholar] [CrossRef]
- Bartels, L.; Ploegh, H.L.; Spits, H.; Wagner, K. Preparation of bispecific antibody-protein adducts by site-specific chemo-enzymatic conjugation. Methods 2019, 154, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Berteau, O.; Guillot, A.; Benjdia, A.; Rabot, S. A new type of bacterial sulfatase reveals a novel maturation pathway in prokaryotes. J. Biol. Chem. 2006, 281, 22464–22470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holder, P.G.; Jones, L.C.; Drake, P.M.; Barfield, R.M.; Banas, S.; de Hart, G.W.; Baker, J.; Rabuka, D. Reconstitution of formylglycine-generating enzyme with copper (II) for aldehyde tag conversion. J. Biol. Chem. 2015, 290, 15730–15745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liebscher, S.; Schöpfel, M.; Aumüller, T.; Sharkhuukhen, A.; Pech, A.; Höss, E.; Parthier, C.; Jahreis, G.; Stubbs, M.T.; Bordusa, F. N-terminal protein modification by substrate-activated reverse proteolysis. Angew. Chem. Int. Ed. Engl. 2014, 53, 3024–3028. [Google Scholar] [CrossRef] [PubMed]
- Liebscher, S.; Kornberger, P.; Fink, G.; Trost-Gross, E.M.; Höss, E.; Skerra, A.; Bordusa, F. Derivatization of antibody Fab fragments: A designer enzyme for native protein modification. Chembiochem 2014, 15, 1096–1100. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.; Liebscher, S.; Bordusa, F. Selective Coupling of Click Anchors to Proteins via Trypsiligase. Bioconjug. Chem. 2016, 27, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Wartner, R.; Böhme, M.; Bordusa, F.; Simon, A.H.; Richter, T. Trypsin Variants with Improved Enzymatic Properties. WO 2020/127808 A1, 25 June 2020. [Google Scholar]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem. Int. Ed. Engl. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Oliveira, B.L.; Guo, Z.; Bernardes, G.J.L. Inverse electron demand Diels-Alder reactions in chemical biology. Chem. Soc. Rev. 2017, 46, 4895–4950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dommerholt, J.; Rutjes, F.P.J.T.; van Delft, F.L. Strain-Promoted 1,3-Dipolar Cycloaddition of Cycloalkynes and Organic Azides. Top. Curr. Chem. 2016, 374, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slamon, D.J.; Clark, G.M.; Wong, S.G.; Levin, W.J.; Ullrich, A.; McGuire, W.L. Human breast cancer: Correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 1987, 235, 177–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slamon, D.J.; Godolphin, W.; Jones, L.A.; Holt, J.A.; Wong, S.G.; Keith, D.E.; Levin, W.J.; Stuart, S.G.; Udove, J.; Ullrich, A. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science 1989, 244, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Wallasch, C.; Weiss, F.; Niederfellner, G.; Jallal, B.; Issing, W.; Ullrich, A. Heregulin-dependent regulation of HER2/neu oncogenic signaling by heterodimerization with HER3. EMBO J. 1995, 14, 4267–4275. [Google Scholar] [CrossRef] [PubMed]
- Pinkas-Kramarski, R.; Soussan, L.; Waterman, H.; Levkowitz, G.; Alroy, I.; Klapper, L.; Lavi, S.; Seger, R.; Ratzkin, B.J.; Sela, M. Diversification of Neu differentiation factor and epidermal growth factor signaling by combinatorial receptor interactions. EMBO J. 1996, 15, 2452–2467. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Greenlee, E.B.; Amick, J.R.; Ligon, G.F.; Lillquist, J.S.; Natoli, E.J., Jr.; Hadari, Y.; Alvarado, D.; Schlessinger, J. Inhibition of ErbB3 by a monoclonal antibody that locks the extracellular domain in an inactive configuration. Proc. Natl. Acad. Sci. USA 2015, 112, 13225–13230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Z.; Carrasco, R.A.; Schifferli, K.; Kinneer, K.; Tammali, R.; Chen, H.; Rothstein, R.; Wetzel, L.; Yang, C.; Chowdhury, P.; et al. A Potent HER3 Monoclonal Antibody That Blocks Both Ligand-Dependent and -Independent Activities: Differential Impacts of PTEN Status on Tumor Response. Mol. Cancer Ther. 2016, 15, 689–701. [Google Scholar] [CrossRef] [Green Version]
- Nusinow, D.P.; Szpyt, J.; Ghandi, M.; Rose, C.M.; McDonald, E.R., 3rd; Kalocsay, M.; Jané-Valbuena, J.; Gelfand, E.; Schweppe, D.K.; Jedrychowski, M.; et al. Quantitative Proteomics of the Cancer Cell Line Encyclopedia. Cell 2020, 180, 387–402.e316. [Google Scholar] [CrossRef] [PubMed]
- Bertelsen, V.; Stang, E. The Mysterious Ways of ErbB2/HER2 Trafficking. Membranes 2014, 4, 424–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giri, D.K.; Ali-Seyed, M.; Li, L.-Y.; Lee, D.-F.; Ling, P.; Bartholomeusz, G.; Wang, S.-C.; Hung, M.-C. Endosomal transport of ErbB-2: Mechanism for nuclear entry of the cell surface receptor. Mol. Cell. Biol. 2005, 25, 11005–11018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordo Russo, R.I.; Béguelin, W.; Díaz Flaqué, M.C.; Proietti, C.J.; Venturutti, L.; Galigniana, N.; Tkach, M.; Guzmán, P.; Roa, J.C.; O’Brien, N.A.; et al. Targeting ErbB-2 nuclear localization and function inhibits breast cancer growth and overcomes trastuzumab resistance. Oncogene 2015, 34, 3413–3428. [Google Scholar] [CrossRef] [Green Version]
- Merrifield, R.B. Solid phase peptide synthesis. I. The synthesis of a tetrapeptide. J. Am. Chem. Soc. 1963, 85, 2149–2154. [Google Scholar] [CrossRef]
- Bycroft, B.W.; Chan, W.C.; Chhabra, S.R.; Hone, N.D. A novel lysine-protecting procedure for continuous flow solid phase synthesis of branched peptides. J. Chem. Soc. 1993, 9, 778–779. [Google Scholar] [CrossRef]
- Lin-Cereghino, J.; Wong, W.W.; Xiong, S.; Giang, W.; Luong, L.T.; Vu, J.; Johnson, S.D.; Lin-Cereghino, G.P. Condensed protocol for competent cell preparation and transformation of the methylotrophic yeast Pichia pastoris. BioTechniques 2005, 38, 44–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Lo, H.-W.; Xia, W.; Wei, Y.; Ali-Seyed, M.; Huang, S.-F.; Hung, M.-C. Novel Prognostic Value of Nuclear Epidermal Growth Factor Receptor in Breast Cancer. Cancer Res. 2005, 65, 338. [Google Scholar] [PubMed]
- Schillaci, R.; Guzmán, P.; Cayrol, F.; Beguelin, W.; Díaz Flaqué, M.C.; Proietti, C.J.; Pineda, V.; Palazzi, J.; Frahm, I.; Charreau, E.H.; et al. Clinical relevance of ErbB-2/HER2 nuclear expression in breast cancer. BMC Cancer 2012, 12, 74. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ErbB3-ECD KD (pM) | ErbB2-ECD KD (pM) | |
|---|---|---|
| anti-ErbB3-Fab-YRH | 99 ± 22 | - |
| anti-ErbB2-Fab-YRH | - | 139 ± 31 |
| anti-ErbB2-RH-Fab | - | 137 ± 43 |
| anti-ErbB2-SPAAC-anti-ErbB3 | 73 ± 21 | 130 ± 28 |
| anti-ErbB3-IEDDA-anti-ErbB2 | 63 ± 19 | 113 ± 20 |
| anti-ErbB3-YRH-anti-ErbB2 | 95 ± 45 | 138 ± 46 |
| anti-ErbB2-linker 5-anti-ErbB3 | 97 ± 33 | 108 ± 20 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Voigt, J.; Meyer, C.; Bordusa, F. Synthesis of Multiple Bispecific Antibody Formats with Only One Single Enzyme Based on Enhanced Trypsiligase. Int. J. Mol. Sci. 2022, 23, 3144. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23063144
Voigt J, Meyer C, Bordusa F. Synthesis of Multiple Bispecific Antibody Formats with Only One Single Enzyme Based on Enhanced Trypsiligase. International Journal of Molecular Sciences. 2022; 23(6):3144. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23063144
Chicago/Turabian StyleVoigt, Johanna, Christoph Meyer, and Frank Bordusa. 2022. "Synthesis of Multiple Bispecific Antibody Formats with Only One Single Enzyme Based on Enhanced Trypsiligase" International Journal of Molecular Sciences 23, no. 6: 3144. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23063144